
Abstract
Erythropoietin (EPO) is known to have numerous biological functions. Its primary function in the body is to increase red blood cell numbers by way of preventing the apoptosis of erythroid progenitor cells via the homodimeric EPO receptor. The discovery that the local production of EPO within the brain in response to hypoxia or ischemia protects neurons against injury via an anti-apoptotic effect formed the basis of the hypothesis that the local generation of EPO limits the extent of injury. Although the hypothesis proved to be true in pre-clinical models of ischemia/reperfusion injury and inflammation, the randomized, controlled clinical trials that followed demonstrated serious adverse events of EPO due to activation of the hematopoietic system. Consequently, derivatives of EPO that lacked erythropoietic activity were discovered to reduce injury in many pre-clinical models associated with ischemia and inflammation. Unfortunately, there are no published clinical trials to determine the efficacy of non-erythropoietic derivatives of EPO in humans.
Similar content being viewed by others
Introduction
For more than a decade now, we have known that the biological properties of erythropoietin (EPO) are not restricted to its erythropoietic effects [1], but also include very important tissue-protective effects. The erythropoietic effects of EPO are mediated by the homodimeric EPO receptor (EPOR), a class 1 cytokine receptor, and are clinically exploited in the treatment of anemia associated with chronic kidney disease, cancer and other chronic illnesses. In 1998, Sakanaka and colleagues [2] reported that the degree of apoptosis caused by brief periods of cerebral ischemia in gerbils was augmented when the effects of endogenous EPO were blocked (by using a soluble EPOR). This discovery formed the basis for the hypothesis that the local production of EPO in response to ischemia limits the progression and extent of the injury caused by noxious stimuli. There is now good evidence that endogenous or exogenous (human recombinant) EPO limits the tissue injury caused by ischemia and inflammation in the brain or other organs [2–8].
The above preclinical studies stimulated the first controlled, phase II clinical study (small case series) aimed at evaluating both the safety and efficacy (in terms of tissue protection) of EPO in patients with stroke (Göttingen EPO Stroke Study) [9]. Evaluation of the safety profile of EPO was deemed important, as EPO has the potential of raising hematocrit and, hence, increasing the likelihood of further transient ischemic attacks. During the 30-day follow-up period hematocrit, hemoglobin, and red blood cell counts all remained normal despite a high dose of EPO (100,000 IU over 3 days), demonstrating that EPO is well tolerated. Clinical assessment using neurological scoring and magnetic resonance imaging showed an improvement in neurological activity and infarct size 1 month following stroke in patients treated with EPO.
The more recent, larger, German Multicenter EPO Stroke Trial, which was designed to reproduce the results of the Göttingen EPO Stroke Study, unexpectedly documented that a combination of EPO and recombinant tissue plasminogen activator (tPA) is not advantageous and may even be detrimental [10]. Patients were infused with 40,000 IU of EPO over 30 minutes within the first 6 hours after the onset of symptoms and EPO (at the same dose) was re-administered 24 and 48 hours after the first dose. During the 90-day follow-up period, patients that had received EPO had an increased risk of intracerebral hemorrhage, brain edema, thromboembolic events and death, highlighting the safety risk associated with EPO treatment. Moreover, patients who received both EPO and tPA (60% of patients) were significantly more likely to die than those receiving EPO only. The authors speculated that it may be possible that the beneficial effects of EPO (observed in the previous trial) may have been abolished by tPA, resulting in a higher mortality.
The potential beneficial and/or adverse effects of EPO were also investigated in a large, multicenter phase II clinical trial in critically ill patients. In patients with trauma-hemorrhage, EPO (40,000 IU per week for 3 weeks) significantly reduced mortality, but also significantly increased the incidence of thrombovascular events [11]. This trial clearly indicated, for the first time, that the tissue-protective effects of EPO may occur in humans and can potentially be exploited to limit tissue injury and to improve outcome.
The above clinical trials highlight the need to be cautious when planning or conducting trials with EPO as increases in hematocrit may lead to excessive thrombosis and significant adverse effects [12]. The above data also indicate that a strategy that delivers the tissue-protective effects of EPO without the known adverse effects may offer clinical benefit in patients with trauma hemorrhage and potentially many other diseases.
Is there a need for an alternative to erythropoietin?
Leist and colleagues [13] first demonstrated that, by modifying the protein backbone of EPO (to produce carbamylated EPO (CEPO)), one is able to retain the tissue-protective effects of EPO. It was found that the concentrations (doses) of CEPO that confer tissue protection were higher than those that caused erythropoiesis, and that its affinity for the 'classical' EPOR was dramatically reduced. This inevitably also implies that a strategy that leads to increased and sustained activation of the EPOR (following multiple administrations of high doses of EPO) will result in an increase in pro-thrombotic events [14, 15]. The Early Intervention with EPO Does Not Affect the Outcome of AKI (EARYARF) trial did not, however, demonstrate a significant increase in serious adverse events associated with EPO treatment when compared to placebo (154 versus 115, respectively) [14]. This is supported by two similar multicenter trials (EPO-2 and EPO-3), which have shown that EPO treatment does not increase the incidence of total serious adverse events when compared to placebo (158 versus 259, respectively) [15]. However, the EPO-3 trial did demonstrate an increase in circulating red cell mass; this combined with EPO-induced platelet aggregability may contribute to poor tissue perfusion, which is undesirable in conditions associated with ischemia/reperfusion. This is supported by pre-clinical data that show that if mice that overexpress endogenous EPO are subjected to cerebral ischemia, the resulting infarct size is larger [16].
Over the past 30 years several other molecules have been actively developed that have tissue-protective properties but are devoid of any erythropoietic activity. The first of these molecules, desialylated-EPO or asialo-EPO, was first discovered whilst assessing the biological activity of EPO [17]. This was later expanded in the late 1980s by enzymatically removing the sialic acid moieties terminating the oligosaccharides of EPO and thus creating a non-erythropoietic EPO analogue (which still has high affinity for the EPOR) with a serum half-life of up to 2 minutes compared to the very long (several hours) half-life of EPO [18, 19]. Since 2003, it has been observed that asialo-EPO reduces the tissue injury caused by cerebral ischemia, spinal cord compression or sciatic nerve crush [20] as well as ischemia-reperfusion injury of the kidney [21], gut [22] and heart [23] (Table 1). Although asialo-EPO is clearly tissue protective, it is still, in principle, able to activate the EPOR and, hence, continuous/multiple administrations of asialo-EPO may still have an impact on the bone marrow and erythropoiesis.
This problem stimulated many to search for EPO analogues that are tissue-protective without being erythropoietic [24–27]. One such modification with potassium cyanate involves the transformation of all the lysine residues in EPO to homocitrulline, which led to the development of CEPO (as described above) in 2004. CEPO has been demonstrated to be non-erythropoietic in vitro (UT-7 cells) and in vivo in the mouse, and, unlike asialo-EPO, is unable to bind to the EPOR [13]. Although, CEPO does not bind to the EPOR, it has a pharmaco-dynamic profile (for example, tissue-protective effects) similar to those of EPO or asialo-EPO in a variety of organs, including the brain [13], heart [28] and kidney [29]. The existence of molecules that confer tissue-protection in the absence of erythropoiesis led to the development of the hypothesis that the tissue-protective effects of EPO are mediated by a receptor that is different from the classical EPOR.
A second erythropoietin receptor that is tissue protective
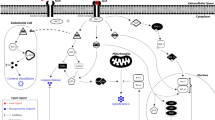
In 2004, Brines and colleagues [30] proposed that the tissue protective effects of EPO and its derivatives may be mediated, in part, by the beta common receptor (βcR). The βcR is primarily a signal transduction subunit, which is shared by the α-chain subunits of the interleukin-5, interleukin-3 and granulocyte-macrophage colony-stimulating factor receptors. The α-chains are able to bind their ligand with low affinity (1 to 100 nM), but the complexes are not able to signal in the absence of the βcR. On the other hand, the βcR does not measurably bind a ligand by itself, but acts as an affinity converter in the presence of an α-chain, allowing ligand binding at concentrations in the picomolar range. On the cell surface the βcR exists as a pre-formed, intertwined dimer [31]. It is not until a ligand binds to its corresponding α-chain subunit that the βcR becomes covalently attached to the α-chain subunit [32]. We know from βcR knock-out mice that the βcR is not required for erythropoiesis as these mice have normal erythrocyte maturation [30]. It has also been previously reported that the EPOR functionally associates with the βcR [33, 34] to generate an EPOR-βcR complex, which gives credibility to the hypothesis of a second EPO receptor. Further evidence to support this hypothesis comes from Campana and colleagues [35], who were able to identify a specific 17 amino acid sequence of EPO that is tissue protective but lacks erythropoietic activity, suggesting that EPO may have two distinct receptor binding domains. Evidence also suggests that the formation of type I cytokine receptors is often by spontaneous self-association of subunits already present in the cell membrane, where the probability of self-assembly is determined by the abundance of each subunit; for example, the probability of EPOR homodimerization increases as the concentration of EPOR in the membrane rises [36]. The EPOR-βcR complex is thought to be locally up-regulated following tissue injury [37, 38], although the expression/regulation of receptor subunits of the βcR family present within the cell membrane is not very well understood and requires further extensive study. In vitro reports using a differentiated human neuroblastoma SH-SY5Y cell line that expresses the EPOR, but not the βcR, demonstrate that the protective effects of EPO against staurosporine-induced apoptosis are mediated by the EPOR homodimer [39]. Conversely, studies to demonstrate the cytoprotective nature of EPO in cells solely expressing the βcR are lacking.
Further structure activity relationship studies have identified that, in aqueous solutions, the tertiary structure of EPO is relatively well defined because of the interaction of the hydrophobic content of its four α-helices (A, B, C and D helices), constraining the molecule into a compact, relatively rigid, globular structure. When EPO is bound to the EPOR [40], helix B and parts of the AB and CD loops face the aqueous medium, away from the binding sites of EPOR (Protein Data Bank (PDB) ID code 1EER). These regions do not contain lysine and, therefore, are not modified by carbamylation of EPO [41]. Further surface-stimulation analysis of EPO found that a select set of amino acids on the aqueous face of helix B possesses pharmacodynamic activity similar to EPO, without having any erythropoietic activity. Based on these studies, a peptide called pyroglutamate helix B surface peptide (pHBSP) has been generated. pHBSP (identical to ARA-290) has been shown to be tissue-protective in numerous preclinical models of ischemia-reperfusion injury (kidney, brain, heart) and trauma hemorrhage [42–44]. There is very recent evidence that the EPOR-βcR complex may be the tissue-protective receptor for EPO and its analogues; for instance, pHBSP produces a sustained inhibition of hypersensitivity after nerve injury in mice, which is abolished in βcR knock-out mice [45]. Similarly, EPO treatment reduced the degree of spinal cord injury in a model of spinal cord compression and this protection was lost in βcR knock-out mice subjected to spinal cord injury [30]. Unfortunately, it is not possible to prove in knock-out animals if the same is true for EPOR as mice with a targeted deletion for EPOR are not viable [46].
Erythropoietin derivatives in clinical trials - where are they?
To date, there have been numerous clinical trials with EPO that have, for the most part, failed to demonstrate any clear efficacy, especially in the setting of acute kidney injury (AKI) [14] or myocardial infarction [47]. For instance, the Reduction of Infarct Expansion and Ventricular Remodeling with EPO After Large Myocardial Infarction (REVEAL) trial [47] included 222 patients with acute ST-segment elevation myocardial infarction who underwent successful percutaneous coronary intervention. EPO was administered at a dose of 60,000 IU within 4 hours of reperfusion of the previously ischemic myocardium. However, EPO did not reduce infarct size (primary endpoint) at any point in the 12-week study period. In fact, the study revealed that older patients (>70 years) receiving EPO had a 41.2% larger infarct size during the first week. In addition, the incidence of both adverse and serious adverse events was significantly higher in the EPO arm (94 of 125 patients) than the placebo arm (50 of 97 patients), with five deaths due to myocardial infarction, stroke, or stent thrombosis occurring in the EPO group (while no deaths occurred in the patients treated with placebo).
The Trial to Reduce Cardiovascular Events with Aranesp Therapy (TREAT) trial [48] included 4,038 patients with diabetes, chronic kidney disease and anemia. EPO was administered based on a hemoglobin target of 13.0 g/dl (that is, a patient with very low hemoglobin would require a higher dose of EPO than a patient with hemoglobin closer to 13.0 g/dl). EPO did not reduce the risk of a cardiovascular event (31.4% versus 29.7%) or renal event (32.4% versus 30.5%) when compared to the placebo group, but did increase the risk of stroke (5% versus 2.6%).
These data tie in well with the German Multicenter EPO Stroke Trial as discussed previously [10], which demonstrated- an increased risk of death with EPO treatment in stroke patients. In addition, the Correction of Hemoglobin and Outcomes in Renal Insufficiency (CHOIR) trial involving 1,432 chronic kidney disease patients was suspended when 125 and 97 patients developed a cardiovascular primary event following EPO treatment when hemoglobin levels were targeted at 13.5 g/dl and 11.3 g/dl, respectively [49]. This, therefore, raises the question of whether the use of EPO analogues that are devoid of erythropoietic effects would be of benefit to patients suffering from anemia?
In contrast, a pilot study to determine the efficacy of EPO to prevent AKI in 71 patients scheduled for elective coronary artery bypass grafting has been assessed. A moderately low dose of EPO (300 IU/kg intravenously) or placebo was randomly administered before surgery. The incidence of AKI in the placebo group was 29%, which was effectively reduced to 8% in the EPO group with no apparent evidence of serious adverse events [50].
It would have been most interesting to see whether or not CEPO would have had a beneficial effect in the above settings as the risks of serious adverse events may be reduced as confirmed by preclinical data demonstrating a tissue protective effect with CEPO in models of myocardial infarction [28, 51, 52] and renal disease [29, 53, 54]. As yet there are no published clinical data of non-erythropoietic derivatives of EPO, although three clinical trials were recently completed with CEPO for the treatment of stroke and neurodegeneration (ClinicalTrials. gov ID: NCT00756249, NCT01016366, NCT00870844).
Conclusion
In the past 25 years, many studies have convincingly demonstrated that EPO (at low doses), in addition to its erythropoietic effects, reduces tissue injury. The erythropoietic effects of EPO are mediated by the classical EPOR, while the tissue-protective effects of EPO may be mediated by a heterocomplex between EPOR and the βcR. Several recent clinical trials with EPO (at high doses) have demonstrated significant adverse effects (secondary to thrombotic events), which outweigh any beneficial effects observed, but it should be mentioned that an increase in survival in trauma-patients treated with EPO was observed in one large trial [11] and in another study there was a reduced incidence of AKI after coronary artery bypass graft surgery [50]. One could argue that the reason for non-efficacy in the majority of EPO trials has been due to the relatively high doses of EPO used, whereas pre-clinical studies have, in the most part, used relatively low doses of EPO. The reason for this disparity is unclear, as patients with anemia are not often treated with exceptionally high doses of EPO. It may well be possible that EPO at low concentrations may only activate the EPOR-βcR complex, but at high concentrations also activates the EPOR homodimer, thus masking any tissue-protective effects that may have been observed following βcR activation. In hindsight, it would have proved useful to dose a third group of patients in clinical trials with an equivalent effective dose seen in pre-clinical experiments. The clinical evaluation of non-erythropoietic derivatives of EPO (CEPO) is ongoing, but currently no data from these trials have been reported.
Abbreviations
- βcR:
-
beta common receptor
- AKI:
-
acute kidney injury
- CEPO:
-
carbamylated erythropoietin
- EPO:
-
erythropoietin
- EPOR:
-
erythropoietin receptor
- pHBSP:
-
pyroglutamate helix B surface peptide
- tPA:
-
tissue plasminogen activator.
References
Sasaki R: Pleiotropic functions of erythropoietin. Intern Med. 2003, 42: 142-149. 10.2169/internalmedicine.42.142.
Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M, Sasaki R: In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci USA. 1998, 95: 4635-4640. 10.1073/pnas.95.8.4635.
Abdelrahman M, Sharples EJ, McDonald MC, Collin M, Patel NS, Yaqoob MM, Thiemermann C: Erythropoietin attenuates the tissue injury associated with hemorrhagic shock and myocardial ischemia. Shock. 2004, 22: 63-69. 10.1097/01.shk.00001276869.21260.9d.
Cuzzocrea S, Mazzon E, Dugo L, Patel NS, Serraino I, Di Paola R, Genovese T, Britti D, De Maio M, Caputi AP, Thiemermann C: Reduction in the evolution of murine type II collagen-induced arthritis by treatment with rosiglitazone, a ligand of the peroxisome proliferator-activated receptor gamma. Arthritis Rheum. 2003, 48: 3544-3556. 10.1002/art.11351.
Heeschen C, Aicher A, Lehmann R, Fichtlscherer S, Vasa M, Urbich C, Mildner-Rihm C, Martin H, Zeiher AM, Dimmeler S: Erythropoietin is a potent physiologic stimulus for endothelial progenitor cell mobilization. Blood. 2003, 102: 1340-1346. 10.1182/blood-2003-01-0223.
Patel NS, Sharples EJ, Cuzzocrea S, Chatterjee PK, Britti D, Yaqoob MM, Thiemermann C: Pretreatment with EPO reduces the injury and dysfunction caused by ischemia/reperfusion in the mouse kidney in vivo. Kidney Int. 2004, 66: 983-989. 10.1111/j.1523-1755.2004.00847.x.
Villa P, Bigini P, Mennini T, Agnello D, Laragione T, Cagnotto A, Viviani B, Marinovich M, Cerami A, Coleman TR, Brines M, Ghezzi P: Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. J Exp Med. 2003, 198: 971-975. 10.1084/jem.20021067.
Vesey DA, Cheung C, Pat B, Endre Z, Gobe G, Johnson DW: Erythropoietin protects against ischaemic acute renal injury. Nephrol Dial Transplant. 2004, 19: 348-355.
Ehrenreich H, Hasselblatt M, Dembowski C, Cepek L, Lewczuk P, Stiefel M, Rustenbeck HH, Breiter N, Jacob S, Knerlich F, Bohn M, Poser W, Rüther E, Kochen M, Gefeller O, Gleiter C, Wessel TC, De Ryck M, Itri L, Prange H, Cerami A, Brines M, Sirén AL: Erythropoietin therapy for acute stroke is both safe and beneficial. Mol Med. 2002, 8: 495-505.
Ehrenreich H, Weissenborn K, Prange H, Schneider D, Weimar C, Wartenberg K, Schellinger PD, Bohn M, Becker H, Wegrzyn M, Jähnig P, Herrmann M, Knauth M, Bähr M, Heide W, Wagner A, Schwab S, Reichmann H, Schwendemann G, Dengler R, Kastrup A, Bartels C, EPO Stroke Trial Group: Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke. 2009, 40: e647-656. 10.1161/STROKEAHA.109.564872.
Corwin HL, Gettinger A, Fabian TC, May A, Pearl RG, Heard S, An R, Bowers PJ, Burton P, Klausner MA, Corwin MJ: Efficacy and safety of epoetin alfa in critically ill patients. N Engl J Med. 2007, 357: 965-976. 10.1056/NEJMoa071533.
Patel NS, Collino M, Yaqoob MM, Thiemermann C: Erythropoietin in the intensive care unit: beyond treatment of anemia. Ann Intensive Care. 2011, 1: 40-10.1186/2110-5820-1-40.
Leist M, Ghezzi P, Grasso G, Bianchi R, Villa P, Fratelli M, Savino C, Bianchi M, Nielsen J, Gerwien J, Kallunki P, Larsen AK, Helboe L, Christensen S, Pedersen LO, Nielsen M, Torup L, Sager T, Sfacteria A, Erbayraktar S, Erbayraktar Z, Gokmen N, Yilmaz O, Cerami-Hand C, Xie QW, Coleman T, Cerami A, Brines M: Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science. 2004, 305: 239-242. 10.1126/science.1098313.
Endre ZH, Walker RJ, Pickering JW, Shaw GM, Frampton CM, Henderson SJ, Hutchison R, Mehrtens JE, Robinson JM, Schollum JB, Westhuyzen J, Celi LA, McGinley RJ, Campbell IJ, George PM: Early intervention with erythropoietin does not affect the outcome of acute kidney injury (the EARLYARF trial). Kidney Int. 2010, 77: 1020-1030. 10.1038/ki.2010.25.
Napolitano LM, Fabian TC, Kelly KM, Bailey JA, Block EF, Langholff W, Enny C, Corwin HL: Improved survival of critically ill trauma patients treated with recombinant human erythropoietin. J Trauma. 2008, 65: 285-297. 10.1097/TA.0b013e31817f2c6e. discussion 297-289
Wiessner C, Allegrini PR, Ekatodramis D, Jewell UR, Stallmach T, Gassmann M: Increased cerebral infarct volumes in polyglobulic mice overexpressing erythropoietin. J Cereb Blood Flow Metab. 2001, 21: 857-864.
Goldwasser E, Kung CK, Eliason J: On the mechanism of erythropoietin-induced differentiation. 13. The role of sialic acid in erythropoietin action. J Biol Chem. 1974, 249: 4202-4206.
Fukuda MN, Sasaki H, Lopez L, Fukuda M: Survival of recombinant erythropoietin in the circulation: the role of carbohydrates. Blood. 1989, 73: 84-89.
Dong YJ, Kung C, Goldwasser E: Receptor binding of asialoerythropoietin. J Cell Biochem. 1992, 48: 269-276. 10.1002/jcb.240480307.
Erbayraktar S, Grasso G, Sfacteria A, Xie QW, Coleman T, Kreilgaard M, Torup L, Sager T, Erbayraktar Z, Gokmen N, Yilmaz O, Ghezzi P, Villa P, Fratelli M, Casagrande S, Leist M, Helboe L, Gerwein J, Christensen S, Geist MA, Pedersen LØ, Cerami-Hand C, Wuerth JP, Cerami A, Brines M: Asialoerythropoietin is a nonerythropoietic cytokine with broad neuroprotective activity in vivo. Proc Natl Acad Sci USA. 2003, 100: 6741-6746. 10.1073/pnas.1031753100.
Okada T, Sawada T, Kubota K: Asialoerythropoietin has strong renoprotective effects against ischemia-reperfusion injury in a murine model. Transplantation. 2007, 84: 504-510. 10.1097/01.tp.0000277672.02783.33.
Mori S, Sawada T, Okada T, Kubota K: Erythropoietin and its derivative protect the intestine from severe ischemia/reperfusion injury in the rat. Surgery. 2008, 143: 556-565. 10.1016/j.surg.2007.12.013.
Sato T, Tanno M, Miki T, Yano T, Shimamoto K, Miura T: Erythropoietin (EPO) affords more potent cardioprotection by activation of distinct signaling to mitochondrial kinases compared with carbamylated EPO. Cardiovasc Drugs Ther. 2010, 24: 401-408. 10.1007/s10557-010-6265-5.
Mun KC, Golper TA: Impaired biological activity of erythropoietin by cyanate carbamylation. Blood Purification. 2000, 18: 13-17. 10.1159/000014403.
Masuda S, Nagao M, Takahata K, Konishi Y, Gallyas F, Tabira T, Sasaki R: Functional erythropoietin receptor of the cells with neural characteristics. Comparison with receptor properties of erythroid cells. J Biol Chem. 1993, 268: 11208-11216.
Grodberg J, Davis KL, Sykowski AJ: Alanine scanning mutagenesis of human erythropoietin identifies four amino acids which are critical for biological activity. Eur J Biochem. 1993, 218: 597-601. 10.1111/j.1432-1033.1993.tb18413.x.
Satake R, Kozutsumi H, Takeuchi M, Asano K: Chemical modification of erythropoietin: an increase in in vitro activity by guanidination. Biochim Biophys Acta. 1990, 1038: 125-129. 10.1016/0167-4838(90)90020-G.
Fiordaliso F, Chimenti S, Staszewsky L, Bai A, Carlo E, Cuccovillo I, Doni M, Mengozzi M, Tonelli R, Ghezzi P, Coleman T, Brines M, Cerami A, Latini R: A nonerythropoietic derivative of erythropoietin protects the myocardium from ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2005, 102: 2046-2051. 10.1073/pnas.0409329102.
Imamura R, Isaka Y, Ichimaru N, Takahara S, Okuyama A: Carbamylated erythropoietin protects the kidneys from ischemia-reperfusion injury without stimulating erythropoiesis. Biochem Biophys Res Commun. 2007, 353: 786-792. 10.1016/j.bbrc.2006.12.099.
Brines M, Grasso G, Fiordaliso F, Sfacteria A, Ghezzi P, Fratelli M, Latini R, Xie QW, Smart J, Su-Rick CJ, Pobre E, Diaz D, Gomez D, Hand C, Coleman T, Cerami A: Erythropoietin mediates tissue protection through an erythropoietin and common beta-subunit heteroreceptor. Proc Natl Acad Sci USA. 2004, 101: 14907-14912. 10.1073/pnas.0406491101.
Carr PD, Gustin SE, Church AP, Murphy JM, Ford SC, Mann DA, Woltring DM, Walker I, Ollis DL, Young IG: Structure of the complete extracellular domain of the common beta subunit of the human GM-CSF, IL-3, and IL-5 receptors reveals a novel dimer configuration. Cell. 2001, 104: 291-300. 10.1016/S0092-8674(01)00213-6.
Stomski FC, Woodcock JM, Zacharakis B, Bagley CJ, Sun Q, Lopez AF: Identification of a Cys motif in the common beta chain of the interleukin 3, granulocyte-macrophage colony-stimulating factor, and interleukin 5 receptors essential for disulfide-linked receptor heterodimerization and activation of all three receptors. J Biol Chem. 1998, 273: 1192-1199. 10.1074/jbc.273.2.1192.
Hanazono Y, Sasaki K, Nitta H, Yazaki Y, Hirai H: Erythropoietin induces tyrosine phosphorylation of the beta chain of the GM-CSF receptor. Biochem Biophys Res Commun. 1995, 208: 1060-1066. 10.1006/bbrc.1995.1442.
Jubinsky PT, Krijanovski OI, Nathan DG, Tavernier J, Sieff CA: The beta chain of the interleukin-3 receptor functionally associates with the erythropoietin receptor. Blood. 1997, 90: 1867-1873.
Campana WM, Misasi R, O'Brien JS: Identification of a neurotrophic sequence in erythropoietin. Int J Mol Med. 1998, 1: 235-241.
Ebie AZ, Fleming KG: Dimerization of the erythropoietin receptor transmembrane domain in micelles. J Mol Biol. 2007, 366: 517-524. 10.1016/j.jmb.2006.11.035.
Brines M, Cerami A: Erythropoietin-mediated tissue protection: reducing collateral damage from the primary injury response. J Intern Med. 2008, 264: 405-432. 10.1111/j.1365-2796.2008.02024.x.
Brines M, Cerami A: The receptor that tames the innate immune response. Mol Med. 2012, 18: 486-496.
Um M, Gross AW, Lodish HF: A "classical" homodimeric erythropoietin receptor is essential for the antiapoptotic effects of erythropoietin on differentiated neuroblastoma SH-SY5Y and pheochromocytoma PC-12 cells. Cell Signal. 2007, 19: 634-645. 10.1016/j.cellsig.2006.08.014.
Syed RS, Reid SW, Li C, Cheetham JC, Aoki KH, Liu B, Zhan H, Osslund TD, Chirino AJ, Zhang J, Finer-Moore J, Elliott S, Sitney K, Katz BA, Matthews DJ, Wendoloski JJ, Egrie J, Stroud RM: Efficiency of signalling through cytokine receptors depends critically on receptor orientation. Nature. 1998, 395: 511-516. 10.1038/26773.
Brines M, Patel NS, Villa P, Brines C, Mennini T, De Paola M, Erbayraktar Z, Erbayraktar S, Sepodes B, Thiemermann C, Ghezzi P, Yamin M, Hand CC, Xie QW, Coleman T, Cerami A: Nonerythropoietic, tissue-protective peptides derived from the tertiary structure of erythropoietin. Proc Natl Acad Sci USA. 2008, 105: 10925-10930. 10.1073/pnas.0805594105.
Patel NS, Nandra KK, Brines M, Collino M, Wong WS, Kapoor A, Benetti E, Goh FY, Fantozzi R, Cerami A, Thiemermann C: A non-erythropoietic peptide that mimics the 3D structure of erythropoietin reduces organ injury/dysfunction and inflammation in experimental hemorrhagic shock. Mol Med. 2011, 17: 883-892.
Ahmet I, Tae HJ, Juhaszova M, Riordon DR, Boheler KR, Sollott SJ, Brines M, Cerami A, Lakatta EG, Talan MI: A small nonerythropoietic helix B surface peptide based upon erythropoietin structure is cardioprotective against ischemic myocardial damage. Mol Med. 2011, 17: 194-200.
Ueba H, Brines M, Yamin M, Umemoto T, Ako J, Momomura S, Cerami A, Kawakami M: Cardioprotection by a nonerythropoietic, tissue-protective peptide mimicking the 3D structure of erythropoietin. Proc Natl Acad Sci USA. 2010, 107: 14357-14362. 10.1073/pnas.1003019107.
Swartjes M, Morariu A, Niesters M, Brines M, Cerami A, Aarts L, Dahan A: ARA290, a peptide derived from the tertiary structure of erythropoietin, produces long-term relief of neuropathic pain: an experimental study in rats and beta-common receptor knockout mice. Anesthesiology. 2011, 115: 1084-1092. 10.1097/ALN.0b013e31822fcefd.
Wu H, Liu X, Jaenisch R, Lodish HF: Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell. 1995, 83: 59-67. 10.1016/0092-8674(95)90234-1.
Najjar SS, Rao SV, Melloni C, Raman SV, Povsic TJ, Melton L, Barsness GW, Prather K, Heitner JF, Kilaru R, Gruberg L, Hasselblad V, Greenbaum AB, Patel M, Kim RJ, Talan M, Ferrucci L, Longo DL, Lakatta EG, Harrington RA, REVEAL Investigators: Intravenous erythropoietin in patients with ST-segment elevation myocardial infarction: REVEAL: a randomized controlled trial. JAMA. 2011, 305: 1863-1872. 10.1001/jama.2011.592.
Pfeffer MA, Burdmann EA, Chen CY, Cooper ME, de Zeeuw D, Eckardt KU, Feyzi JM, Ivanovich P, Kewalramani R, Levey AS, Lewis EF, McGill JB, McMurray JJ, Parfrey P, Parving HH, Remuzzi G, Singh AK, Solomon SD, Toto R, TREAT Investigators: A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009, 361: 2019-2032. 10.1056/NEJMoa0907845.
Singh AK, Szczech L, Tang KL, Barnhart H, Sapp S, Wolfson M, Reddan D: Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006, 355: 2085-2098. 10.1056/NEJMoa065485.
Song YR, Lee T, You SJ, Chin HJ, Chae DW, Lim C, Park KH, Han S, Kim JH, Na KY: Prevention of acute kidney injury by erythropoietin in patients undergoing coronary artery bypass grafting: a pilot study. Am J Nephrol. 2009, 30: 253-260. 10.1159/000223229.
Xu X, Cao Z, Cao B, Li J, Guo L, Que L, Ha T, Chen Q, Li C, Li Y: Carbamylated erythropoietin protects the myocardium from acute ischemia/reperfusion injury through a PI3K/Akt-dependent mechanism. Surgery. 2009, 146: 506-514. 10.1016/j.surg.2009.03.022.
Xu K, George I, Klotz S, Hay I, Xydas S, Zhang G, Cerami A, Wang J: Erythropoietin derivate improves left ventricular systolic performance and attenuates left ventricular remodeling in rats with myocardial infarct-induced heart failure. J Cardiovasc Pharmacol. 2010, 56: 506-512. 10.1097/FJC.0b013e3181f4f05a.
Imamura R, Okumi M, Isaka Y, Ichimaru N, Moriyama T, Imai E, Nonomura N, Takahara S, Okuyama A: Carbamylated erythropoietin improves angiogenesis and protects the kidneys from ischemia-reperfusion injury. Cell Transplant. 2008, 17: 135-141. 10.3727/000000008783907044.
Cassis P, Azzollini N, Solini S, Mister M, Aiello S, Cugini D, Scudeletti P, Gagliardini E, Abbate M, Gallon L, Remuzzi G, Noris M: Both darbepoetin alfa and carbamylated erythropoietin prevent kidney graft dysfunction due to ischemia/reperfusion in rats. Transplantation. 2011, 92: 271-279. 10.1097/TP.0b013e3182241106.
Wang X, Zhu C, Wang X, Gerwien JG, Schrattenholz A, Sandberg M, Leist M, Blomgren K: The nonerythropoietic asialoerythropoietin protects against neonatal hypoxia-ischemia as potently as erythropoietin. J Neurochem. 2004, 91: 900-910. 10.1111/j.1471-4159.2004.02769.x.
Grasso G, Sfacteria A, Erbayraktar S, Passalacqua M, Meli F, Gokmen N, Yilmaz O, La Torre D, Buemi M, Iacopino DG, Coleman T, Cerami A, Brines M, Tomasello F: Amelioration of spinal cord compressive injury by pharmacological preconditioning with erythropoietin and a nonerythropoietic erythropoietin derivative. J Neurosurg Spine. 2006, 4: 310-318. 10.3171/spi.2006.4.4.310.
Mennini T, De Paola M, Bigini P, Mastrotto C, Fumagalli E, Barbera S, Mengozzi M, Viviani B, Corsini E, Marinovich M, Torup L, Van Beek J, Leist M, Brines M, Cerami A, Ghezzi P: Nonhematopoietic erythropoietin derivatives prevent motoneuron degeneration in vitro and in vivo. Mol Med. 2006, 12: 153-160.
Yokomaku Y, Sugimoto T, Kume S, Araki S, Isshiki K, Chin-Kanasaki M, Sakaguchi M, Nitta N, Haneda M, Koya D, Uzu T, Kashiwagi A: Asialoerythropoietin prevents contrast-induced nephropathy. J Am Soc Nephrol. 2008, 19: 321-328. 10.1681/ASN.2007040481.
Price CD, Yang Z, Karlnoski R, Kumar D, Chaparro R, Camporesi EM: Effect of continuous infusion of asialoerythropoietin on short-term changes in infarct volume, penumbra apoptosis and behaviour following middle cerebral artery occlusion in rats. Clin Exp Pharmacol Physiol. 2010, 37: 185-192. 10.1111/j.1440-1681.2009.05257.x.
Nakazawa J, Isshiki K, Sugimoto T, Araki S, Kume S, Yokomaku Y, Chin-Kanasaki M, Sakaguchi M, Koya D, Haneda M, Kashiwagi A, Uzu T: Renoprotective effects of asialoerythropoietin in diabetic mice against ischaemia-reperfusion-induced acute kidney injury. Nephrology (Carlton). 2010, 15: 93-101. 10.1111/j.1440-1797.2009.01170.x.
Yamashita T, Nonoguchi N, Ikemoto T, Miyatake S, Kuroiwa T: Asialoerythropoietin attenuates neuronal cell death in the hippocampal CA1 region after transient forebrain ischemia in a gerbil model. Neurol Res. 2010, 32: 957-962. 10.1179/016164110X12700393823336.
Ogino A, Takemura G, Kawasaki M, Tsujimoto A, Kanamori H, Li L, Goto K, Maruyama R, Kawamura I, Takeyama T, Kawaguchi T, Watanabe T, Moriguchi Y, Saito H, Fujiwara T, Fujiwara H, Minatoguchi S: Erythropoietin receptor signaling mitigates renal dysfunction-associated heart failure by mechanisms unrelated to relief of anemia. J Am Coll Cardiol. 2010, 56: 1949-1958. 10.1016/j.jacc.2010.04.068.
Sasaki N, Sekiguchi M, Kikuchi S, Konno S: Effects of asialo-erythropoietin on pain-related behavior and expression of phosphorylated-p38 map kinase and tumor necrosis factor-alpha induced by application of autologous nucleus pulposus on nerve root in rat. Spine (Phila Pa 1976). 2011, 36: E86-94. 10.1097/BRS.0b013e3181f137a8.
Acknowledgements
NSAP is supported by a Kidney Research UK Post-Doctoral Fellowship (PDF4/09). KKN is supported by a British Heart Foundation Studentship (FS/10/57/28485). This work forms part of the research themes contributing to the translational research portfolio of Barts and the London Cardiovascular Biomedical Research Unit, which is supported and funded by the National Institute of Health Research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
About this article
Cite this article
Patel, N.S., Nandra, K.K. & Thiemermann, C. Bench-to-bedside review: Erythropoietin and its derivatives as therapies in critical care. Crit Care 16, 229 (2012). https://doi.org/10.1186/cc11315
Published:
DOI: https://doi.org/10.1186/cc11315