
Abstract
The concept of acute kidney syndromes has shifted in recent years from acute renal failure to acute kidney injury (AKI). AKI implies injury or damage but not necessarily dysfunction. The human kidney has an important glomerular function reserve, and dysfunction becomes evident only when more than 50% of the renal mass is compromised. Recent AKI classifications include even slight changes in serum creatinine, acknowledging that this condition is associated with worse outcomes. This, however, still represents a functional criterion for AKI and implies a glomerular filtration rate alteration that may be a late phenomenon in the time course of the syndrome. An early diagnosis of AKI by using tubular damage biomarkers preceding filtration function loss is possible today. Some studies have shown evidence that there is an additional value of new biomarkers not only because they allow a diagnosis to be made earlier but also because they allow a kidney injury to be diagnosed even in the absence of subsequent dysfunction. Only recently, tubular damage without glomerular function loss was demonstrated to be associated with worse renal and overall outcomes. For this condition, the term 'subclinical' AKI has been introduced, challenging the traditional view that a kidney problem is clinically relevant, only when a loss of filtration function becomes apparent. A new domain of AKI diagnosis could then include functional criteria and damage criteria. This may have an impact on the epidemiology, prevention, and management of AKI.
Similar content being viewed by others
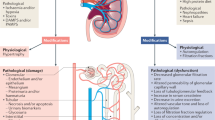


Many terms have been used to describe acute events occurring to or involving the kidneys, such as acute renal failure, acute kidney diseases, acute kidney syndromes, or acute kidney injury (AKI). Indeed, the spectrum of such disorders has been expanding over the last decades.
The diagnosis and management of acute syndromes involving the kidneys has become a multidisciplinary field concerning not only nephrology and urology but also critical care medicine, cardiology, radiology, and other fields. With this evolution, the term acute renal failure, used for many years in clinical practice, has been replaced with the term AKI. The new term implies potentially reversible kidney injury or damage occurring in a time frame of hours or days and characterizing the disorder as 'acute'. Although the term 'injury' would not necessarily encompass kidney dysfunction without damage, the diagnosis of AKI syndrome is still made on the basis of a change in serum creatinine or urine output, both likely deriving from an altered glomerular filtration and therefore identifying more a dysfunction than damage. Histopathological changes during AKI have been studied [1], but mostly in animal experiments. In fact, histopathological criteria are difficult to apply to the diagnosis of AKI in critically ill patients in whom kidney biopsy is usually considered to carry an inappropriate risk-to-benefit ratio. Only recently, some attention has been given to signs of structural damage to the nephrons and its clinical relevance [2].
There are many reasons why a distinction should be made between kidney injury and dysfunction. Indeed, human kidneys have an important functional reserve; thus, dysfunction, especially when defined as an altered glomerular filtration, becomes evident only when more than 50% of the renal mass is compromised. The rise in serum creatinine is a sign of a glomerular filtration alteration that has been ongoing for hours or days. In the case of a transient decrease in glomerular filtration rate(GFR), creatinine may never rise significantly. On the other hand, a rise in serum creatinine may occur in the presence of a constant creatinine generation when glomerular filtration has been impaired for at least 24 to 48 hours. The new steady state and the new level of serum creatinine will be set by the new level of glomerular filtration; at the new steady state, excretion will equal generation, but the required excretion will occur at the expense of a significantly higher creatinine level. Standard AKI classifications such as the RIFLE (Risk, Injury, Failure, Loss, and End-stage kidney disease) criteria [3] or modified RIFLE criteria [4] include as a diagnostic criterion even slight changes in serum creatinine (as low as 0.3 mg/dL), and validation studies of these criteria show that such conditions are associated with worse outcomes [5]. Once again, these approaches, though refined in comparison with previous criteria of diagnosis (often described by doubling serum creatinine or the need for dialysis), are still based on functional criteria and imply an altered GFR. Such alteration may have occurred long after the injury has taken place and long before the creatinine has reached the new higher steady level. This explains why a rise in serum creatinine is often a sign of severe kidney damage even if the rise in creatinine is minimal. In such a condition, information on the magnitude of tubular damage and other kidney tissues and functions is often not available.
Very recently, significant contributions to the literature have shown that an early diagnosis of AKI can be made by using a single structural or functional biomarker (or a combination thereof) capable of detecting kidney injury almost in real time [6]. Many studies with several thousands of patients have shown evidence that there is an additional value of new biomarkers not only because they allow a diagnosis to be made earlier [7] but also because they allow a kidney injury to be diagnosed even in the absence of a subsequent manifest dysfunction [2, 8]. What are the ideal characteristics for such biomarkers? They must be specific for kidney injury, sensitive enough to detect even less severe insults, easy and rapid to measure, and inexpensive enough to make their use sustainable. A series of molecules have been evaluated over the years, and significant advances have been made in the field. Molecules such as NGAL (neutrophil gelatinase-associated lipocalin), KIM-1 (kidney injury molecule-1), and Cys-C (cystatin-C) have demonstrated a peculiar capacity to detect an injury to the kidney well before the rise in serum creatinine can be observed. Thus, a new diagnostic approach to acute kidney syndromes can be envisaged. Such a biomarker-driven approach raises the question of whether AKI could be diagnosed even in the absence of the classic signs that have characterized the syndrome in the past. In other words, if we compare AKI with the acute coronary syndrome, in which an increase in troponin may be a sufficient trigger for a diagnosis and a therapeutic intervention, the diagnosis of AKI could be made even in the absence of oliguria or increased creatinine levels. The fact that AKI is not clinically manifest does not necessarily mean that the kidney is intact and that the function is perfect. A subclinical entity may be unveiled by the new biomarkers and we should still call this entity AKI. In these circumstances, revised RIFLE or Acute Kidney Injury Network (AKIN) criteria should integrate one or more injury markers with creatinine, urine output, and/or other functional markers. This concept was recently proposed, expanding the spectrum of clinical conditions under the umbrella of AKI [9]. Today, we do not have sufficient data to characterize the severity of the syndrome on the basis of structural biomarker levels alone and therefore the grading of the syndrome still relies on renal function markers. Nevertheless, this should not mean that we can neglect a clinical condition characterized by positive bio marker and negative creatinine findings. In practice, we have a new class or stage of AKI that is diagnosed by tubular damage biomarkers such as NGAL and that may or may not evolve into a clinically manifest syndrome characterized by decreased GFR and increased serum creatinine. At this point, one would be tempted to call this AKIN stage 0 but this would imply no AKI. The same is true for a possible RIFLE 0 condition. In this substantial and not merely semantic discussion, we may speculate that AKIN stages better describe the severity of the syndrome, not because they significantly differ from RIFLE stages but because the terms 'risk' and 'injury', typical of RIFLE, may lead to confusion. In fact, if a biomarker-positive, creatinine-negative patient is observed, he or she has probably received an 'injury' to his kidney but may still be in a preclinical (according to creatinine) phase. For this reason, we propose an additional criterion to the classic RIFLE or AKIN criteria since both are basically function-driven: the new criterion is injury-driven and identifies asubclinical AKI that, however, is definitely an AKI. The new criterion also enables a timely diagnosis of 'prerenal' AKI and a more precise distinction of the term since during such a condition tubular damage biomarkers are negative but simultaneously a loss of filtration function is evident [6, 10]. For example, hypovolemia due to reduced fluid intake or increased fluid loss may cause a reduction in GFR, but the integrity and polarity of tubular cells may be preserved in the absence of inflammation or ischemia. Nonetheless, a prolonged and severe hypovolemia in patients with comorbidities may contribute to tubular damage, especially when hypoperfusion develops.
Isolated function loss or isolated damage, when occurring alone, is each sufficient to constitute AKI, and the two occurring together - regardless of which one occurs first - may progress to a combined function loss and damage. Table 1 presents the entities of AKI syndrome on the basis of renal filtration function and tubular damage markers.
This new approach has several implications. First, the diagnosis of AKI may include a larger spectrum of conditions, and the epidemiology of the syndrome may well change in terms of incidence and prevalence. In fact, a larger number of individuals may fall within the definition of AKI if function and injury criteria are used for diagnosis. Second, the concept of prevention or early organ protection or both may change, and we might be able to rewrite several sections of the chapter on preventive measures for AKI. Several interventions, in fact, have failed to provide positive results, even when previous laboratory and animal data were extremely encouraging. This may have to do with the timing in which the prevention/protection measure is applied. The condition in which a tubular damage biomarker is positive, even in the absence of a rise in serum creatinine (subclinical AKI), may become sufficient to trigger interventions and protective strategies for the kidney. (This was not possible before, because we used only biomarkers of dysfunction, like creatinine, which are a mirror of a late phenomenon. Thus, interventions done in the past were always done too late.) Third, we may need to re-establish a term for renal function loss (for example, acute renal dysfunction), distinguishing it from AKI with tubular damage, or at least to provide a new staging classification (subclinical AKI for patients who are biomarker-positive and creatinine-negative). Fourth, the accuracy and timeliness of differential diagnosis of the underlying cause of AKI (prerenal versus nonprerenal) may be improved.
More considerations on the concept of 'biomarker positivity' are needed. Biomarkers are becoming more and more reliable in terms of sensitivity and specificity, and even high-sensitivity assays with extended range are becoming available. These new assays have improved the capability of damage detection even below the classic cutoff values [11]. In such circumstances, sequential measurements may be indicated to build biomarker curves capable of identifying trends characteristic of a transient or ongoing renal insult. This may open up a new frontier in the diagnosis of AKI and its consequences in terms of prevention and therapeutic strategies.
In conclusion, we are living in a post-creatinine world where creatinine should not be abandoned but we should move beyond it. What today is considered subclinical AKI (tubular damage biomarker positivity without dysfunction) can, thanks to the new biomarkers, be defined as AKI and we cannot afford to neglect such conditions when they are associated with negative outcomes. The accuracy of new biomarkers is crucial in this process, and we should start considering not only cutoff values but also trends and biomarker curves, especially now that high-sensitivity assays are becoming available. In other words, we must consider that subclinical AKI is still AKI.
Abbreviations
- AKI:
-
acute kidney injury
- AKIN:
-
Acute Kidney Injury Network
- GFR:
-
glomerular filtration rate
- NGAL:
-
NGAL neutrophil gelatinase-associated lipocalin
- RIFLE:
-
Risk: Injury: Failure: Loss: and End-stage kidney disease.
References
Chvojka J, Sykora R, Krouzecky A, Radej J, Varnerova V, Karvunidis T, Hes O, Novak I, Radermacher P, Matejovic M: Renal haemodynamic, microcirculatory, metabolic and histopathological responses to peritonitis-induced septic shock in pigs. Crit Care 2008, 12: R164.
Haase M, Devarajan P, Haase-Fielitz A, Bellomo R, Cruz DN, Wagener G, Koyner JL, Murray P, Zappitelli M, Goldstein SL, Makris K, Ronco C, Martensson J, Martling CR, Venge P, Siew E, Ware LB, Ikizler TA, Mertens PR: The outcome of neutrophil gelatinase-associated lipocalin-positive subclinical acute kidney injury: a multicenter pooled analysis of prospective studies. J Am Coll Cardiol 2011, 57: 1752-1761. 10.1016/j.jacc.2010.11.051
Bellomo R, Ronco C, Kellum JA, Mehta RL, Palevsky P, Acute Dialysis Quality Initiative workgroup: Acute renal failure: definition, outcome measures, animal models, fluid therapy and information technology needs. The Second International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI) Group. Crit Care 2004, 8: R204-R212. 10.1186/cc2872
Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, Levin A: Acute Kidney Injury Network: Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care 2007, 11: R31. 10.1186/cc5713
Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW: Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 2005, 16: 3365-3370. 10.1681/ASN.2004090740
Paragas N, Qiu A, Zhang Q, Samstein B, Deng SX, Schmidt-Ott KM, Viltard M, Yu W, Forster CS, Gong G, Liu Y, Kulkarni R, Mori K, Kalandadze A, Ratner AJ, Devarajan P, Landry DW, D'Agati V, Lin CS, Barasch J: The Ngal reporter mouse detects the response of the kidney to injury in real time. Nat Med 2011, 17: 216-222. 10.1038/nm.2290
Haase M, Bellomo R, Devarajan P, Schlattmann P, Haase-Fielitz A, NGAL Metaanalysis Investigator Group: Accuracy of neutrophil gelatinase-associated lipocalin (NGAL) in diagnosis and prognosis in acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis 2009, 54: 1012-1024. 10.1053/j.ajkd.2009.07.020
Nickolas TL, Schmidt-Ott KM, Canetta P, Forster C, Singer E, Sise M, Elger A, Maarouf O, Sola-Del Valle DA, O'Rourke M, Sherman E, Lee P, Geara A, Imus P, Guddati A, Polland A, Rahman W, Elitok S, Malik N, Giglio J, El-Sayegh S, Devarajan P, Hebbar S, Saggi SJ, Hahn B, Kettritz R, Luft FC, Barasch J: Diagnostic and Prognostic Stratification in the Emergency Department Using Urinary Biomarkers of Nephron Damage. A Multicenter Prospective Cohort Study. J Am Coll Cardiol 2012, 59: 246-255. 10.1016/j.jacc.2011.10.854
Murugan R, Kellum JA: Acute kidney injury: what's the prognosis? Nat Rev Nephrol 2011, 7: 209-217. 10.1038/nrneph.2011.13
Nickolas TL, O'Rourke MJ, Yang J, Sise ME, Canetta PA, Barasch N, Buchen C, Khan F, Mori K, Giglio J, Devarajan P, Barasch J: Sensitivity and specificity of a single emergency department measurement of urinary neutrophil gelatinase-associated lipocalin for diagnosing acute kidney injury. Ann Intern Med 2008, 148: 810-819.
Ronco C, Cruz D, Noland BW: Neutrophil gelatinase-associated lipocalin curve and neutrophil gelatinase-associated lipocalin extended-range assay: a new biomarker approach in the early diagnosis of acute kidney injury and cardio-renal syndrome. Semin Nephrol 2012, 32: 121-128. 10.1016/j.semnephrol.2011.11.015
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
CR has received speaker honoraria from Gambro (Stockholm, Sweden), Alere (Waltham, MA, USA), Abbott (Abbott Park, IL, USA), General Electric Company (Fairfield, CT, USA), and Merck (Darmstadt, Germany) and has done consulting for CardioBridge (Hechingen, Germany), EFG (Dublin Ireland), and Asahi Kasei Medical Co. Ltd (Tokyo, Japan). JK has received grants from Gambro, Baxter (Deerfield, IL, USA), Alere, and Astute Medical, Inc. (San Diego, CA, USA) and has done consulting for Gambro, Baxter, Alere, Abbott, Astute, and Fresenius (Bad Homburg, Germany). MH has received lecture fees from Abbott, Alere, Fresenius, and Roche (Basel, Switzerland).
Rights and permissions
About this article
Cite this article
Ronco, C., Kellum, J.A. & Haase, M. Subclinical AKI is still AKI. Crit Care 16, 313 (2012). https://doi.org/10.1186/cc11240
Published:
DOI: https://doi.org/10.1186/cc11240