
Abstract
A relative immunosuppression is observed in patients after sepsis, trauma, burns, or any severe insults. It is currently proposed that selected patients will benefit from treatment aimed at boosting their immune systems. However, the host immune response needs to be considered in context with pathogen-type, timing, and mainly tissue specificity. Indeed, the immune status of leukocytes is not universally decreased and their activated status in tissues contributes to organ failure. Accordingly, any new immune-stimulatory therapeutic intervention should take into consideration potentially deleterious effects in some situations.
Similar content being viewed by others

Introduction
Refinements of supportive care of patients with severe sepsis have decreased their overall mortality, but no adjuvant drug therapy has emerged despite strenuous efforts in the field. Twenty years have passed since the first patients with sepsis were included in clinical trials based on the understanding that TNF orchestrates the inflammatory response and should be the target for therapeutic intervention. In response to the failure of therapies aiming to target either the up-stream microbial activators or the effector molecules of the inflammatory cascade, a new concept has emerged of boosting the immune system to counter immunosuppression that develops in patients who survive the initial, hyperinflammatory period of sepsis [1].
Inflammation is a highly sophisticated and complex response that fundamentally ‘aims’ to protect the host. In this review, we argue against the promulgation of what we believe is a misleading perception of sepsis inducing secondary immunosuppression. The possible negative consequences of immune system-boosting therapy are so great that we believe such an approach should be considered with great caution.
It is important to realize that events occurring in patients with sepsis are not a simple dichotomy resulting from a balance between pro-inflammatory and anti-inflammatory mediators. Oversimplification may have led to previous therapeutic failures. Maintaining such an over-simplistic analysis of inflammation may lead to further failed trials, compounding our current state of therapeutic futility in sepsis. By reviewing the immunological events of sepsis, we also attempt to understand why we have already spent over 20 fruitless years trying to reverse it with targeted adjuvant therapies.
Arbitrarily classifying cytokines as pro- or anti-inflammatory mediators is unreliable
Classifying cytokines as pro- or anti-inflammatory has led to oversimplification of the inflammatory response. Consequentially, especially in a disorder as complex as sepsis, simplistic therapeutic approaches have been considered. The inappropriateness of this classification is widely illustrated by studies revealing some paradoxical behaviors of so-called pro- and anti-inflammatory cytokines. In regard to TNF, the prototypic ‘pro-inflammatory’ cytokine, TNF-treated murine macrophages produced less IL-12 and IL-23 after IFNγ and lipopolysaccharide (LPS) stimulation [2]. In vivo, TNF induces extra-adrenal production of immunoregulatory glucocorticoids in the intestinal mucosa during acute intestinal inflammation [3]. Similar observations have been reported for other pro-inflammatory cytokines such as IL-1 and IFNγ. In a dextran sulphate sodium-induced colitis model, mice deficient in IL-1RI signaling showed increased susceptibility to and failed to mount a protective type I interferon response after Toll-like receptor (TLR) 9 ligand administration [4]. Furthermore, IFNγ knockout (KO) mice had significantly greater endotoxin-induced uveitis as compared with wild-type mice, and the injection of murine IFNγ suppressed the severity of endotoxin-induced uveitis in both wild-type and KO mice [5].
Paradoxical properties have also been reported for IL-10, the prototypic anti-inflammatory cytokine. Its pro-inflammatory activity been established in human volunteers receiving endotoxin injection [6]. Our own in vitro studies showed that adherence of human monocytes modulated the effect of IL-10 on expression of 16 genes, including ‘suppressor of cytokine stimulation’ (SOCS) molecules, in the opposite direction as compared with non-adherent cells [7]. These observations illustrate the statement by Moore and colleagues that ‘IL-10 can effect very different outcomes depending on timing, dose, and location of expression. In some scenarios, the expected immuno-suppressive activities are observed, while in others, IL-10 enhances immune or inflammatory responses’ [8].
Among other cytokines classified as anti-inflammatory, transforming growth factor-beta (TGFβ) may behave as pro-inflammatory mediator as TGFβ-transgenic mice are more sensitive to LPS-induced shock [9] and some of its inflammatory activities reflect its capacity to favor the differentiation of T helper (Th)17 and production of the pro-inflammatory IL-17. The classification of non-cytokine inflammatory mediators also relies on an overly simplistic division between pro- and anti-inflammatory properties. This is well illustrated by prostaglandin E2 (PGE2), a key mediator of infectious immunopathology. On one hand, PGE2 induces fever, increases vascular permeability, increases vasodilatation, and causes pain while also inhibiting production of TNF, increasing production of IL-6, inhibiting 5-lipoxygenase and leukotriene A4 generation, and inducing 15-lipoxygenase and the generation of the lipoxins involved in inflammation resolution. On the other hand, PGE2 has inhibitory properties on macrophages, neutrophils, Th1 lymphocytes, natural killer (NK) cells, and cytotoxic lymphocytes but activates mast cells, Th2, Th17, and regulatory T lymphocytes (Treg) [10]. This panoply of PGE2-stimulated events amply demonstrates the inability to simply characterize the activities of this and the other molecules mentioned as pro- or anti-inflammatory.
Sometimes-ambiguous roles of cytokines in infection and sepsis
In an early anti-TNF monoclonal antibody intervention study, a significant improvement in day 3 survival was observed between the antibody-treated group and the placebo group [11]. Although this was not a pre-specified primary outcome, it is interesting to see that the treatment targeting TNF consisting of a single early injection was beneficial within a short period of time after sepsis onset, reinforcing the idea that TNF plays a key deleterious role in the early events of sepsis. Once anti-TNF treatments were better targeted to the sickest patients by adding biological inclusion parameters (plasma IL-6 level), survival was significantly improved on day 28 [12].
Synergistic effects between immune modulators are a key characteristic of their effect. This explains how a non-lethal dose of one cytokine can lead to mortality when injected with a non-lethal dose of another cytokine. Similarly, it may explain how the removal of some inflammatory mediators by coupled plasma filtration-adsorption was protective in an endotoxin-shock model while levels of circulating bio-active TNF were unaffected [13].
Clear demonstrations of cytokine-mediated tissue damage exist. Nevertheless, because of their ambiguous role mentioned above, identification of their precise role during sepsis has led to controversy. In animal models of sepsis, the role of TNF may vary depending upon the type of infection [14]. Many model parameters influence conclusions of the relative role of the different mediators studied. Identical cytokines have been found to be protective or deleterious depending upon the model. This has been the case for IFNγ [15] and granulocyte-macrophage colony-stimulating factor (GM-CSF) [16] among others (for example, IL-17, IL-33, ‘TNF-related apoptosis-inducing ligand’ (TRAIL), and TGFβ).
Opposing effects of immune cells in sepsis
Host-protective innate immune responses and consequent inflammation are inextricably linked and overlapping. Consequently, the same cellular actors are key elements defending the host against infection while simultaneously contributing to deleterious events. For example, neutrophil extracellular traps that catch and kill bacteria and fungi are associated with the release of elements such as histones and mitochondria that behave like damage-associated molecular patterns perpetuating the inflammatory process.
Beneficial or deleterious roles of the same leukocyte subset have been reported depending upon the experimental model. For example, a peritonitis model using nude mice (lacking T cells) suggested that T lymphocytes contribute to protective immune responses [17]. By contrast, in an Escherichia coli sepsis murine model, T lymphocytes markedly contributed to severity [18]. Similarly, Treg improved survival in polymicrobial sepsis [19] whereas, in another report, reduced Treg activity led to improved survival [20]. The ‘half angel/half devil’ role of NK cells during severe infection is also described. NK cells contribute to systemic inflammation during polymicrobial sepsis but play a critical protective role in host defense against Staphylococcus aureus lung infection (as reviewed in [21]). Although apoptosis of dendritic cells (DCs) is particularly increased during sepsis, they are protective in murine polymicrobial sepsis [22]. Transcriptomic analysis of DCs in trauma patients shows a large number of upregulated inflammatory genes, suggesting their contribution to systemic inflammation and organ failure [23].
Apoptosis of lymphocytes, DCs, and NK cells is a hallmark of sepsis. Hotchkiss and colleagues [24] provided key experiments demonstrating that lymphocyte apoptosis was deleterious and its prevention highly protective. In addition to the depletion of apoptotic lymphocytes that contribute to the alteration of the immune status, apoptotic T cells themselves can further produce an immunosuppressive milieu following their release of TGFβ [25]. In contrast, the apoptosis of neutrophils is reduced. Interestingly, injection of apoptotic neutrophils in LPS-challenged mice with or undergoing cecal ligature puncture improved outcomes [26]. This may be due to the capacity of apoptotic neutrophils to limit the production of IL-1 and TNF by LPS-activated monocytes and to favor the production of IL-10 and TGFβ [27]. Favoring neutrophil apoptosis while differentially preventing that of lymphocytes and DCs would represent a considerable interventional challenge!
Inflammatory mediators and their effects in various organ compartments promote organ failure
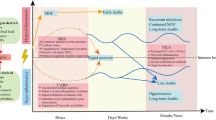
In sepsis, apoptosis does not only affect immune cells. Apoptosis of epithelial cells, endothelial cells, neurons, and cardiac myocytes is reported with crucial effects of loss of altered barrier function (Figure 1): in the lungs - acute lung injury and adult respiratory response syndrome [28]; in the kidneys - acute kidney injury [29]. Enhanced translocation of bacteria and bacterial products occurs consequent on intestinal epithelial cell apoptosis [30], contributing to the concept of the gut as the motor of multiple organ failure (MOF). Sepsis-induced cardiac myocyte apoptosis produces altered contractility and cardiac dysfunction [31]. Apoptosis of endothelial cells [32] induces vascular leakage. Finally, microglial and neuronal apoptosis may follow autonomic failure that precedes shock and MOF [33].

During sepsis, many types of cells (but not neutrophils) display enhanced apoptosis, leading to various deleterious consequences. AKI, acute kidney injury; ALI/ARDS, acute lung injury/acute respiratory distress syndrome; NK, natural killer.
In addition to epithelial apoptosis, tight junction alterations enhance organ dysfunction. It has been demonstrated that nitric oxide favors disruption of epithelial cell tight junctions in numerous organs, including liver, gut, and lung. Leukotrienes favor protein extravasation as shown in the kidney of septic mice [34].
Still, cytokines remain the main orchestrators of these tissue injuries. In a model of acute kidney injury, it was nicely demonstrated that inflammatory cytokines, including TNF and IL-17, cause small intestine and liver injury [35]. Among others, IL-17A is critical for generation of intestinal ischemia/reperfusion injury and subsequent liver and kidney injury [36]. All together, the altered functions of epithelial cells, endothelial cells, neurons, and cardiac myocytes contribute to MOF that may influence outcomes in sepsis more than altered immune status.
The concomitant occurrence of inflammation, anti-infectious response, and altered immune status in sepsis
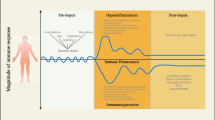
When Roger Bone coined the concepts of systemic inflammatory response syndrome (SIRS) and compensatory anti-inflammatory response syndrome (CARS), he conceived that one or the other would be predominating [37]. However, we contend that CARS should be considered an adapted compartmentalized response with the aim of silencing some acute pro-inflammatory genes and maintaining the expression of certain genes involved in the anti-infectious process. Despite our views [38], authors still propose a two-wave concept with SIRS appearing before CARS, although they admit that ‘rigorous examination of previous studies provides evidence that both pro-inflammatory and opposing anti-inflammatory response(s) occur concomitantly in sepsis’ [1].
Tamayo and colleagues [39] studied a large panel of circulating cytokines in patients with SIRS or sepsis, concluding that both pro- and anti-inflammatory mediators play roles from the very beginning of this life-threatening condition. Similarly, meta-analysis of 12 transcriptomic studies including 784 individuals led to the conclusion that ‘the arbitrary distinction of separating sepsis into pro-inflammatory and anti-inflammatory phases is not supported by gene-expression data’ [40].
Immune status has been studied frequently by measuring TNF or other inflammatory cytokine production by circulating monocytes in response to LPS [41]. We studied patients undergoing abdominal aortic surgery, showing that reduced expression of human leukocyte antigen (HLA)-DR on CD14HIGH monocytes occurs during surgery [42]. Similarly, HLA-DR expression was already reduced on monocytes taken very soon after severe trauma at accident scenes [43]. Altered TNF production capacity of circulating cells in response to TLR2 or TLR4 agonists is also observed very soon after injurious insults, such as on admission of patients after cardiac arrest [44]. Even if soon after the initial insult the intensity of the inflammatory response reaches its peak, there is a persistent inflammation associated with altered immune status in surviving patients [45]. The more severe the insult, the more profound is the alteration and the more chance the patients have to develop adverse clinical outcome.
The immune status of leukocytes during sepsis and SIRS varies depending on the compartment in which they reside
Terms such as ‘immunoparalysis, immunosuppression, and anergy’ are far too extreme to describe the immune status of circulating leukocytes in patients with sepsis or SIRS. Altered immune status of circulating leukocytes is not globally present. Indeed, some functions like phagocytosis remain unaltered [46], and ex vivo cytokine production in response to heat-killed S. aureus (HKSA) remains unchanged in patients with sepsis [47] compared with healthy controls. This is in full agreement with the observation that LPS primes HKSA-induced TNF production in macrophage cell lines instead of leading to cross-tolerance [48]. While the concept of endotoxin tolerance is considered to partially mimic the alteration of immune status in sepsis, it is worth mentioning that cross-tolerance between microbial agonists is not invariant. For example, Candida albicans and fungal cell wall β-glucan also prime LPS-induced pro-inflammatory cytokine production [49].
These observations led us to propose the concept of leukocyte reprogramming [50] to explain the fact that tolerised macrophages retain anti-infectious properties. In addition, in tissues, there are numerous examples to illustrate the hyper-activity of these cells. For example, in mice with polymicrobial sepsis alone or as a ‘second hit’ after traumatic hemorrhage, it was nicely demonstrated by Chaudry’s group [51] that the ex vivo production of TNF or IL-6 after LPS activation was significantly reduced among peripheral blood mononuclear cells and splenic macrophages but that it was enhanced in alveolar and Kupffer cells. Similarly, in a murine model of trauma, the cytokine productive capacity of Kupffer cells and alveolar macrophages was enhanced [52]. Indeed, macrophage functions differ depending on the compartment from which they derive. We established [53] that the specific cytokine and cellular microenvironment within the lung was responsible for this particular resistance of alveolar macrophages to endotoxin tolerance, which can also be observed in human alveolar macrophages [54]. Similarly, in kidneys, in response to a second challenge with LPS, the expression of TNF and inducible nitric oxide synthase was further enhanced [55]. This may explain why unilateral nephrectomy could be protective in a murine peritonitis model and after LPS injection [56]. Most importantly, despite the fashionable concept of M1/M2 macrophages, the response of macrophages to IL-4 and IFNγ is in fact completely different depending upon their origin [57]. As a consequence of this great heterogeneity of immune cells within different compartments, each tissue behaves independently, contributing to the global inflammatory response with a specific pattern, as illustrated by differential cytokine expression in liver, lungs, heart, brain, muscle, kidney, intestine, and spleen [58]. Another example of the different behavior of leukocytes in various compartments is the frequent occurrence of hemophagocytosis (>60%) directly observed in the bone marrow of the critically ill [59]. This phenomenon is associated with extreme production of inflammatory cytokines. Accordingly, it has been proposed that when hemophagocytosis is diagnosed in critical care patients, aggressive immunosuppressive therapy be undertaken without delay [59].
Differences between cells harvested from different compartments after sepsis have also been reported for spleen and peritoneal myeloid DCs [60]. The major differences between compartments are further illustrated by the fact that gene deficiency may differentially affect outcomes of infection. For example, IL-10 deficiency protects against Francisella tularensis pulmonary infection but aggravates cutaneous infection [61]. Similarly, we showed that scavenger receptor-A (SR-A), ‘macrophage associated receptor with a collagenous base’ (MARCO), CD36, or TLR2 deficiency protect mice against peritoneal S. aureus infection while these deficiencies aggravated pneumonia [62]. Interestingly, when Streptococcus pneumoniae was the pathogen used to colonize the murine nasopharynx, MARCO KO mice (but not SR-A KO mice) had significantly impaired clearance of pneumococcal colonization [63].
Furthermore, inflammatory foci cells may not behave similarly to cells from other healthy compartments. For example, it was shown that neutrophils derived from sputum of patients with chronic bronchitis or cystic fibrosis are insensitive to inhibitory effects of IL-10 in contrast to circulating neutrophils [64].
Murine models poorly mimic the clinical settings
The concomitant presence of inflammation within tissues and altered immune status within the hematopoietic compartment is short-lived in murine models rendering them inappropriate to study patients with concomitant sepsis and CARS [65]. In addition, mice are highly resistant to bacteria like S. aureus and their serum contains factors that limit inflammatory response intensity as compared with human serum [66]. A most provocative report comparing transcriptomic patterns of circulating cells from trauma patients, human endotoxemia-model participants, and murine-model equivalents revealed total absence of correlation [67]. When most therapeutic approaches have been validated in preclinical studies performed with murine models, one understands why those were not the most appropriate ones.
The scientific community needs to reconsider models used to validate therapeutic approaches. If murine responses do not resemble human processes, maybe other species, like the pig, should be preferred. Porcine monocytes and LPS-activated macrophages are closer to their human counterparts than murine cells [68]. Of course, murine models remain valuable to further decipher the mechanisms of sepsis. The best example is the two-hit model, which demonstrated that the nature of the first hit and its severity, the nature of the infection, and the route of infection may influence the outcome in a completely opposite direction [69].
Are patients with sepsis dying of immune failure - dissecting the arguments used to describe compensatory immunosuppression occurring after sepsis?
The clinical observations used to argue that immunosuppression occurs in sepsis patients surviving the initial inflammatory cascade [1] are in essence that patients develop nosocomial infections due to opportunistic pathogens, including reactivated chronic viral infections, and that patients who die after sepsis have unresolved foci of infection. These underpinning observations require further consideration.
Representing bacteria such as Enterococcus faecium, Stentrophomonas maltophilia, and Pseudomonas aeruginosa along with Candida as ‘opportunistic pathogens’ overstates the role of sepsis-induced immune dysregulation as the primary cause of nosocomial infection in these patients. These multiply-instrumented, high-intensity care, bed-bound, vulnerable patients often have breaches in their integument and mucous membranes (airways, surgical sites, indwelling catheters) and perturbed microbiomes from antibiotic treatments. Overgrowth of antibiotic-resistant microorganisms and barrier defects predispose them to secondary infections, even without overt defects in their immune defenses [70]. These are all organisms of normal virulence that cause nosocomial infections in sepsis patients because of the selection pressure of potent antibiotics and the presence of biofilm affected/colonized intravascular and urinary catheters.
Additionally, reactivation of herpes simplex virus (HSV) and cytomegalovirus (CMV) may have some clinical relevance in critically ill patients. CMV-emia is quite common in patients with sepsis (30% in some studies) and is at least associated with worse outcome in ICU patients in recent meta-analyses. Whether CMV could cause immune compromise itself, be a reflection of immune compromise, or simply be an indicator of poor outcome in patients with sepsis remains unclear [71]. Reactivation of oro-labial HSV is extremely common in sepsis, and HSV can frequently be detected in respiratory secretions. However, only one study has reliably investigated lower respiratory tract infection in critically ill, immunocompetent patients, showing that 21% of patients with ventilator-associated pneumonia (VAP) had bronchopneumonitis due to HSV [72]. In 55% of these patients, the VAP appeared to be due to HSV alone. However, acyclovir treatment had no impact on the outcome in patients with HSV bronchopneumonitis [72]. Of greater relevance to predisposition to nosocomial infection in sepsis patients remaining in ICUs for prolonged periods are physical breaches in innate immune system barriers. Intravascular catheters, endotracheal tubes with consequently increased dead space, and increased gastric pH due to peptic ulcer prophylaxis regimens are all, along with broad-spectrum antibiotics, potent promoters of nosocomial infection.
Post-mortems (PMs) identifying unresolved infection foci are not reliable proof that patients are dying of sepsis. Pneumonia is frequently present in patients in whom supportive care is withdrawn due to failure to thrive. Where pneumonia has been found more frequently at PM than was appreciated ante-mortem, the extent of pulmonary involvement was not quantified [73]. In this series, there was clear agreement by clinical and PM assessment that MOF was the commonest cause of death [73]. These data call into question the relevance of unresolved, PM infection in patients dying in the ICU as a direct indicator of immunosuppression following as a direct consequence of previous sepsis. If the patients die with infectious foci and altered immune status, it does not mean they die because of them.
Boosting the immune system
Because of the monocyte deactivation in sepsis, it was proposed to restore it with the use of either IFNγ or GM-CSF, two cytokines that counteract endotoxin tolerance. The first attempt was successfully performed in nine septic patients who received subcutaneous IFNγ that restored ex vivo cytokine production and HLA-DR expression by monocytes [74]. The authors claimed that overall mortality was lower in the treated group compared with historical controls. In mechanically ventilated trauma patients, IFNγ was aerosolized. However, in a previous phase III study in burn patients, IFNγ had failed to protect patients from infection or decrease mortality [75]. We must recall that IFNγ injection increases mortality in animal models of polymicrobial infection [15]. All together, these data have limited the routine use of IFNγ in ICU patients, although a Dutch clinical trial is ongoing.
GM-CSF has been demonstrated to be able to restore some immune status parameters. However, a meta-analysis concluded that GM-CSF did not significantly reduce in-hospital mortality, although it significantly increased infection recovery [76]. Although no adverse effects were reported, it is worth recalling a case report of a patient who developed a fatal adult respiratory distress syndrome after GM-CSF treatment [77]. In animal models, GM-CSF favors LPS-induced lung inflammation, amplifying LPS-induced bronchoconstriction [78]. GM-CSF favors production of TNF and IL-1. In a recent study, it was confirmed that GM-CSF synergizes with LPS, promoting IL-1β secretion [79]. Lethal injection of LPS in GM-CSF receptor KO mice led to far lower mortality among these mice as compared to wild type mice. Given all the efforts made by some authors to convince the scientific community of the use of GM-CSF, it is challenging to read the conclusion of this present paper given that GM-CSF has been previously underestimated as a target for therapeutic intervention in many bacterial infections and inflammatory disorders associated with the production of IL-1β.
IL-7 is another cytokine that is promoted for the treatment of sepsis and that is supported by murine and human ex vivo tissue data [1, 80]. One can conjecture that systemic treatment with IL-7 may act in undesired places, as illustrated by the following: IL-7 worsens graft-versus-host-induced tissue inflammation [81]; favors inflammation in colitis [82], contributes to arthritis severity [83]; upregulates chemokines, IFNγ, macrophage recruitment, and lung inflammation [84]; and, finally, increases production of inflammatory cytokines by monocytes and T cells [85].
Many other cytokines (for example, IL-2, IL-12, IL-15, and TNF) can boost the immune system and are reported to be beneficial in murine sepsis models. However, one wonders whether systemic treatment with any immunostimulating cytokine may act on tissue leukocytes boosting the inflammatory process while boosting immune status as well. In this perspective, the attempt to treat peripheral mononuclear cells of sepsis patients ex vivo with IL-2 before re-injecting them is an interesting approach that prevents the delivery of this cytokine to the bloodstream, allowing it to act strictly on the desired cells [86]. In this study, the mortality was 8% in the extracorporeally treated group of patients (n = 121) but was 21% in the patients receiving standard treatment (n = 52).
Approaches for innovative therapeutic interventions
Rather than repeating the mistakes of past experimental treatments for sepsis in which therapies were developed after successful preclinical models that may be far from mimicking human disease, it would be ideal to proceed in the future with new treatments in which extensive human data are available prior to embarking on expensive licensure studies. Furthermore, identifying currently licensed drugs with tolerable safety profiles as potential sepsis agents leap-frogs costly drug development and early-phase human studies.
In animal models, extant licensed drugs, such as chloroquine [87] and androstenenediol [51], have successfully restored immune status. Most interestingly, in the latter case, the treatment protected mice against polymicrobial sepsis and boosted altered ex vivo cytokine production observed with peripheral blood cells and spleen macrophages, dampening production observed with alveolar macrophages and Kupffer cells. A similar compartmentalized adapted specificity was reported with estradiol [88].
Other approaches involve pro-resolving lipid mediators [89], although it is uncertain whether they may also adversely boost immune status. The recently recognized aspirin-triggered lipoxins, anti-inflammatory mediators of inflammation resolution, make aspirin a possible inexpensive agent for both prevention and treatment of sepsis. Considerable observational cohort data show improvements in mortality in patients with sepsis pretreated with aspirin [90]. This approach is being prospectively studied as part of an aspirin primary prevention trial.
Could other immunomodulatory approaches be considered with less putative dangerous consequences on inflamed tissues. This may be the case of thymosin-α1. Indeed, a very promising study demonstrated its efficiency to improve clinical outcome in patients with severe sepsis [91], after a preliminary investigation had demonstrated a better performance with respect to organ failure scores in thymosin-α1-treated patients with sepsis arising from intra-abdominal infection due to carbapenem-resistant bacteria [92]. However, one must call for caution since thymosin-α1 can also favor the production of inflammatory cytokines and nitric oxide and further increases the percentage of Treg cells [93, 94]. Still, very little is known of its effect on leukocytes present in different compartments.
The cell surface molecules containing in their intracytoplasmic domain an immunoreceptor tyrosine-based inhibition motif - such as programmed death-1 (PD-1), B and T lymphocyte attenuator (BTLA), and cytotoxic T-lymphocyte antigen 4 (CTLA-4) - could also be interesting targets for new therapeutic approaches. The expression of PD-1 on T cells and its ligand (PD-L1) on monocytes is upregulated in critically ill [95] or septic shock [96] patients. Increased expressions were associated with increased occurrence of secondary nosocomial infections and mortality after septic shock [97]. Not only are PD-1-deficient mice markedly protected from the lethality of sepsis, accompanied by a decreased bacterial burden and suppressed inflammatory cytokine response [98], but also blockade of PD-1 or PD-L1 improves survival in a murine model of sepsis, reverses immune dysfunction, inhibits lymphocyte apoptosis, and attenuates organ dysfunction [99–101]. The relevance of these observations in human settings is still needed. CTLA-4 is a high-avidity receptor for CD80 and CD86. Enhanced CTLA-4 expression was demonstrated more frequently in patients with sepsis than in non-infected critically ill patients or control subjects [102], and blocking CTLA-4 improved survival in bacterial and fungal experimental sepsis [103, 104]. However, the use of such an approach seems tricky since, in animal models at high dose, anti-CTLA-4 could worsen survival [103], and the use of Abatacept (a soluble CTLA-4 dimerized with an Fc fragment of immunoglobulin) led to increased survival in invasive pneumococcal infection [105]. Similarly, BTLA expression is enhanced in patients with SIRS or sepsis [106] and, in a murine model of sepsis, BTLA-deficient mice displayed an enhanced resistance [107]. In contrast, these mice displayed enhanced susceptibility to endotoxin-induced shock [108]. Accordingly, the exact role of BTLA needs to be further deciphered before strategies targeting BTLA could be proposed to treat patients with sepsis.
Conclusions
New therapeutic approaches to treat sepsis should take into consideration that the immune status of leukocytes in the peripheral blood might be quite different from those present in inflamed tissues. We believe that a systemic approach to immune stimulation is not appropriate if immune cells are boosted generally, independent of their location. An ideal drug would limit the overzealous inflammatory process that leads to organ failure and favor homeostatic responsiveness of leukocytes (Figure 2). This is the challenge we have to address if we wish to avoid further decades of disillusionment.

New therapeutic interventions should address both the events in the tissues that lead to organ failure and the altered immune status of leukocytes restricted to some specific compartments.
Abbreviations
- BTLA:
-
B and T lymphocyte attenuator
- CARS:
-
Compensatory anti-inflammatory response syndrome
- CMV:
-
Cytomegalovirus
- CTLA-4:
-
Cytotoxic T-lymphocyte antigen 4
- DC:
-
Dendritic cell
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- HKSA:
-
Heat-killed Staphylococcus aureus
- HLA:
-
Human leukocyte antigen
- HSV:
-
Herpes simplex virus
- IFNγ:
-
Interferon-gamma
- IL:
-
Interleukin
- KO:
-
Knockout
- LPS:
-
Lipopolysaccharide
- MARCO:
-
Macrophage-associated receptor with a collagenous base
- MOF:
-
Multiple organ failure
- NK:
-
Natural killer
- PD-1:
-
Programmed death-1
- PD-L1:
-
Programmed death-1 ligand
- PGE2:
-
Prostaglandin E2
- PM:
-
Post-mortem
- SIRS:
-
Systemic inflammatory response syndrome
- SR-A:
-
Scavenger receptor-A
- TGFβ:
-
Transforming growth factor-beta
- Th:
-
T helper
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- Treg:
-
Regulatory T lymphocyte
- VAP:
-
Ventilator-associated pneumonia.
References
Hotchkiss RS, Monneret G, Payen D: Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis 2013, 13: 260-268. 10.1016/S1473-3099(13)70001-X
Zakharova M, Ziegler HK: Paradoxical anti-inflammatory actions of TNF-alpha: inhibition of IL-12 and IL-23 via TNF receptor 1 in macrophages and dendritic cells. J Immunol 2005, 175: 5024-5033.
Noti M, Corazza N, Mueller C, Berger B, Brunner T: TNF suppresses acute intestinal inflammation by inducing local glucocorticoid synthesis. J Exp Med 2010, 207: 1057-1066. 10.1084/jem.20090849
González-Navajas JM, Law J, Nguyen KP, Bhargava M, Corr MP, Varki N, Eckmann L, Hoffman HM, Lee J, Raz E: Interleukin 1 receptor signaling regulates DUBA expression and facilitates toll-like receptor 9-driven antiinflammatory cytokine production. J Exp Med 2010, 207: 2799-2807. 10.1084/jem.20101326
Fukushima A, Ozaki A, Ishida W, Fukata K, Ueno H: Systemic interferon-gamma suppresses the development of endotoxin-induced uveitis in mice. Curr Eye Res 2005, 30: 7-12. 10.1080/02713680490522461
Lauw FN, Pajkrt D, Hack CE, Kurimoto M, van Deventer SJH, van der Poll T: Proinflammatory effects of IL-10 during human endotoxemia. J Immunol 2000, 165: 2783-2789.
Petit-Bertron AF, Pedron T, Gross U, Coppée JY, Sansonetti PJ, Cavaillon JM, Adib-Conquy M: Adherence modifies the regulation of gene expression induced by interleukin-10. Cytokine 2005, 29: 1-12.
Moore KW, De Waal Malefyt R, Coffman RL, O’Garra A: Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 2001, 19: 683-765. 10.1146/annurev.immunol.19.1.683
Garcia-Lazaro JF, Thieringer F, Lüth S, Czochra P, Meyer E, Renteria IB, Galle PR, Lohse AW, Herkel J, Kanzler S: Hepatic over-expression of TGF-beta1 promotes LPS-induced inflammatory cytokine secretion by liver cells and endotoxemic shock. Immunol Lett 2005, 101: 217-222. 10.1016/j.imlet.2005.06.003
Kalinski P: Regulation of immune responses by prostaglandin E2. J Immunol 2012, 188: 21-28. 10.4049/jimmunol.1101029
Abraham E, Wunderink R, Silverman H, Perl TM, Nasraway S, Levy H, Bone R, Wenzel RP, Balk R, Allred R, Pennington JE, Wherry JC, Bellamy P, Cryer H, Busuttil R, Winston D, Perry C, Leeper KV, Jones C, Martin M, Tuma P, Baird I, Brooks J, Baird R, Rangel S, Wagner N, Costigan M, Gutierrez G, Johnson P, et al.: Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome. A randomized, controlled, double-blind, multicenter clinical trial. TNF-alpha MAb sepsis study group. JAMA 1995, 273: 934-941. 10.1001/jama.1995.03520360048038
Reinhart K, Wiegand-Löhnert C, Grimminger F, Kaul M, Withington S, Treacher D, Eckart J, Willatts S, Bouza C, Krausch D, Stockenhuber F, Eiselstein J, Daum L, Kempeni J: Assessment of the safety and efficacy of the monoclonal anti-tumor necrosis factor antibody-fragment, MAK 195 F, in patients with sepsis and septic shock: a multicenter, randomized, placebo-controlled, dose-ranging study. Crit Care Med 1996, 24: 733-742. 10.1097/00003246-199605000-00003
Tetta C, Gianotti L, Cavaillon JM, Wratten ML, Fini M, Braga M, Bisagni P, Giavaresi G, Bolzani R, Giardino R: Continuous plasmafiltration coupled with sorbent adsorption in a rabbit model of endotoxic shock. Crit Care Med 2000, 28: 1526-1533. 10.1097/00003246-200005000-00045
Lorente JA, Marshall JC: Neutralization of tumor necrosis factor in preclinical models of sepsis. Shock 2005, 24: 107-119. 10.1097/01.shk.0000191343.21228.78
Miles RH, Paxton TP, Dries DJ, Gamelli RL: Interferon-gamma increases mortality following cecal ligation and puncture. J Trauma 1994, 36: 607-611. 10.1097/00005373-199405000-00001
Spight D, Trapnell B, Zhao B, Berclaz P, Shanley TP: Granulocyte-macrophage-colony-stimulating factor-dependent peritoneal macrophage responses determine survival in experimentally induced peritonitis and sepsis in mice. Shock 2008, 30: 434-442. 10.1097/SHK.0b013e3181673543
Reim D, Westenfelder K, Kaiser-Moore S, Schlautkotter S, Holzmann B, Weighardt H: Role of T cells for cytokine production and outcome in a model of acute septic peritonitis. Shock 2009, 31: 245-250. 10.1097/SHK.0b013e31817fd02c
van Schaik SM, Abbas AK: Role of T cells in a murine model of Escherichia coli sepsis. Eur J Immunol 2007, 37: 3101-3110. 10.1002/eji.200737295
Heuer JG, Zhang T, Zhao J, Ding C, Cramer M, Justen KL, Vonderfecht SL, Na S: Adoptive transfer of in vitro-stimulated CD4 + CD25+ regulatory T cells increases bacterial clearance and improves survival in polymicrobial sepsis. J Immunol 2005, 174: 7141-7146.
Hiraki S, Ono S, Tsujimoto H, Kinoshita M, Takahata R, Miyazaki H, Saitoh D, Hase K: Neutralization of interleukin-10 or transforming growth factor-beta decreases the percentages of CD4+ CD25+ Foxp3+ regulatory T cells in septic mice, thereby leading to an improved survival. Surgery 2012, 151: 313-322. 10.1016/j.surg.2011.07.019
Souza-Fonseca-Guimaraes F, Adib-Conquy M, Cavaillon JM: Natural killer (NK) cells in antibacterial innate immunity: angels or devils? Mol Med 2012, 18: 270-285.
Scumpia PO, McAuliffe PF, O’Malley KA, Ungaro R, Uchida T, Matsumoto T, Remick DG, Clare-Salzler MJ, Moldawer LL, Efron PA: CD11c + dendritic cells are required for survival in murine polymicrobial sepsis. J Immunol 2005, 175: 3282-3286.
Maier M, Wutzler S, Bauer M, Trendafilov P, Henrich D, Marzi I: Altered gene expression patterns in dendritic cells after severe trauma: implications for systemic inflammation and organ injury. Shock 2008, 30: 344-351. 10.1097/SHK.0b013e3181673eb4
Hotchkiss RS, Swanson PE, Knudson CM, Chang KC, Cobb JP, Osborne DF, Zollner KM, Buchman TG, Korsmeyer SJ, Karl IE: Overexpression of Bcl-2 in transgenic mice decreases apoptosis and improves survival in sepsis. J Immunol 1999, 162: 4148-4156.
Chen W, Frank M, Jin W, Wahl S: TGF-beta released by apoptotic T cells contributes to an immunosuppressive milieu. Immunity 2001, 14: 715-725. 10.1016/S1074-7613(01)00147-9
Ren Y, Xie Y, Jiang G, Fan J, Yeung J, Li W, Tam PK, Savill J: Apoptotic cells protect mice against lipopolysaccharide-induced shock. J Immunol 2008, 180: 4978-4985.
Byrne A, Reen DJ: Lipopolysaccharide induces rapid production of IL-10 by monocytes in the presence of apoptotic neutrophils. J Immunol 2002, 168: 1968-1977.
Perl M, Chung CS, Lomas-Neira J, Rachel TM, Biffl WL, Cioffi WG, Ayala A: Silencing of Fas, but not caspase-8, in lung epithelial cells ameliorates pulmonary apoptosis, inflammation, and neutrophil influx after hemorrhagic shock and sepsis. Am J Pathol 2005, 167: 1545-1559. 10.1016/S0002-9440(10)61240-0
Glynne PA, Evans TJ: Inflammatory cytokines induce apoptotic and necrotic cell shedding from human proximal tubular epithelial cell monolayers. Kidney Int 1999, 55: 2573-2597. 10.1046/j.1523-1755.1999.00456.x
Husain KD, Stromberg PE, Woolsey CA, Turnbull IR, Dunne WM, Javadi P, Buchman TG, Karl IE, Hotchkiss RS, Coopersmith CM: Mechanisms of decreased intestinal epithelial proliferation and increased apoptosis in murine acute lung injury. Crit Care Med 2005, 33: 2350-2357. 10.1097/01.CCM.0000182797.89252.A3
Buerke U, Carter JM, Schlitt A, Russ M, Schmidt H, Sibelius U, Grandel U, Grimminger F, Seeger W, Mueller-Werdan U, Werdan K, Buerke M: Apoptosis contributes to septic cardiomyopathy and is improved by simvastatin therapy. Shock 2008, 29: 497-503. 10.1097/SHK.0b013e318142c434
Zhou M, Simms HH, Wang P: Adrenomedullin and adrenomedullin binding protein-1 attenuate vascular endothelial cell apoptosis in sepsis. Ann Surg 2004, 240: 321-330. 10.1097/01.sla.0000133253.45591.5b
Sharshar T, Gray F, Lorin De La Grandmaison G, Hopkinson NS, Ross E, Dorandeu A, Orlikowski D, Raphael JC, Gajdos P, Annane D: Apoptosis of neurons in cardiovascular autonomic centres triggered by inducible nitric oxide synthase after death from septic shock. Lancet 2003, 362: 1799-1805. 10.1016/S0140-6736(03)14899-4
Benjamim CF, Canetti C, Cunha FQ, Kunkel SL, Peters-Golden M: Opposing and hierarchical roles of leukotrienes in local innate immune versus vascular responses in a model of sepsis. J Immunol 2005, 174: 1616-1620.
Park SW, Chen SW, Kim M, Brown KM, Kolls JK, D’Agati VD, Lee HT: Cytokines induce small intestine and liver injury after renal ischemia or nephrectomy. Lab Invest 2011, 91: 63-84. 10.1038/labinvest.2010.151
Lee HT, Kim M, Kim JY, Brown KM, Ham A, D’Agati VD, Mori-Akiyama Y: Critical role of interleukin-17A in murine intestinal ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol 2013, 304: G12-G25. 10.1152/ajpgi.00201.2012
Bone RC, Grodzin CJ, Balk RA: Sepsis: a new hypothesis for pathogenesis of the disease process. Chest 1997, 121: 235-243.
Cavaillon J-M, Adib-Conquy M, Cloëz-Tayarani I, Fitting C: Immunodepression in sepsis and SIRS assessed by ex vivo cytokine production is not a generalized phenomenon: a review. J Endotoxin Res 2001, 7: 85-93.
Tamayo E, Fernández A, Almansa R, Carrasco E, Heredia M, Lajo C, Goncalves L, Gómez-Herreras JI, de Lejarazu RO, Bermejo-Martin JF: Pro- and anti-inflammatory responses are regulated simultaneously from the first moments of septic shock. Eur Cytokine Netw 2011, 22: 82-87.
Tang BM, Huang SJ, McLean AS: Genome-wide transcription profiling of human sepsis: a systematic review. Crit Care 2010, 14: R237. 10.1186/cc9392
Muñoz C, Carlet J, Fitting C, Misset B, Bleriot JP, Cavaillon JM: Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest 1991, 88: 1747-1754. 10.1172/JCI115493
Kim OY, Monsel A, Bertrand M, Coriat P, Cavaillon JM, Adib-Conquy M: Differential down-regulation of HLA-DR on monocyte subpopulations during systemic inflammation. Crit Care 2010, 14: R61. 10.1186/cc8959
Kox M, Timmermans K, Veaneker M, Scheffer G, Pickkers P: Immune paralysis in trauma patients, implications for pre-hospital intervention. Crit Care 2013, 17: 9. 10.1186/cc11933
Adrie C, Adib-Conquy M, Laurent I, Monchi M, Vinsonneau C, Fitting C, Fraisse F, Dinh-Xuan AT, Carli P, Spaulding C, Dhainaut JF, Cavaillon JM: Successful cardiopulmonary resuscitation after cardiac arrest as a ‘sepsis like’ syndrome. Circulation 2002, 106: 562-568. 10.1161/01.CIR.0000023891.80661.AD
Gentile LF, Cuenca AG, Efron PA, Ang D, Bihorac A, McKinley BA, Moldawer LL, Moore FA: Persistent inflammation and immunosuppression: a common syndrome and new horizon for surgical intensive care. J Trauma Acute Care Surg 2012, 72: 1491-1501. 10.1097/TA.0b013e318256e000
Danikas DD, Karakantza M, Theodorou GL, Sakellaropoulos GC, Gogos CA: Prognostic value of phagocytic activity of neutrophils and monocytes in sepsis. Correlation to CD64 and CD14 antigen expression. Clin Exp Immunol 2008, 154: 87-97. 10.1111/j.1365-2249.2008.03737.x
Adib-Conquy M, Adrie C, Fitting C, Gattoliat O, Beyaert R, Cavaillon JM: Up-regulation of MyD88s and SIGIRR, molecules inhibiting Toll-like receptor signaling, in monocytes from septic patients. Crit Care Med 2006, 34: 2377-2385. 10.1097/01.CCM.0000233875.93866.88
Peck OM, Fan H, Tempel GE, Teti G, Halushka PV, Cook JA: Staphylococcus aureus and lipopolysaccharide induce homologous tolerance but heterologous priming: role of interferon-gamma. Shock 2004, 21: 254-260. 10.1097/01.shk.0000111662.09279.59
Quintin J, Saeed S, Martens JH, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, Jacobs L, Jansen T, Kullberg BJ, Wijmenga C, Joosten LA, Xavier RJ, van der Meer JW, Stunnenberg HG, Netea MG: Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe 2012, 12: 223-232. 10.1016/j.chom.2012.06.006
Cavaillon JM, Adrie C, Fitting C, Adib-Conquy M: Reprogramming of circulatory cells in sepsis and SIRS. J Endotoxin Res 2005, 11: 311-320. 10.1177/09680519050110050901
Suzuki T, Shimizu T, Szalay L, Choudhry MA, Rue LW 3rd, Bland KI, Chaudry IH: Androstenediol ameliorates alterations in immune cells cytokine production capacity in a two-hit model of trauma-hemorrhage and sepsis. Cytokine 2006, 34: 76-84. 10.1016/j.cyto.2006.04.007
Neunaber C, Oestern S, Andruszkow H, Zeckey C, Mommsen P, Kutter D, Stöfen M, Krettek C, Hildebrand F: Cytokine productive capacity of alveolar macrophages and Kupffer cells after femoral fracture and blunt chest trauma in a murine trauma model. Immunol Lett 2013, 152: 159-166. 10.1016/j.imlet.2013.05.012
Philippart F, Fitting C, Cavaillon JM: Lung microenvironment contributes to the resistance of alveolar macrophages to develop tolerance to endotoxin*. Crit Care Med 2012, 40: 2987-2996. 10.1097/CCM.0b013e31825b8d57
Hoogerwerf JJ, de Vos AF, van’t Veer C, Bresser P, de Boer A, Tanck MW, Draing C, van der Zee JS, van der Poll T: Priming of alveolar macrophages upon instillation of lipopolysaccharide in the human lung. Am J Respir Cell Mol Biol 2010, 42: 349-356. 10.1165/rcmb.2008-0362OC
Zager RA, Johnson AC, Lund S: ‘Endotoxin tolerance’: TNF-alpha hyper-reactivity and tubular cytoresistance in a renal cholesterol loading state. Kidney Int 2007, 71: 496-503. 10.1038/sj.ki.5002092
Zurovsky Y, Eligal Z: Reduction of risk following endotoxin injection in unilaterally nephrectomized rats. Exp Toxicol Pathol 1996, 48: 41-46. 10.1016/S0940-2993(96)80092-3
Zhang S, Kim CC, Batra S, McKerrow JH, Loke P: Delineation of diverse macrophage activation programs in response to intracellular parasites and cytokines. PLoS Negl Trop Dis 2010, 4: e648. 10.1371/journal.pntd.0000648
Hacham M, Cristal N, White RM, Segal S, Apte RN: Complementary organ expression of IL-1 vs IL-6 and CSF-1 activities in normal and LPS injected mice. Cytokine 1996, 8: 21-31. 10.1006/cyto.1995.0004
Raschke RA, Garcia-Orr R: Hemophagocytic lymphohistiocytosis: a potentially underrecognized association with systemic inflammatory response syndrome, severe sepsis, and septic shock in adults. Chest 2011, 140: 933-938. 10.1378/chest.11-0619
Ding Y, Chung CS, Newton S, Chen Y, Carlton S, Albina JE, Ayala A: Polymicrobial sepsis induces divergent effects on splenic and peritoneal dendritic cell function in mice. Shock 2004, 22: 137-144. 10.1097/01.shk.0000131194.80038.3f
Metzger DW, Salmon SL, Kirimanjeswara G: Differing effects of interleukin-10 on cutaneous and pulmonary Francisella tularensis live vaccine strain infection. Infect Immun 2013, 81: 2022-2027. 10.1128/IAI.00024-13
Blanchet C, Jouvion G, Fitting C, Cavaillon JM, Adib-Conquy M: Protective or deleterious role of scavenger receptors SR-A and CD36 on host resistance to Staphylococcus aureus depends on the site of infection. PloS One 2014, 9: e87927. 10.1371/journal.pone.0087927
Dorrington MG, Roche AM, Chauvin SE, Tu Z, Mossman KL, Weiser JN, Bowdish DM: MARCO is required for TLR2- and Nod2-mediated responses to Streptococcus pneumoniae and clearance of pneumococcal colonization in the murine nasopharynx. J Immunol 2013, 190: 250-258. 10.4049/jimmunol.1202113
Pang G, Ortega M, Zighang R, Reeves G, Clancy R: Autocrine modulation of IL-8 production by sputum neutrophils in chronic bronchial sepsis. Am J Respir Crit Care Med 1997, 155: 726-371. 10.1164/ajrccm.155.2.9032219
Muenzer JT, Davis CG, Chang K, Schmidt RE, Dunne WM, Coopersmith CM, Hotchkiss RS: Characterization and modulation of the immunosuppressive phase of sepsis. Infect Immun 2010, 78: 1582-15492. 10.1128/IAI.01213-09
Warren HS, Fitting C, Hoff E, Adib-Conquy M, Beasley-Topliffe L, Tesini B, Liang X, Valentine C, Hellman J, Hayden D, Cavaillon JM: Resilience to bacterial infection: difference between species could be due to proteins in serum. J Infect Dis 2010, 201: 223-232. 10.1086/649557
Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, López CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, et al.: Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 2013, 110: 3507-3512. 10.1073/pnas.1222878110
Kapetanovic R, Fairbairn L, Beraldi D, Sester DP, Archibald AL, Tuggle CK, Hume DA: Pig bone marrow-derived macrophages resemble human macrophages in their response to bacterial lipopolysaccharide. J Immunol 2012, 188: 3382-3394. 10.4049/jimmunol.1102649
Takahashi H, Tsuda Y, Takeuchi D, Kobayashi M, Herndon DN, Suzuki F: Influence of systemic inflammatory response syndrome on host resistance against bacterial infections. Crit Care Med 2004, 32: 1879-1885. 10.1097/01.CCM.0000139606.34631.61
Otto GP, Sossdorf M, Claus RA, Rödel J, Menge K, Reinhart K, Bauer M, Riedemann NC: The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care 2011, 15: R183. 10.1186/cc10332
Osawa R, Singh N: Cytomegalovirus infection in critically ill patients: a systematic review. Crit Care 2009, 13: R68. 10.1186/cc7875
Luyt CE, Combes A, Deback C, Aubriot-Lorton MH, Nieszkowska A, Trouillet JL, Capron F, Agut H, Gibert C, Chastre J: Herpes simplex virus lung infection in patients undergoing prolonged mechanical ventilation. Am J Respir Crit Care Med 2007, 175: 935-942. 10.1164/rccm.200609-1322OC
Torgersen C, Moser P, Luckner G, Mayr V, Jochberger S, Hasibeder WR, Dünser MW: Macroscopic postmortem findings in 235 surgical intensive care patients with sepsis. Anesth Analg 2009, 108: 1841-1847. 10.1213/ane.0b013e318195e11d
Döcke WD, Randow F, Syrbe U, Krausch D, Asadullah K, Reinke P, Volk HD, Kox W: Monocyte deactivation in septic patients: restoration by IFNg treatment. Nature Med 1997, 3: 678-681. 10.1038/nm0697-678
Wasserman D, Ioannovich JD, Hinzmann RD, Deichsel G, Steinmann GG: Interferon-gamma in the prevention of severe burn-related infections: a European phase III multicenter trial. The Severe Burns Study Group. Crit Care Med 1998, 26: 434-439. 10.1097/00003246-199803000-00010
Bo L, Wang F, Zhu J, Li J, Deng X: Granulocyte-colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) for sepsis: a meta-analysis. Crit Care 2011, 15: R58. 10.1186/cc10031
Verhoef G, Boogaerts M: Treatment with granulocyte-macrophage colony stimulating factor and the adult respiratory distress syndrome. Am J Hematol 1991, 36: 285-287. 10.1002/ajh.2830360413
Bozinovski S, Jones J, Vlahos R, Hamilton J, Anderson G: Granulocyte/macrophage-colony-stimulating factor (GM-CSF) regulates lung innate immunity to lipopolysaccharide through Akt/Erk activation of NFkappa B and AP-1 in vivo. J Biol Chem 2002, 277: 42808-42814. 10.1074/jbc.M207840200
Khameneh HJ, Isa SA, Min L, Nih FW, Ruedl C: GM-CSF signalling boosts dramatically IL-1 production. PLoS One 2011, 6: e23025. 10.1371/journal.pone.0023025
Venet F, Foray AP, Villars-Méchin A, Malcus C, Poitevin-Later F, Lepape A, Monneret G: IL-7 restores lymphocyte functions in septic patients. J Immunol 2012, 189: 5073-5081. 10.4049/jimmunol.1202062
Sinha ML, Fry TJ, Fowler DH, Miller G, Mackall CL: Interleukin 7 worsens graft-versus-host disease. Blood 2002, 100: 2642-2649. 10.1182/blood-2002-04-1082
Willis CR, Seamons A, Maxwell J, Treuting PM, Nelson L, Chen G, Phelps S, Smith CL, Brabb T, Iritani BM, Maggio-Price L: Interleukin-7 receptor blockade suppresses adaptive and innate inflammatory responses in experimental colitis. J Inflamm (Lond) 2012, 9: 39. 10.1186/1476-9255-9-39
Hartgring SA, Willis CR, Alcorn D, Nelson LJ, Bijlsma JW, Lafeber FP, van Roon JA: Blockade of the interleukin-7 receptor inhibits collagen-induced arthritis and is associated with reduction of T cell activity and proinflammatory mediators. Arthritis Rheum 2010, 62: 2716-2725. 10.1002/art.27578
Jin JO, Yu Q: Systemic administration of TLR3 agonist induces IL-7 expression and IL-7-dependent CXCR3 ligand production in the lung. J Leukoc Biol 2013, 93: 413-425. 10.1189/jlb.0712360
Alderson MR, Tough TW, Ziegler SF, Grabstein KH: Interleukin 7 induces cytokine secretion and tumoricidal activity by human peripheral blood monocytes. J Exp Med 1991, 173: 923-930. 10.1084/jem.173.4.923
Ostatin A, Paltsev A, Leplina O, Shevela Y, Chernykh H: The experience of surgical infections treatment with extracorporal immunotherapy. Medicinskaya Immunol 2000, 2: 43-51.
Ertel W, Morrison MH, Ayala A, Chaudry IH: Chloroquine attenuates hemorrhagic shock-induced immunosuppression and decreases susceptibility to sepsis. Arch Surg 1992, 127: 70-75. 10.1001/archsurg.1992.01420010084012
Suzuki T, Shimizu T, Yu HP, Hsieh YC, Choudhry MA, Schwacha MG, Chaudry IH: Tissue compartment-specific role of estrogen receptor subtypes in immune cell cytokine production following trauma-hemorrhage. J Appl Physiol 2007, 102: 163-168.
Chiang N, Fredman G, Bäckhed F, Oh SF, Vickery T, Schmidt BA, Serhan CN: Infection regulates pro-resolving mediators that lower antibiotic requirements. Nature 2012, 484: 524-528. 10.1038/nature11042
Eisen DP, Reid D, McBryde ES: Acetyl salicylic acid usage and mortality in critically ill patients with the systemic inflammatory response syndrome and sepsis. Crit Care Med 2012, 40: 1761-1767. 10.1097/CCM.0b013e318246b9df
Wu J, Zhou L, Liu J, Ma G, Kou Q, He Z, Chen J, Ou-Yang B, Chen M, Li Y, Wu X, Gu B, Chen L, Zou Z, Qiang X, Chen Y, Lin A, Zhang G, Guan X: The efficacy of thymosin alpha 1 for severe sepsis (ETASS): a multicenter, single-blind, randomized and controlled trial. Crit Care 2013, 17: R8. 10.1186/cc11932
Zhang Y, Chen H, Li YM, Zheng SS, Chen YG, Li LJ, Zhou L, Xie HY, Praseedom RK: Thymosin alpha1- and ulinastatin-based immunomodulatory strategy for sepsis arising from intra-abdominal infection due to carbapenem-resistant bacteria. J Infect Dis 2008, 198: 723-730. 10.1086/590500
Sodhi A, Paul S: Involvement of mitogen-activated protein kinases in the signal transduction pathway of bone marrow-derived macrophage activation in response to in vitro treatment with thymosin alpha 1. Int Immunopharmacol 2002, 2: 47-58. 10.1016/S1567-5769(01)00139-4
Yang X, Qian F, He HY, Liu KJ, Lan YZ, Ni B, Tian Y, Fu XL, Zhang J, Shen ZG, Li J, Yin Y, Li JT, Wu YZ: Effect of thymosin alpha-1 on subpopulations of Th1, Th2, Th17, and regulatory T cells (Tregs) in vitro. Braz J Med Biol Res 2012, 45: 25-32. 10.1590/S0100-879X2011007500159
Monaghan SF, Thakkar RK, Tran ML, Huang X, Cioffi WG, Ayala A, Heffernan DS: Programmed death 1 expression as a marker for immune and physiological dysfunction in the critically ill surgical patient. Shock 2012, 38: 117-122. 10.1097/SHK.0b013e31825de6a3
Zhang Y, Li J, Lou J, Zhou Y, Bo L, Zhu J, Zhu K, Wan X, Cai Z, Deng X: Upregulation of programmed death-1 on T cells and programmed death ligand-1 on monocytes in septic shock patients. Crit Care 2011, 15: R70. 10.1186/cc10059
Guignant C, Lepape A, Huang X, Kherouf H, Denis L, Poitevin F, Malcus C, Chéron A, Allaouchiche B, Gueyffier F, Ayala A, Monneret G, Venet F: Programmed death-1 levels correlate with increased mortality, nosocomial infection and immune dysfunctions in septic shock patients. Crit Care 2011, 15: R99. 10.1186/cc10112
Huang X, Venet F, Wang YL, Lepape A, Yuan Z, Chen Y, Swan R, Kherouf H, Monneret G, Chung CS, Ayala A: PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc Natl Acad Sci U S A 2009, 106: 6303-6308. 10.1073/pnas.0809422106
Brahmamdam P, Inoue S, Unsinger J, Chang KC, McDunn JE, Hotchkiss RS: Delayed administration of anti-PD-1 antibody reverses immune dysfunction and improves survival during sepsis. J Leukoc Biol 2010, 88: 233-240. 10.1189/jlb.0110037
Zhang Y, Zhou Y, Lou J, Li J, Bo L, Zhu K, Wan X, Deng X, Cai Z: PD-L1 blockade improves survival in experimental sepsis by inhibiting lymphocyte apoptosis and reversing monocyte dysfunction. Crit Care 2010, 14: R220. 10.1186/cc9354
Zhu W, Bao R, Fan X, Tao T, Zhu J, Wang J, Li J, Bo L, Deng X: PD-L1 blockade attenuated sepsis-induced liver injury in a mouse cecal ligation and puncture model. Mediators Inflamm 2013, 2013: 361501.
Manjuck J, Saha DC, Astiz M, Eales LJ, Rackow EC: Decreased response to recall antigens is associated with depressed costimulatory receptor expression in septic critically ill patients. J Lab Clin Med 2000, 135: 153-160. 10.1067/mlc.2000.104306
Inoue S, Bo L, Bian J, Unsinger J, Chang K, Hotchkiss RS: Dose-dependent effect of anti-CTLA-4 on survival in sepsis. Shock 2011, 36: 38-44. 10.1097/SHK.0b013e3182168cce
Chang KC, Burnham CA, Compton SM, Rasche DP, Mazuski R, Smcdonough J, Unsinger J, Korman AJ, Green JM, Hotchkiss RS: Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit Care 2013, 17: R85. 10.1186/cc12711
LeMessurier K, Häcker H, Tuomanen E, Redecke V: Inhibition of T cells provides protection against early invasive pneumococcal disease. Infect Immun 2010, 78: 5287-5294. 10.1128/IAI.00431-10
Shubin NJ, Monaghan SF, Heffernan DS, Chung CS, Ayala A: B and T lymphocyte attenuator expression on CD4+ T-cells associates with sepsis and subsequent infections in ICU patients. Crit Care 2013, 17: R276. 10.1186/cc13131
Shubin NJ, Chung CS, Heffernan DS, Irwin LR, Monaghan SF, Ayala A: BTLA expression contributes to septic morbidity and mortality by inducing innate inflammatory cell dysfunction. J Leukoc Biol 2012, 92: 593-603. 10.1189/jlb.1211641
Kobayashi Y, Iwata A, Suzuki K, Suto A, Kawashima S, Saito Y, Owada T, Kobayashi M, Watanabe N, Nakajima H: B and T lymphocyte attenuator inhibits LPS-induced endotoxic shock by suppressing Toll-like receptor 4 signaling in innate immune cells. Proc Natl Acad Sci U S A 2013, 110: 5121-5126. 10.1073/pnas.1222093110
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
J-MC and DE wrote the review. DA read and amended it. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Cavaillon, JM., Eisen, D. & Annane, D. Is boosting the immune system in sepsis appropriate?. Crit Care 18, 216 (2014). https://doi.org/10.1186/cc13787
Published:
DOI: https://doi.org/10.1186/cc13787