
Abstract
Introduction
Nuclear factor (NF)-κB is central to the pathogenesis of inflammation in acute lung injury, but also to inflammation resolution and repair. We wished to determine whether overexpression of the NF-κB inhibitor IκBα could modulate the severity of acute and prolonged pneumonia-induced lung injury in a series of prospective randomized animal studies.
Methods
Adult male Sprague-Dawley rats were randomized to undergo intratracheal instillation of (a) 5 × 109 adenoassociated virus (AAV) vectors encoding the IκBα transgene (5 × 109 AAV-IκBα); (b) 1 × 1010 AAV-IκBα; (c) 5 × 1010 AAV-IκBα; or (d) vehicle alone. After intratracheal inoculation with Escherichia coli, the severity of the lung injury was measured in one series over a 4-hour period (acute pneumonia), and in a second series after 72 hours (prolonged pneumonia). Additional experiments examined the effects of IκBα and null-gene overexpression on E. coli-induced and sham pneumonia.
Results
In acute pneumonia, IκBα dose-dependently decreased lung injury, improving arterial oxygenation and lung static compliance, reducing alveolar protein leak and histologic injury, and decreasing alveolar IL-1β concentrations. Benefit was maximal at the intermediate (1 × 1010) IκBα vector dose; however, efficacy was diminished at the higher (5 × 1010) IκBα vector dose. In contrast, IκBα worsened prolonged pneumonia-induced lung injury, increased lung bacterial load, decreased lung compliance, and delayed resolution of the acute inflammatory response.
Conclusions
Inhibition of pulmonary NF-κB activity reduces early pneumonia-induced injury, but worsens injury and bacterial load during prolonged pneumonia.
Similar content being viewed by others


Introduction
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are life-threatening disorders, for which no specific therapy is known. When ARDS occurs in the setting of multisystem organ failure, mortality rates more than 60% have been reported, with significant morbidity in 50% of survivors [1]. ALI and ARDS develop most commonly in the context of severe sepsis [2], particularly infection with gram-negative bacilli such as Escherichia coli (E. coli) [3], and sepsis-induced ARDS has the worst outcome [4].
Nuclear factor kappa B (NF-κB) is a key transcriptional regulator in the setting of inflammation and injury and plays a role in diverse inflammatory disorders, including acute lung injury [5]. Activation of NF-κB occurs in response to diverse stimuli, such as endotoxin, which bind to cell-surface receptors that in turn activate the canonic and/or noncanonic signaling pathway. This signaling cascade ultimately results in the phosphorylation and inactivation of the cytosolic inhibitor IκB complex, which then dissociates, allowing NF-κB to translocate to the nucleus to initiate gene transcription [5]. Inhibition of NF-κB reduces injury in preclinical models of ALI, including ischemia-reperfusion [6], endotoxemia [7], and cecal ligation and puncture-induced sepsis [8]. However, NF-κB also exerts important cytoprotective effects, promoting cell survival, resolution of inflammation, and wound repair [9]. Of interest, NF-κB signaling plays a central role in the host response to lung bacterial infection [10]. Consequently, inhibition of NF-κB may constitute a double-edged sword, particularly in pneumonia-induced ALI/ARDS, in which immune competence is essential to eradication of the infectious agent [11].
We wished to determine the potential for inhibition of pulmonary NF-κB activity to modulate the severity of pneumonia-induced lung injury. We used a gene-based therapy approach, via intrapulmonary delivery of three different doses of adenoassociated viral vector encoding the NF-κB inhibitor IκBα gene (AAV-IκBα), to modulate the NF-κB signaling pathway in the lung. We hypothesized that pulmonary overexpression of the NF-κB inhibitor IκBα would (a) attenuate the severity of the lung injury induced by acute E. coli pneumonia; but would (b) worsen the severity of prolonged E. coli pneumonia-induced lung injury; and (c) a dose-response relation would exist, with higher AAV-IκBα doses having the greatest effect.
Materials and methods
Specific-pathogen-free adult male Sprague-Dawley rats (350 to 450 g) were used in all studies. The experimental model was based on those previously reported [12–14]. All work was approved by the National University of Ireland Galway Research Ethics Committee and conducted under license from the Department of Health, Ireland.
Preparation of AAV vectors
AAV-vector production was carried out as previously described, with several modifications [15]. The IκBα-SuperRepressor (IκBα-SR) gene (1,566 bp) was ligated into the pAAV-MCS vector (Agilent Technologies Inc., Santa Clara, CA, USA), and plasmid size confirmed by gel electrophoresis and validated by sequencing (Eurofins MWG Operon, Ebersberg, Germany). The IκBα- FLAG plasmid DNA and AAV serotype 6 envelope were generated and sent to Virapur for AAV production (VIRAPUR, 6160 Lusk Blvd., Suite C-101, San Diego, CA, USA). AAV serotype 6 was chosen because AAV has reduced immunogenicity, the virus plasmid size is sufficient for the IκBα-SuperRepressor (IκBα-SR) gene, and serotype 6 has demonstrated tropism for lung epithelial cells.
Viral vector particle titers were determined with quantitative real-time polymerase chain reaction (qRT-PCR) and aliquoted and stored at -80oC. As required, an aliquot was thawed and added to 75 μl of the porcine surfactant Curosurf (120 mg/ml) (Trinity-Chiesi Pharmaceuticals Limited, Cheadle, UK), and a final instillate volume of 300 μl was made up with PBS. For those animals receiving vehicle only, the instillate was 75 μl of Curosurf mixed with 225 µl of phosphate-buffered saline (PBS). Curosurf surfactant was added to each instillate, as it was demonstrated in prior studies to enhance spread and improve transgene expression [16].
Acute and prolonged E. coli-induced lung injury
Experimental design
Animals were randomized to intratracheal instillation of the following: (a) vehicle alone; (b) 5 × 109 drp AAV IκBα-SR; (c) 1 × 1010 drp AAV IκBα-SR; or (d) 5 × 1010 drp AAV IκBα-SR, and to undergo subsequent acute (Series 1) or prolonged (Series 2) pneumonia-induced ALI. Additional experimental series examined the effects of 1 × 1010 AAV-IκBα versus 1 × 1010 AAV-Null in both acute and prolonged E. coli and sham pneumonia.
Vector instillation
Animals were anesthetized by inhalational induction with isoflurane and an intraperitoneal injection of 40 mg/kg ketamine (Pfizer, Kent, UK). After confirmation of depth of anesthesia, laryngoscopy was performed (Welch Allyn Otoscope; Buckinghamshire, UK), and the trachea intubated with a size 16 intravenous catheter (BD Insyte; Becton Dickinson Ltd., Oxford, UK). After instillation of vector or vehicle, depending on the specific series, animals were extubated and allowed to recover from anesthesia.
E. coli/vehicle instillation
The E. coli used in these experiments is E5162 (serotype: O9 K30 H10) and was supplied by the National Collection of Type Cultures, Central Public Health Laboratory, London, England. The E. coli were stored on preservative beads (Protect, Lancashire, England) at -80°C. Beads were placed in 3-ml vials of peptone water (Cruinn Diagnostics, Dublin, Ireland) and incubated at 37°C for 18 to 24 hours to allow bacterial concentrations to reach a plateau. The bacterial suspension was then centrifuged and washed in phosphate-buffered saline to produce the inoculum. The bacterial load in each inoculum was determined by plating serial dilutions on agar plates. Preliminary experiments were performed to determine the bacterial load of intratracheal E. coli required to produce a lung injury over a 4-hour and over a 72-hour period.
Acute pneumonia protocol
Ninety-six hours after virus instillation, animals were anesthetized with intraperitoneal 80 mg/kg ketamine and 8 mg/kg xylazine, and anesthesia maintained with Alfaxalone (Alfaxadone 0.9% and alfadadolone acetate 0.3%). A tracheostomy tube was inserted, and intraarterial access was sited in the carotid artery. Muscle relaxation was induced with cisatracurium besylate, and the lungs were mechanically ventilated with 30% O2 in 70% N2. After 20 minutes, arterial blood gas measurement was performed and 1 × 1011 E. coli in a 300-μl PBS suspension (or vehicle alone) instilled via the tracheostomy [12, 13]. Animals were ventilated for 4 hours, with systemic arterial blood pressure, peak airway pressure, and body temperature continually measured. Lung compliance and arterial blood gas analysis was measured hourly, and body temperature was maintained at 36°C to 37.5°C.
Prolonged pneumonia protocol
Animals were anesthetized by inhalational induction with isoflurane and intraperitoneal 40-mg/kg ketamine (Pfizer, Kent, UK). After confirmation of anesthesia depth, 5 × 109 E. coli in a 300-μl PBS suspension was instilled into the trachea under direct vision, and the animals allowed to recover [17]. Animals were monitored closely for 72 hours after E. coli instillation, and then reanesthetized, tracheostomized, and mechanical ventilation was instituted, and injury severity assessed, as described [18].
Postmortem analyses
At the end of the protocols, the animals were killed by exsanguination under anesthesia, and the plasma snap-frozen for later analysis. The heart-lung block was dissected from the thorax, bronchoalveolar lavage (BAL) was performed, and BAL fluid differential leukocyte counts and lung bacterial colony counts were completed. BAL fluid was centrifuged, and the supernatant was snap-frozen and stored at -80°C. BAL concentrations of IL-1β, TNF-a, IL-6, CINC-1, IL-10, and KGF were determined by using ELISA (R&D Systems, Abingdon, UK), and BAL protein concentrations were measured (Micro BCA Protein assay kit; Pierce, Rockford, IL, USA) [16]. The left lung was isolated and fixed, and the extent of histologic lung damage determined by using quantitative stereologic techniques [18].
Assessment of transgene expression and efficacy
IκBα-FLAG transgene expression was determined in lung homogenates with real-time PCR and Western blotting, as previously described [16, 19]. In brief, RNA was extracted from lung tissue, cDNA was synthesised, and quantitative PCR was performed for IκBα-FLAG, normalized against a GAPDH control product. For Western blot IκBα-FLAG analysis, total cell protein was extracted, protein concentration was determined, and samples were electrophoresed on an SDS-PAGE gel and transferred to nitrocellulose [20]. Primary anti-human IκBα-FLAG monoclonal antibody (Sigma-Aldrich, St. Louis, MO, USA) was used, with secondary antibody conjugated to horseradish peroxidase (Cell Signaling Technology, Danvers, MA, USA), and the membrane incubated with a chemiluminescent substrate (SuperSignal West Pico; Pierce).
The effect of IκBα overexpression on the activation of the NF-κB pathway was assessed by measurement of nuclear accumulation of the activated P65 subunit of NF-κB [19]. Nuclear extracts were performed on homogenized rat lung tissue by using a NE-PER Nuclear and Cytoplasmic Extraction Kit (Fisher Scientific Ireland, Dublin, Ireland), and NF-κB (p65) measured by using an NF-κB Transcription Factor Assay Kit (Cayman Chemical Company, Ann Arbor, MI, USA).
Data presentation and analysis
Continuous responsive variables are summarized by using mean (SD) and median (interquartile range, IQR) as necessary. The proportion of animals surviving was analyzed by using the χ2 test. All other data were analyzed with one-way ANOVA, followed by the Dunnett test or by Kruskal-Wallis, followed by the Dunn test, with the vehicle group used as the reference group for all comparisons. The assumptions underlying all models were checked by using suitable residual plots. A P value of <0.05 was considered statistically significant.
Results
Acute pneumonia-induced ALI
Sixty-eight animals were entered into these experimental series. In the first set of experiments, 48 animals were randomized to receive (a) vehicle alone (n = 12); (b) 5 × 109 AAV-IκBα n = 12); (c) 1 × 1010 AAV-IκBα (n = 12); (d) 5 × 1010 AAV-IκBα (n = 12), and all animals subsequently underwent intratracheal E. coli instillation. No differences were noted between the groups at baseline (Table 1).
IκBα expression and function Pulmonary instillation of AAV-IκBα produced a dose-dependent increase in lung IκBα gene transcription (Figure 1A) and protein production (Figure 1B, C). Densitometry of IκBα-FLAG Western blot (n = 3 per group) confirmed this dose-dependent increase in IκBα protein (Figure 1C). IκBα overexpression decreased E. coli-induced NF-κB activation, as measured by nuclear accumulation of the activated P65 subunit of NF-κB (Figure 1D).

Overexpression of IκBα-reduced NF-κB activity. Delivery of the adeno-associated viral vector encoding IκBβ resulted in a dose-dependent increase in lung homogenate IκBβ FLAG mRNA expression (A) and IκBβ protein concentrations, as demonstrated by the representative Western blot (B), and by densitometry of Western blots (n = 3 per group) (C). IκBβ decreased Escherichia coli-induced nuclear accumulation of the activated P65 subunit of NF-κB (D). Vehicle, animals that received intratracheal vehicle alone; IκBβ 5 × 109, animals that received 5 × 109 AAV6 particles encoding IκBβ; IκBβ 1 × 1010, animals that received 1 × 1010 AAV6 particles encoding IκBβ; IκBβ 5 × 1010, animals that received 5 × 1010 AAV6 particles encoding IκBβ. *Significantly different from vehicle group (P < 0.05, ANOVA).
Survival and bacterial load IκBα overexpression enhanced animal survival, with survival rates of 33% in the Vehicle group, 58% with 5 × 109 IκBα, and 92% with 1 × 1010 IκBα Of concern, the highest IκBα (5 × 1010) dose abolished the survival benefit (Table 1). IκBα overexpression also enhanced the duration of animal survival, which followed this same pattern, with the effect greatest with the1 × 1010 IκBα dose (Table 1). Importantly, IκBα did not alter lung bacterial loads (Table 1).
IκBα modulated lung-injury severity
The E. coli instillation induced a severe acute lung injury (Figure 2). IκBα overexpression reduced the E. coli-induced decrement in arterial oxygenation, with benefit greatest at intermediate (1 × 1010) IκBα vector dose, but abolished at the higher (5 × 1010) IκBα dose (Figure 2A and Table 1). A similar pattern was seen with regard to the decrement in static compliance (Figure 2B), and pulmonary permeability, as assessed by protein leak into the BAL fluid (Figure 2C), but no effect was seen in regard to BAL neutrophil or mononuclear cell counts (Figure 2D and Table 1). IκBα reduced E. coli-induced histologic evidence of lung injury, with the intermediate (1 × 1010) vector dose of IκBα again most effective (Figure 3A through E).

IκBα modulated severity of acute pneumonia-induced ALI. Overexpression of IκBα attenuated the decrement in arterial oxygenation (A), and in static lung compliance (B), and decreased BAL protein concentrations (C), after Escherichia coli-induced ALI. Beneficial effects were greatest with the intermediate IκBα (1 × 1010 particles) dose but were abolished by the higher IκBα (5 × 1010 particles) dose. No effect of IκBα was noted on BAL neutrophil infiltration (D). The gray bars represent sham (that is, uninfected) animals. Vehicle, animals that received intratracheal vehicle alone; IκBα 5 × 109, animals that received 5 × 109 AAV6 particles encoding IκBα; IκBα 1 × 1010, animals that received 1 × 1010 AAV6 particles encoding IκBα; IκBα 5 × 1010, animals that received 5 × 1010 AAV6 particles encoding IκBα. Significantly different from vehicle group (P < 0.05, ANOVA).

IκBα modulated structural lung injury induced by acute pneumonia. Overexpression of IκBα attenuated the decrement in alveolar airspace and the increase in alveolar tissue (A) after pneumonia-induced lung injury. (B) Image from a pneumonia-injured lung that received intratracheal vehicle, demonstrating increased wall thickness and inflammatory cell infiltrate. (C through E) Images from pneumonia-injured lungs that received increasing doses of intratracheal vector encoding the IκBα transgene. The animals that received the intermediate (5 × 1010) dose demonstrated reduced injury and inflammatory cell infiltration. Vehicle, animals that received intratracheal vehicle alone; IκBα 5 × 109, animals that received 5 × 109 AAV6 particles encoding IκBα; IκBα 1 × 1010, animals that received 1 × 1010 AAV6 particles encoding IκBα; IκBα 5 × 1010, animals that received 5 × 1010 AAV6 particles encoding IκBα. Significantly different from vehicle group (P < 0.05, ANOVA). S cale bar, 200 μm.
IκBα modulates pulmonary inflammation
The E. coli instillation activated the inflammatory response. IκBα significantly decreased alveolar IL-1β, but did not alter BAL TNF-α or interleukin-6 concentrations (Figure 4A through C). IκBα dose-dependently modulated BAL CINC-1 and KGF, but did not alter BAL IL-10 concentrations (Figure 4D, F).

IκBα dose-dependently modulated the inflammatory response to acute pneumonia-induced ALI. Overexpression of IκBα attenuated the increase in BAL interleukin-1β (A), but did not alter BAL TNF-α (B), or interleukin-6 concentrations (C), after Escherichia coli-induced ALI. IκBα dose dependently decreased BAL CINC-1 (D), did not alter BAL IL-10 concentrations (E), and dose dependently decreased BAL KGF concentrations (F). These effects were greatest with the highest IκBα (5 × 1010 particles) dose. The gray bars represent sham (uninfected) animals. Vehicle, animals that received intratracheal vehicle alone; IκBα 5 × 109, animals that received 5 × 109 AAV6 particles encoding IκBα; IκBα 1 × 1010, animals that received 1 × 1010 AAV6 particles encoding IκBα; IκBα 5 × 1010, animals that received 5 × 10109 AAV6 particles encoding IκBα, BAL, bronchoalveolar lavage; IL-1β, interleukin-1β; TNF-α, tumor necrosis factor-α; CINC-1, cytokine-induced neutrophil chemoattractant-1; KGF, keratinocyte growth factor. Significantly different from vehicle group (P < 0.05, ANOVA).
Sham acute pneumonia and null-transgene series
Additional experiments were performed to determine the effect of overexpression of a null transgene (versus vehicle; n = 4 per group) on the severity of E. coli pneumonia, and to determine the effect of IκBα and null-gene overexpression in sham pneumonia (versus vehicle; n = 4 per group). In these studies, the null transgene did not alter the severity of E. coli-induced lung injury (Table 2). In addition, overexpression the null or IκBα transgenes in sham-injured animals did not alter lung function (Table 3).
Prolonged pneumonia-induced ALI
Fifty-six animals were entered into these experimental series. In the first set of experiments, 44 animals were randomized to receive (a) vehicle (n = 11); (b) 5 × 109 AAV-IκBα (n = 11); (c) 1 × 1010 AAV-IκBα (n = 11); (d) 5 × 1010 AAV-IκBα (n = 11), and underwent prolonged E. coli-induced lung injury. No differences were found between the groups at baseline (Table 4).
Animal Survival and Bacterial Load
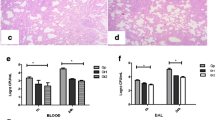
IκBα did not alter the number or duration of animal survival (Table 4). However, IκBα substantially increased lung E. coli bacterial loads at all three IκBα doses (Table 4).
IκBα worsens lung injury
E. coli instillation induced a severe lung injury compared with sham pneumonia (Figure 5). IκBα overexpression did not alter the decrement in arterial oxygenation (Figure 5A). IκBα overexpression dose dependently worsened static lung compliance (Figure 5B). IκBα did not alter alveolar protein leak (Figure 5C), but did increase the proportion of neutrophils in the alveolar infiltrate (Figure 5D), while decreasing alveolar mononuclear cells (Table 4). IκBα worsened E. coli- induced histologic injury, with the intermediate (1 × 1010) and higher (5 × 1010) dose of IκBα, resulting in significantly greater injury (Figure 6A through E).

IκBα worsened ALI induced by prolonged pneumonia. Overexpression of IκBα did not alter the decrement in arterial oxygenation (A). IκBα worsened the Escherichia coli-induced decrement in static lung compliance, with effect greatest with 5 × 1010 IκBα particles (B). IκBα did not alter BAL protein concentrations (C), but did significantly increase the percentage of neutrophils in the alveolar infiltrate (D). The gray bars represent sham (uninfected) animals. Vehicle, animals that received intratracheal vehicle alone; IκBα 5 × 109, animals that received 5 × 109 AAV6 particles encoding IκBα; IκBα 1 × 1010, animals that received 1 × 1010 AAV6 particles encoding IκBα; IκBα 5 × 1010, animals that received 5 × 1010 AAV6 particles encoding IκBα. Significantly different from vehicle group (P < 0.05, ANOVA).

IκBα worsened structural lung injury induced by prolonged pneumonia. Overexpression of IκBα worsened the decrement in alveolar airspace and the increase in alveolar tissue (A) after pneumonia-induced lung injury. (B) Image from a prolonged pneumonia-injured lung that received intratracheal vehicle, demonstrating increased wall thickness and inflammatory cell infiltrate. (C through E) Images from a pneumonia-injured lung that received increasing doses of intratracheal vector encoding the IκBα transgene. The animals that received 1 to 5 × 1010 doses demonstrated greater injury. Vehicle, animals that received intratracheal vehicle alone; IκBα 5 × 109, animals that received 5 × 109 AAV6 particles encoding IκBα; IκBα 1 × 1010, animals that received 1 × 1010 AAV6 particles encoding IκBα; IκBα 5 × 1010, animals that received 5 × 1010 AAV6 particles encoding IκBα. *Significantly different from vehicle group (P < 0.05, ANOVA). Scale bar, 200 μm.
IκBα modulates pulmonary inflammation
Prolonged E. coli pneumonia activated the inflammatory response compared with sham pneumonia (Figure 7). IκBα overexpression dose-dependently increased alveolar IL-1β (Figure 7A) and TNF-α (Figure 7B), with concentrations highest after 5 × 1010 IκBα. In contrast, no effect was seen on alveolar IL-6 (Figure 7C) or CINC-1 (Figure 7D) concentrations. IκBα increased alveolar IL-10 (Figure 7E) and KGF concentrations, with a step-wise dose effect seen (Figure 7F).

IκBα dose-dependently modulated the inflammatory response to prolonged pneumonia. Overexpression of IκBα dose-dependently increased BAL interleukin-1β (A) and TNF-α (B) concentrations compared with vehicle. No effect of IκBα was seen on interleukin-6 (C) or CINC-1 (D) concentrations. IκBα dose-dependently increased BAL IL-10 (E), and KGF concentrations (F). These effects were greatest with the highest IκBα (5 × 1010 particles) dose. The gray bars represent sham (uninfected) animals. Vehicle, animals that received intratracheal vehicle alone; IκBα 5 × 109, animals that received 5 × 109 AAV6 particles encoding IκBα; IκBα 1 × 1010, animals that received 1 × 1010 AAV6 particles encoding IκBα; IκBα 5 × 1010, animals that received 5 × 1010 AAV6 particles encoding IκBα. *Significantly different from vehicle group (P < 0.05, ANOVA).
Sham Prolonged Pneumonia
In a subsequent experiment, 12 animals were randomized to receive: (a) Vehicle (n = 4); (b) 1 × 1010 AAV-Null (n = 4); and (3) 1 × 1010 AAV-IκBα and to undergo subsequent sham (vehicle) instillation. The null or IκBα transgenes did not alter lung function in sham infected animals (Table 5).
Discussion
Pulmonary overexpression of IκBα attenuated acute pneumonia-induced lung injury, decreasing the severity of the decrement in lung function, whereas also decreasing the inflammatory response. In contrast, IκBα worsened lung injury and inflammation induced by prolonged pneumonia, increasing lung bacterial load and delaying resolution of the inflammatory response. These findings provide novel insights into the effects of NF-κB in pneumonia-induced lung injury, and raise concerns regarding therapeutic potential of inhibiting NF-κB, particularly in prolonged untreated pneumonia.
E. coli acute and prolonged pneumonia
Infection with gram-negative bacilli such as E. coli is the commonest cause of ARDS [21, 22] and is also a very common complication of ARDS due to other causes [23]. We studied E. coli-induced pneumonia, a well-characterized animal model that mimics the clinical development of ARDS very closely [23–28]. Intratracheal instillation of 1 × 1011 E. coli resulted in physiological and pathologic changes consistent with a severe lung injury over a 6-hour period, similar to that previously reported [12, 13]. Instillation of a lower dose of 5 × 109 E. coli produced a more gradually evolving injury over a 72-hour period, as previously described [14, 17].
NF-κB: role in lung inflammation
The role of the NF-κB signaling in the host immune response to lung injury is increasingly well understood [19]. NF-κB activation pathway gene polymorphisms alter the susceptibility to [29] and severity [30] of clinical ARDS. NF-κB is a dimer of a number of related proteins, including RelA (also known as P65), p50, p52, RelB, and cRel, with the RelA and p50 heterodimer, the most common form. On cell activation, by stimuli such as gram-negative bacterial endotoxin, diverse signaling pathways are activated, which converge to phosphorylate and activate the IκB kinase complex proteins (IKK), which then phosphorylate and inactivate the IκB proteins, which dissociates from NF-κB. The active NF-κB translocates to the nucleus and binds to specific cognate-binding sequences in the promoter or enhancer regions of different target genes to initiate transcription [5].
The therapeutic potential of strategies to inhibit NF-κB is evident from the demonstration that NF-κB inhibition decreases injury in nonseptic ALI models, including pulmonary [6] and systemic reperfusion, and endotoxemia [7]. Pulmonary overexpression of the RelB member of the NF-κB family decreases cigarette smoke-induced lung injury [31].
ARDS, pneumonia, and NF-κB
The effects of modulation of the NF-κB in the setting of lung bacterial infection are less well understood [19]. NF-κB decoy oligodeoxynucleotides reduce ALI in mice in the early phases of cecal ligation and puncture-induced sepsis [8]. Selective inhibition of vascular endothelial NF-κB activity in endotoxemic transgenic mice reduced lung inflammation and increased survival [32]. Of interest, this approach improved survival and systemic organ function, but did not alter bacterial clearance, in septic mice [32]. However, others have found inhibition of NF-κB signaling to exert detrimental effects in the setting of infection. Inhibition of hepatocyte NF-κB activity reduced Listeria monocytogenes clearance, decreasing murine survival [11]. In contrast, clearance of pseudomonas bacteria from the mouse lung was enhanced by pulmonary overexpression of the RelA subunit of NF-κB [33], suggesting that the NF-κB pathway plays a pivotal role in maintaining immune competence and is essential to eradication of the infectious agent [11].
NF-κB: role in lung inflammation, injury, and repair
NF-κB also promotes cell survival, resolution of inflammation, and repair after injury. Inhibition of NF-κB signaling retards pulmonary [20] and intestinal [34] epithelial wound healing. Maturational differences in lung NF-κB activation profiles exist, with NF-κB activation protecting the lung against hyperoxia [35] and endotoxemia [36] in neonatal rats. Consequently strategies to inhibit NF-κB, particularly if not spatially or temporally targeted, may have detrimental effects.
Targeting pulmonary NF-κB
We wished to determine whether inhibition of pulmonary NF-κB activity could modulate the severity of pneumonia-induced lung injury. We found that pulmonary overexpression of the IκBα gene did reduce acute pneumonia-induced injury. IκBα decreased the decrement in arterial oxygenation and lung static compliance, decreased alveolar protein leak, and decreased histologic injury, compared with vehicle. IκBα modulated the cytokine response to E. coli instillation, decreasing alveolar IL-1β but increasing CINC-1 concentrations. Of importance, these effects were dose dependent, with benefit maximal at the intermediate (1 × 1010) IκBα dose, and a loss of efficacy at the higher (1 × 1010) IκBα concentration.
In contrast, in prolonged untreated E. coli pneumonia, pulmonary IκBα gene overexpression worsened the lung injury, and increased lung bacterial load. IκBα delayed the resolution of the acute inflammatory response, increasing the proportion of alveolar neutrophils while decreasing alveolar mononuclear cells, which comprise lymphocytes and macrophages and are considered important to repair. IκBα increased alveolar concentrations of TNF-α and IL-1β, cytokines implicated in the early phase of the response to septic insult. IκBα increased alveolar IL-10 concentrations, which may partly explain the ineffective response to the bacterial insult.
NF-κB and sepsis: an integrated paradigm
The contrasting effects of NF-κB inhibition in acute versus prolonged pneumonia generate a number of important insights. First, the finding that NF-κB inhibition reduced the severity of acute pneumonia and decreased the host response induced ALI suggests that any benefit in this setting is mediated via a decreased host response to the instillation of E. coli [37]. This is consistent with the concept that in the early phases of pneumonia, secreted toxins, and the host immune response may be the predominant source of injury [37, 38]. This is supported by the fact that NF-κB inhibition is protective in nonseptic inflammatory ALI models. Second, this effect was dose dependent, with lower and intermediate IκBα doses protective and the beneficial effects ablated at higher doses. This suggests a U-shaped dose-effect curve, reducing the therapeutic utility of approaches to inhibit NF-κB in the setting of acute pneumonia. Third, IκBα worsened the severity of prolonged pneumonia, substantially increasing lung bacterial loads, and resulting in a persisting pulmonary inflammatory response. IκBα, by inhibiting the early host response to E. coli instillation, may have led to a persistence of infection, ultimately worsening lung injury. Finally, the beneficial effects of NF-κB inhibition in acute pneumonia were lost at the highest dose. The reasons for this is unclear, but may include NF-κB inhibition mediated reduction in lung epithelial repair [20], apoptosis of airway epithelial cells [16] and effects on lung macrophage function [39].
Conclusions
IκBα overexpression reduces injury severity and inflammation in early pneumonia, but slows resolution of inflammation and bacterial clearance and worsens injury during prolonged pneumonia. These findings raise serious questions regarding the utility of strategies that inhibit the innate immune response, such as NF-κB, in live bacterial pneumonia.
Key Message: Inhibition of pulmonary NF-κB activity decreases the severity of early pneumonia-induced lung injury, but worsens injury severity and bacterial load during prolonged pneumonia.
Abbreviations
- AAV:
-
adenoassociated virus
- ALI:
-
acute lung injury
- ANOVA:
-
analysis of variance
- ARDS:
-
acute respiratory distress syndrome
- BAL:
-
bronchoalveolar lavage
- CINC-1:
-
cytokine-induced neutrophil chemoattractant-1
- DNA:
-
deoxyribonucleic acid
- IL:
-
interleukin
- IQR:
-
interquartile range
- KGF:
-
keratinocyte growth factor
- qRT-PCR:
-
quantitative real-time polymerase chain reaction
- NF-κB:
-
nuclear factor kappa B
- PBS:
-
phosphate-buffered saline
- RNA:
-
ribonucleic acid
- TNF-α:
-
tumor necrosis factor-α
References
Rubenfeld GD: Epidemiology of acute lung injury. Crit Care Med 2003, 31: S276-284. 10.1097/01.CCM.0000057904.62683.2B
Zilberberg MD, Epstein SK: Acute lung injury in the medical ICU: comorbid conditions, age, etiology, and hospital outcome. Am J Respir Crit Care Med 1998, 157: 1159-1164. 10.1164/ajrccm.157.4.9704088
Markowicz P, Wolff M, Djedaini K, Cohen Y, Chastre J, Delclaux C, Merrer J, Herman B, Veber B, Fontaine A, et al.: Multicenter prospective study of ventilator-associated pneumonia during acute respiratory distress syndrome: incidence, prognosis, and risk factors: ARDS Study Group. Am J Respir Crit Care Med 2000, 161: 1942-1948. 10.1164/ajrccm.161.6.9909122
TenHoor T, Mannino DM, Moss M: Risk factors for ARDS in the United States: analysis of the 1993 National Mortality Followback Study. Chest 2001, 119: 1179-1178. 10.1378/chest.119.4.1179
Park GY, Christman JW: Nuclear factor kappa B is a promising therapeutic target in inflammatory lung disease. Curr Drug Targets 2006, 7: 661-668. 10.2174/138945006777435317
Ishiyama T, Dharmarajan S, Hayama M, Moriya H, Grapperhaus K, Patterson GA: Inhibition of nuclear factor kappaB by IkappaB superrepressor gene transfer ameliorates ischemia-reperfusion injury after experimental lung transplantation. J Thorac Cardiovasc Surg 2005, 130: 194-201. 10.1016/j.jtcvs.2005.02.040
Matsuda N, Hattori Y, Takahashi Y, Nishihira J, Jesmin S, Kobayashi M, Gando S: Therapeutic effect of in vivo transfection of transcription factor decoy to NF-kappaB on septic lung in mice. Am J Physiol 2004, 287: L1248-1255.
Matsuda N, Hattori Y, Jesmin S, Gando S: Nuclear factor-kappaB decoy oligodeoxynucleotides prevent acute lung injury in mice with cecal ligation and puncture-induced sepsis. Mol Pharmacol 2005, 67: 1018-1025. 10.1124/mol.104.005926
Rahman A, Fazal F: Blocking NF-kappaB: an inflammatory issue. Proc Am Thorac Soc 2011, 8: 497-503. 10.1513/pats.201101-009MW
Batra S, Balamayooran G, Sahoo MK: Nuclear factor-kappaB: a key regulator in health and disease of lungs. Arch Immunol Ther Exp 2011, 59: 335-351. 10.1007/s00005-011-0136-z
Lavon I, Goldberg I, Amit S, Landsman L, Jung S, Tsuberi BZ, Barshack I, Kopolovic J, Galun E, Bujard H, et al.: High susceptibility to bacterial infection, but no liver dysfunction, in mice compromised for hepatocyte NF-kappaB activation. Nat Med 2000, 6: 573-577. 10.1038/75057
O'Croinin DF, Hopkins NO, Moore MM, Boylan JF, McLoughlin P, Laffey JG: Hypercapnic acidosis does not modulate the severity of bacterial pneumonia-induced lung injury. Crit Care Med 2005, 33: 2606-2612. 10.1097/01.CCM.0000186761.41090.C6
Ni Chonghaile M, Higgins BD, Costello JF, Laffey JG: Hypercapnic acidosis attenuates severe acute bacterial pneumonia-induced lung injury by a neutrophil-independent mechanism. Crit Care Med 2008, 36: 3135-3144. 10.1097/CCM.0b013e31818f0d13
Chonghaile MN, Higgins BD, Costello J, Laffey JG: Hypercapnic acidosis attenuates lung injury induced by established bacterial pneumonia. Anesthesiology 2008, 109: 837-848. 10.1097/ALN.0b013e3181895fb7
Sen S, Conroy S, Hynes SO, McMahon J, O'Doherty A, Bartlett JS, Akhtar Y, Adegbola T, Connolly CE, Sultan S, et al.: Gene delivery to the vasculature mediated by low-titre adeno-associated virus serotypes 1 and 5. J Gene Med 2008, 10: 143-151. 10.1002/jgm.1133
Hassett P, Curley GF, Contreras M, Masterson C, Higgins BD, O'Brien T, Devaney J, O'Toole D, Laffey JG: Overexpression of pulmonary extracellular superoxide dismutase attenuates endotoxin-induced acute lung injury. Intensive Care Med 2011, 37: 1680-1687. 10.1007/s00134-011-2309-y
O'Croinin DF, Nichol AD, Hopkins N, Boylan J, O'Brien S, O'Connor C, Laffey JG, McLoughlin P: Sustained hypercapnic acidosis during pulmonary infection increases bacterial load and worsens lung injury. Crit Care Med 2008, 36: 2128-2135. 10.1097/CCM.0b013e31817d1b59
Laffey JG, Honan D, Hopkins N, Hyvelin JM, Boylan JF, McLoughlin P: Hypercapnic acidosis attenuates endotoxin-induced acute lung injury. Am J Respir Crit Care Med 2004, 169: 46-56. 10.1164/rccm.200205-394OC
Contreras M, Ansari B, Curley G, Higgins BD, Hassett P, O'Toole D, Laffey JG: Hypercapnic acidosis attenuates ventilation-induced lung injury by a nuclear factor-kappaB-dependent mechanism. Crit Care Med 2012, 40: 2622-2630. 10.1097/CCM.0b013e318258f8b4
O'Toole D, Hassett P, Contreras M, Higgins B, McKeown S, McAuley D, O'Brien T, Laffey J: Hypercapnic acidosis attenuates pulmonary epithelial wound repair by an NF-kB dependent mechanism. Thorax 2009, 64: 976-982. 10.1136/thx.2008.110304
Fein AM, Lippmann M, Holtzman H, Eliraz A, Goldberg SK: The risk factors, incidence, and prognosis of ARDS following septicemia. Chest 1983, 83: 40-42. 10.1378/chest.83.1.40
Martin MA, Silverman HJ: Gram-negative sepsis and the adult respiratory distress syndrome. Clin Infect Dis 1992, 14: 1213-1228. 10.1093/clinids/14.6.1213
Markowicz P, Wolff M, Djedaini K, Cohen Y, Chastre J, Delclaux C, Merrer J, Herman B, Veber B, Fontaine A, et al.: Multicenter prospective study of ventilator-associated pneumonia during acute respiratory distress syndrome: incidence, prognosis, and risk factors: ARDS Study Group. Am J Respir Crit Care Med 2000, 161: 1942-1948. 10.1164/ajrccm.161.6.9909122
Freeman BD, Correa R, Karzai W, Natanson C, Patterson M, Banks S, Fitz Y, Danner RL, Wilson L, Eichacker PQ: Controlled trials of rG-CSF and CD11b-directed MAb during hyperoxia and E. coli pneumonia in rats. J Appl Physiol 1996, 80: 2066-2076.
Karzai W, von Specht BU, Parent C, Haberstroh J, Wollersen K, Natanson C, Banks SM, Eichacker PQ: G-CSF during Escherichia coli versus Staphylococcus aureus pneumonia in rats has fundamentally different and opposite effects. Am J Respir Crit Care Med 1999, 159: 1377-1382. 10.1164/ajrccm.159.5.9806082
Russo TA, Bartholomew LA, Davidson BA, Helinski JD, Carlino UB, Knight PR, Beers MF, Atochina EN, Notter RH, Holm BA: Total extracellular surfactant is increased but abnormal in a rat model of gram-negative bacterial pneumonia. Am J Physiol Lung Cell Mol Physiol 2002, 283: L655-663.
Song GW, Robertson B, Curstedt T, Gan XZ, Huang WX: Surfactant treatment in experimental Escherichia coli pneumonia. Acta Anaesthesiol Scand 1996, 40: 1154-1160. 10.1111/j.1399-6576.1996.tb05580.x
Zeni F, Parent C, Correa R, Natanson C, Freeman B, Fontana J, Quezado M, Danner RL, Fitz Y, Richmond S, et al.: ICAM-1 and CD11b inhibition worsen outcome in rats with E. coli pneumonia. J Appl Physiol 1999, 87: 299-307.
Zhai R, Zhou W, Gong MN, Thompson BT, Su L, Yu C, Kraft P, Christiani DC: Inhibitor kappaB-alpha haplotype GTC is associated with susceptibility to acute respiratory distress syndrome in Caucasians. Crit Care Med 2007, 35: 893-898. 10.1097/01.CCM.0000256845.92640.38
Adamzik M, Frey UH, Rieman K, Sixt S, Beiderlinden M, Siffert W, Peters J: Insertion/deletion polymorphism in the promoter of NFKB1 influences severity but not mortality of acute respiratory distress syndrome. Intensive Care Med 2007, 33: 1199-1203. 10.1007/s00134-007-0649-4
McMillan DH, Baglole CJ, Thatcher TH, Maggirwar S, Sime PJ, Phipps RP: Lung-targeted overexpression of the NF-kappaB member RelB inhibits cigarette smoke-induced inflammation. Am J Pathol 2011, 179: 125-133. 10.1016/j.ajpath.2011.03.030
Ye X, Ding J, Zhou X, Chen G, Liu SF: Divergent roles of endothelial NF-kappaB in multiple organ injury and bacterial clearance in mouse models of sepsis. J Exp Med 2008, 205: 1303-1315. 10.1084/jem.20071393
Sadikot RT, Zeng H, Joo M, Everhart MB, Sherrill TP, Li B, Cheng DS, Yull FE, Christman JW, Blackwell TS: Targeted immunomodulation of the NF-kappaB pathway in airway epithelium impacts host defense against Pseudomonas aeruginosa . J Immunol 2006, 176: 4923-4930.
Egan LJ, Eckmann L, Greten FR, Chae S, Li ZW, Myhre GM, Robine S, Karin M, Kagnoff MF: IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci USA 2004, 101: 2452-2457. 10.1073/pnas.0306734101
Yang G, Abate A, George AG, Weng YH, Dennery PA: Maturational differences in lung NF-kappaB activation and their role in tolerance to hyperoxia. J Clin Invest 2004, 114: 669-678.
Alvira CM, Abate A, Yang G, Dennery PA, Rabinovitch M: Nuclear factor-kappaB activation in neonatal mouse lung protects against lipopolysaccharide-induced inflammation. Am J Respir Crit Care Med 2007, 175: 805-815. 10.1164/rccm.200608-1162OC
Curley G, Contreras MM, Nichol AD, Higgins BD, Laffey JG: Hypercapnia and acidosis in sepsis: a double-edged sword? Anesthesiology 2010, 112: 462-472. 10.1097/ALN.0b013e3181ca361f
Laffey JG, O'Croinin D, McLoughlin P, Kavanagh BP: Permissive hypercapnia: role in protective lung ventilatory strategies. Intensive Care Med 2004, 30: 347-356. 10.1007/s00134-003-2051-1
Groesdonk HV, Schlottmann S, Richter F, Georgieff M, Senftleben U: Escherichia coli prevents phagocytosis-induced death of macrophages via classical NF-kappaB signaling, a link to T-cell activation. Infect Immun 2006, 74: 5989-6000. 10.1128/IAI.00138-06
Acknowledgements
The authors acknowledge the help of Dr. Shirley Hanley in regard to the FACS analyses, and of Dr Jeremy A. Scott for his critical review of the manuscript. Data from this manuscript were presented at the American Thoracic Society Annual Scientific Meeting, Denver, Colorado, May 2011.
This study was funded by the Health Research Board, Ireland and by European Research Council. Dr Devaney is a postdoctoral researcher with the Health Research Board, Ireland (grant RP/2008/193). Dr O'Toole is a Research Fellow with the European Research Council (grant ERC-2007-StG 207777). Dr Laffey is supported by a Merit Award from the Department of Anesthesia at the University of Toronto.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JD performed the animal experiments, assays, and histologic analyses, analyzed the data, drafted the manuscript, and agreed to the final submitted version. GFC and MH assisted with animal experiments, contributed to drafting the manuscript, and agreed to the final submitted version. CM, BA, and DOT performed assays and histologic analyses, contributed to drafting the manuscript, and agreed to the final submitted version. TOB conceived and designed the experiments, provided viral vector expertise, drafted the manuscript, and agreed to the final submitted version. JL conceived and designed the experiments, analyzed the data, drafted the manuscript, and agreed to the final submitted version.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Devaney, J., Curley, G.F., Hayes, M. et al. Inhibition of pulmonary nuclear factor kappa-B decreases the severity of acute Escherichia coli pneumonia but worsens prolonged pneumonia. Crit Care 17, R82 (2013). https://doi.org/10.1186/cc12696
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/cc12696