
Abstract
Diaphragmatic function is a major determinant of the ability to successfully wean patients from mechanical ventilation. However, the use of controlled mechanical ventilation in animal models results in a major reduction of diaphragmatic force-generating capacity together with structural injury and atrophy of diaphragm muscle fibers, a condition termed ventilator-induced diaphragmatic dysfunction (VIDD). Increased oxidative stress and exaggerated proteolysis in the diaphragm have been linked to the development of VIDD in animal models, but much less is known about the extent to which these phenomena occur in humans undergoing mechanical ventilation in the ICU. In the present review, we first briefly summarize the large body of evidence demonstrating the existence of VIDD in animal models, and outline the major cellular mechanisms that have been implicated in this process. We then relate these findings to very recently published data in critically ill patients, which have thus far been found to exhibit a remarkable degree of similarity with the animal model data. Hence, the human studies to date have indicated that mechanical ventilation is associated with increased oxidative stress, atrophy, and injury of diaphragmatic muscle fibers along with a rapid loss of diaphragmatic force production. These changes are, to a large extent, directly proportional to the duration of mechanical ventilation. In the context of these human data, we also review the methods that can be used in the clinical setting to diagnose and/or monitor the development of VIDD in critically ill patients. Finally, we discuss the potential for using different mechanical ventilation strategies and pharmacological approaches to prevent and/or to treat VIDD and suggest promising avenues for future research in this area.
Similar content being viewed by others
Introduction
Mechanical ventilation (MV) is one of the most frequently used forms of supportive therapy in the ICU [1]. It is currently employed in the ICU management of a wide variety of conditions leading to respiratory failure as well as other life threatening pathologies such as stroke, cardiac failure, postoperative illness and others. Although it is life-saving in ICU patients, MV is also associated with numerous potential complications. Indeed, MV can be associated with lung injury, the severity of which depends on the levels of alveolar stress and strain as well as the inherent susceptibility of a previously damaged lung [2, 3]. This phenomenon is called ventilator-induced lung injury (VILI) [4], and research initially performed in animal models led to the important discovery that VILI can be mitigated and clinical outcomes in patients with lung injury improved by ventilating lungs with limited pressure and/or low tidal volume (so-called protective ventilation) [5–7].
In the 1980s and early 1990s, there was a frequently held view among intensivists that many patients who developed ventilatory failure when faced with an increased work of breathing were suffering from a state of diaphragmatic fatigue. This led to the belief that a period of 'diaphragmatic rest', achieved through the use of controlled MV to completely suppress diaphragmatic activity, might be a useful measure. This idea has now been completely turned on its head with the demonstration that controlled MV is associated with adverse affects on multiple aspects of diaphragmatic structure and function. This condition, which was initially termed ventilator-induced diaphragmatic dysfunction (VIDD) by Vassilakopoulos and Petrof [8], appears to affect other respiratory muscles as well as the diaphragm [9, 10], and has important implications for how patients undergoing MV should be managed.
The purpose of this article is to briefly outline the key data that have unequivocally demonstrated the adverse effects of controlled MV on the diaphragm in animal models. We will then turn our major focus to several very recently published human studies [11–15] that have largely confirmed the observations about VIDD made in the animal studies. Finally, we will offer some clinical as well as future research perspectives on the problem of VIDD in ICU patients.
Brief review of animal model data
For the purposes of this review, VIDD refers to MV-induced changes in diaphragmatic function that arise from alterations outside of the central or peripheral nervous systems. Indeed, in animal models of VIDD the contractility of isolated diaphragmatic strips is severely reduced despite normal nervous impulse transmission and neuromuscular junction [16]. In addition, a few hours of totally controlled MV (without any spontaneous breathing cycles) promotes decreases of diaphragmatic contractile force both in vitro and in vivo [10, 16–25]. Endurance, as defined by the capacity to maintain contractile force when facing an inspiratory resistive load, is also significantly impaired [10, 17].
In addition to decreased diaphragmatic force, several histological and biochemical modifications have been reported in the diaphragms of animals with VIDD. Among the most prominent changes is muscle fiber atrophy [17, 19, 21, 22, 25], which is the consequence of decreased protein synthesis [26, 27] as well as increased proteolysis [27–31]. Muscle fiber remodeling [25] and muscle fiber injury are also observed [9, 24, 31, 32]. Several of these histological and biochemical changes have been linked to an increased level of oxidative stress in the diaphragm, which can be observed by 6 hours after MV initiation in rats [33] and after 72 hours in piglets [20]. Interestingly, the cellular targets of diaphragmatic protein oxidation may involve elements of the contractile machinery, such as myosin and actin [33]. Proteolysis by the calpain (calcium-dependent proteases) [34] and caspase systems has been reported to be involved in sarcomeric protein breakdown [35] by cleaving and disassembling myofilament proteins, which can then be more readily processed and degraded by the ubiquitin-proteasome system [36]. These same proteolytic systems have been shown to be activated in the diaphragm during the development of VIDD in animal models [27, 29, 30].
What do the clinical studies in humans show?
Alterations of diaphragmatic function in mechanically ventilated patients
One of the major challenges in determining whether VIDD occurs in humans is the difficulty of accurately evaluating respiratory muscle function in critically ill patients undergoing MV. The American Thoracic and European Respiratory Societies have extensively reviewed the methodologies for respiratory muscle evaluation in patients [37], and we will only briefly summarize tests that can be easily applied in the ICU. Measurements of maximum inspiratory pressure (Pi, max) are relatively easy to perform, but a low value often reflects a submaximal effort due to poor patient coordination or cooperation rather than true respiratory muscle weakness. Respiratory frequency and tidal volume are also easily monitored, and a rapid shallow breathing pattern is associated with weaning failure [38], but it is non-specific and does not necessarily indicate respiratory muscle dysfunction. To overcome the lack of specificity of the above tests, measurements of transdiaphragmatic pressure (Pdi) can be performed in ICU patients. This requires simultaneous recordings of esophageal (Pes) and gastric (Pga) pressures, with Pdi thus being determined as the pressure difference across the diaphragm.
To avoid the difficulties in interpretation of Pdi measurements related to variable levels of patient co-operation, bilateral phrenic nerve stimulation can be performed. As initially described by the team of Similowski and colleagues [39], this is well tolerated by most patients when using transcervical magnetic (as opposed to electrical) stimulation of the phrenic nerves. Using this method, Laghi and colleagues [40] measured Pdi before and after a weaning trial, and compared the values in critically ill patients with weaning success versus failure. Twitch Pdi values revealed considerable diaphragmatic weakness in almost all ventilated patients, but the study suggested that weaning failure was not accompanied by low-frequency fatigue of the diaphragm. A high prevalence of diaphragmatic weakness, as determined by magnetic stimulation of the phrenic nerves, was also reported by Watson and colleagues [41] in a population of 33 critically ill patients. Recently, Hermans and colleagues [11] assessed diaphragmatic force production in 25 mechanically ventilated ICU patients using cervical magnetic stimulation of the phrenic nerves in a prospective study performed at a single center. Seven of the patients were evaluated more than once during their ICU stay. The authors reported that values obtained with the phrenic nerve stimulation technique were reproducible, and that a longer duration of MV was associated with more severe diaphragmatic force loss. They also found that diaphragmatic force impairment was correlated with the amount of sedation received. However, the authors were unable to determine whether sedative use is an independent risk factor for diaphragmatic weakness in mechanically ventilated patients, or simply a marker of more prolonged periods of MV.
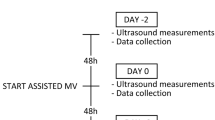
The magnitude of the negative deflection in tracheal pressure during twitch stimulation of the phrenic nerves can be used as a surrogate of the Pdi in intubated patients [42], and has the major advantage of not requiring the placement of esophageal and gastric balloon catheters. Watson and colleagues [41] first described the feasibility and the reproducibility of using twitch tracheal airway pressure (TwPtr) in this manner in intubated ICU patients. In our ICU, we also measure TwPtr using bilateral, supramaximal transcervical magnetic stimulation of the phrenic nerves in patients with unexplained weaning failure as part of our routine clinical care. In some cases, sedation is necessary to achieve adequate patient relaxation during the airway occlusion procedure. In addition, when using TwPtr to evaluate changes in diaphragmatic force-generating capacity over time, it is important to ensure that respiratory mechanics and end expiratory lung volume remain relatively stable during the measurement period. In a recently published study from our group [13], we performed serial measurements of TwPtr in critically ill patients who were ventilated for approximately one week. TwPtr decreased progressively during the period of MV, with a mean reduction of 32 ± 6% at 6 days. In summary, the human studies published to date are consistent in showing that a major degree of diaphragmatic weakness is frequently present in mechanically ventilated ICU patients. Furthermore, the magnitude of diaphragmatic weakness appears to be directly correlated with the duration of MV.
Alterations of diaphragmatic structure and biochemistry in mechanically ventilated patients
To better understand the mechanisms involved in the diaphragmatic weakness reported in critically ill patients during MV, diaphragmatic biopsies from mechanically ventilated patients are required. The first study to obtain such tissue samples [43], published in the late 1980 s, was a post-mortem analysis of neonates, in which diffuse diaphragmatic muscle fiber atrophy was found in patients who received ventilatory assistance for 12 days or more immediately before death. Such changes were not present in extradiaphragmatic muscles from the same patients or diaphragms of infants ventilated for 7 days or less [43].
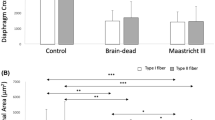
Another strategy that has been used to study the diaphragms of mechanically ventilated patients is to obtain the diaphragmatic tissue samples from brain-dead organ donors at the time of organ procurement. In comparison to other ICU patients, organ donors are often relatively free of the confounding effects of multiple organ system failure, and these patients also undergo an evaluation to rule out sepsis prior to organ donation. In a landmark study, Levine and colleagues [15] evaluated diaphragm biopsy specimens from 14 adult brain-dead organ donors who had undergone MV for variable periods of time (18 to 69 hours) prior to organ harvest, and compared them to specimens obtained from eight control patients who were undergoing thoracic surgery for benign lesions or localized lung cancer (MV for 2 to 3 hours). The authors reported a marked atrophy of both slow- and fast-twitch fibers in the diaphragms of the MV group compared to the controls, whereas no such differences were observed in the pectoralis muscle. Muscle fiber atrophy in the diaphragms of patients in the MV group was associated with signs of increased oxidative stress (decreased levels of glutathione, an antioxidant molecule) and an increase in muscle proteolysis biomarkers (caspase-3 and the E3 ubiquitin ligases atrogin-1 and MuRF-1).
Using a similar experimental paradigm, Hussain and colleagues [12] compared diaphragms from nine brain-dead organ donors (MV for 15 to 276 hours) and nine control patients (lung surgery, MV for 2 to 4 hours). These authors directly demonstrated that prolonged MV is associated with increased oxidative stress in the diaphragm, as indicated by oxidative modifications to diaphragmatic proteins. In addition to confirming the increase in E3 ubiquitin ligases, this paper also reported an upregulation of the autophagic system in the mechanically ventilated diaphragm. Autophagy (literally 'self-eating') is a catabolic pathway characterized by the formation of vesicles (autophagosomes) that engulf cytoplasmic organelles and proteins, and then fuse with lysosomes that degrade their contents. This process is a major regulated mechanism for degrading long-lived proteins and the only known pathway for degrading organelles. It occurs at low basal levels to perform homeostatic functions, but can be rapidly upregulated when cells need to generate energy. In their article, Hussain and colleagues [12] demonstrated that prolonged MV triggers autophagy, and this was associated with an upregulation of the transcription factor FOXO-1. Importantly, these changes were relatively specific to the diaphragm and not observed to the same degree in the control limb muscle (quadriceps) from the same patients. Therefore, the authors concluded that MV itself likely activates diaphragm autophagy via oxidative stress and induction of FOXO-1, thus contributing to the development of diaphragm muscle fiber atrophy and dysfunction.
Levine and colleagues [14] also examined several indices of the ubiquitin-proteasome and Akt-FOXO atrophy pathways, as well as the content of contractile proteins, in diaphragms of 18 brain-dead organ donors (MV for 18 to 72 hours) and 11 short-term (lung surgery, MV for 2 to 4 hours) ventilated patients. They found similar results to Hussain and colleagues [12] regarding the changes in these atrophy biomarkers, and also reported that these modifications were significantly correlated with the duration of MV. Once again, there were no significant differences between the MV and control groups in the reference control muscle (pectoralis) obtained from the same subjects [14].
We have recently reported the results of a histobiochemical evaluation of diaphragmatic biopsies obtained from 15 brain-dead organ donors (MV for 24 to 249 hours) and 10 control patients who underwent thoracic surgery for stage 1A lung cancers (MV for 2 to 3 hours). In this study by Jaber and colleagues [13], all subjects were required to have undergone MV via an endotracheal tube in fully controlled mode, that is, without significant spontaneous breathing efforts, during the MV period. Diaphragmatic biopsies were analyzed for ultrastructural changes, atrophy, and the expression of proteolysisrelated proteins (ubiquitin, NF-kB and calpains). Compared to the control group, the long- term MV patient diaphragms showed significantly lower values for muscle fiber cross-sectional area (that is, atrophy), an increase in the global levels of ubiquitinated proteins, higher expression of p65 NF-κB, and greater protein expression levels of the calpain isoforms (1, 2, and 3) in the diaphragm.
A particularly interesting finding in our study was the presence of ultrastructural injury in diaphragm muscle fibers, characterized primarily by disruption of the normal myofibrillar organization [13]. Although this observation had been made in the past in animal models [9, 24, 31, 32], this is the first report of this phenomenon in mechanically ventilated humans. Ultrastructural injury was observed with five to six times greater frequency in long-term MV patients compared to the control group diaphragms [13]. In addition, the potential role that the calcium-activated calpains might play in this process is especially intriguing, given their known role in promoting the degradation of structural proteins of the sarcomere [34]. Finally, it should be noted that both ultrastructural injury and atrophy of diaphragmatic fibers in the long-term MV group were significantly correlated with the total duration of MV.
To summarize, studies performed on the diaphragms of mechanically ventilated ICU patients have largely confirmed animal model data with respect to the structural and biochemical changes that take place in the diaphragm during MV. Hence, findings that are common to both the animal and human studies include increased oxidative stress, muscle fiber atrophy and injury, as well as the activation of several major proteolytic pathways (ubiquitin-proteasome, caspases, calpains). In addition, recently published human data point to an important degree of upregulation of the lysosomally mediated autophagy pathway in the diaphragm during MV.
Potential therapeutic strategies
Mechanical ventilation approaches
Several animal studies have demonstrated that maintaining spontaneous respiratory efforts during mechanical ventilation alleviates VIDD at either a functional or cellular structure/biochemical level [18, 21, 44, 45]. This suggests that clinicians should encourage persistent diaphragmatic activity, as long as this is compatible with adequate patient comfort and gas exchange. Further work is needed to ascertain the optimal level of diaphragmatic effort and it is not known whether the specific method of promoting diaphragmatic effort during MV (for example, spontaneous breathing trials, assist-control, and pressure-support) has any impact upon the risk of developing VIDD. The effects of standard MV settings, such as tidal volume and level of applied positive end-expiratory pressure (PEEP), should also be evaluated. In addition, it would be interesting to determine whether newer modes of mechanical ventilation, such as neurally adjusted ventilatory assist (NAVA), adaptive support ventilation (ASV), and proportional assist ventilation (PAV), have any advantages over the more traditional forms of ventilatory assistance. Another possible method for ensuring adequate levels of muscle activity during MV is through the use of daily intermittent electrical stimulation, an approach that has been reported to preserve muscle mass in the lower extremities of ICU patients [46, 47]. In theory, this could also be applied to the diaphragm as a preventive strategy for VIDD, and this approach deserves further investigation.
Pharmacological approaches
It is important to recognize that even when using partial support modes of MV or allowing for intermittent periods of spontaneous breathing, animal studies have found evidence of persistent oxidative stress [18] as well as a residual deficit of diaphragmatic force production [45, 48], suggesting that additional measures beyond altering the ventilator settings or mode, such as pharmacological agents, may also be needed to completely prevent VIDD in humans. As discussed earlier, both animal and human data indicate that MV is associated with an increase in markers of oxidative stress in the diaphragm. Furthermore, treatment with an antioxidant (the vitamin E analogue Trolox) during MV mitigated diaphragmatic proteolysis and also prevented the loss of diaphragmatic force in animals [49]. This same antioxidant also attenuated diaphragmatic fiber atrophy [50]. In one study of critically ill surgical patients, an anti-oxidant supplement containing vitamins E and C was reported to reduce the duration of MV in comparison to non-supplemented patients [51].
Animal studies have begun to shed light on the main sources of the reactive oxygen species (ROS) generated in the diaphragm during MV. NADPH oxidase, which can generate superoxide within muscle fibers [52], was only found to be minimally increased in the mechanically ventilated diaphragm [53]. In addition, administration of the NADPH oxidase inhibitor apocynin, while beneficial in mitigating VIDD in rats, appeared to exert these effects by upregulating an endogenous calpain inhibitor (calpastatin) rather than through a specific antioxidant action [26]. Xanthine oxidase, another potential source of superoxide, is also upregulated in the diaphragm during MV, and its inhibition modestly improved diaphragmatic contractility but failed to have an impact upon atrophy [54]. Given the above, it appears that mitochondria could be the main source of excessive ROS production in the diaphragm during MV. In this regard, mitochondria isolated from mechanically ventilated rats release significantly more ROS and also exhibit biochemical evidence of oxidative damage [36]. Furthermore, abnormalities of diaphragmatic mitochondrial respiration have been found in several animal species during MV [36, 55, 56]. Therefore, future studies should focus their attention not only on the use of broad-based antioxidants for the prevention or treatment of VIDD, but also upon the potential use of mitochondria-targeted forms of antioxidant therapy.
There is good evidence that the calpain, caspase, and ubiquitin-proteasome proteolysis pathways all play significant roles in the development of MV-induced atrophy. Therefore, these proteolytic systems are also logical targets for therapeutic intervention. In animals, a single administration of leupeptin (inhibitor of calpain/cathepsin) at the onset of MV not only blocked atrophy but also prevented intrinsic contractile impairment in the rat diaphragm [29]. Acute high-dose corticosteroid administration was also found to prevent calpain up-regulation and mitigate VIDD in rats [57], although corticosteroids are unlikely to be a viable treatment for VIDD given their association with critical illness myopathy [58]. Neuromuscular blocking agents can synergize with MV to exacerbate VIDD in animals, with attendant increases in activation of the calpain and ubiquitin-proteasome systems [59, 60]. Somewhat surprisingly, in a recent multicenter randomized study, Papazian and colleagues [61] showed that in patients with severe acute respiratory distress syndrome (PaO2/FiO2 < 150 mmHg), an early and limited period (24 to 48 hours) of cisatracurium administration actually improved survival and increased time off the ventilator without increasing muscle weakness or delaying weaning from mechanical ventilation. Nonetheless, cisatracurium administration, although perhaps less deleterious than rocuronium [59, 60], should still be used with care in patients undergoing long-term MV (that is, >48 hours), as the risk of VIDD is likely to be increased.
Implications and conclusions
In addition to the areas for research into therapeutic interventions mentioned above, in the future it will be important to determine the influence upon VIDD of the prior state of the diaphragm (for example, if fatigued or injured) and other co-morbid conditions that could affect diaphragmatic properties. For example, in the diaphragms of patients with chronic obstructive pulmonary disease, multiple abnormalities have been reported, including decreased myosin content, fiber injury, atrophy, and increased protein oxidation [62, 63]. There is also very little information available regarding the interaction between VIDD and metabolic stresses commonly found in ICU patients, such as hyperglycemia and sepsis. With regard to the latter, one study has reported that short-term MV in rats (4 hours) is actually protective against endotoxin-induced diaphragmatic dysfunction [64]. Further studies are also needed to evaluate the recovery response of the diaphragm from VIDD once spontaneous respiratory efforts have resumed or after MV has been discontinued entirely.
In this review we have attempted to emphasize the key features of VIDD that have been ascertained from recent studies, with a particular focus on the human data (Figure 1). It is important to point out that almost all of the available animal model data describing VIDD were thus far performed in previously healthy animals. This is obviously very different from the usual scenario in ICU patients, in whom multiple confounding factors (for example, previous co-morbidities, different severities and causes of ICU admission, drugs, nutritional status, and so on) are generally present and could very well contribute to reductions in diaphragmatic function. Indeed, due to the presence of these multiple confounding factors, it is generally impossible for the practicing clinician to assign a definitive diagnosis of VIDD to any given patient. Hence, in critically ill humans who undergo long-term MV, it is likely that several factors converge with VIDD to produce diaphragmatic weakness. Given this fact, the similarity of findings between the animal model data and the human studies of mechanically ventilated diaphragms is all the more remarkable. As was the case with VILI, the integration of information gained from studies of animal models and human patients should allow us to develop a conceptual framework for understanding the problem of VIDD and applying this knowledge to patient management.

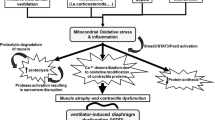
Mechanistic pathways implicated in the human studies of ventilator-induced diaphragmatic dysfunction in critically ill patients. Diaphragmatic inactivity induced by mechanical ventilation is associated with an increased generation of reactive oxygen species (ROS) and a downregulation of the Akt pathway. This leads to increased proteolysis in the diaphragm via activation of the caspase, calpain, ubiquitin-proteasome, and lysosomal-autophagic pathways. In addition, downregulation of the Akt pathway also inhibits protein synthesis (dotted line). Collectively, all of the above contribute to atrophy and injury of diaphragmatic muscle fibers, resulting in functional impairment of the diaphragm. Akt-P, phosphorylated (activated) form of protein kinase B.
Abbreviations
- MV:
-
mechanical ventilation
- NF:
-
nuclear factor
- Pdi:
-
transdiaphragmatic pressure
- ROS:
-
reactive oxygen species
- TwPtr:
-
twitch tracheal airway pressure
- VIDD:
-
ventilator-induced diaphragmatic dysfunction
- VILI:
-
ventilator-induced lung injury.
References
Wunsch H, Linde-Zwirble WT, Angus DC, Hartman ME, Milbrandt EB, Kahn JM: The epidemiology of mechanical ventilation use in the United States. Crit Care Med 2010, 38: 1947-1953.
Pinheiro de Oliveira R, Hetzel MP, dos Anjos Silva M, Dallegrave D, Friedman G: Mechanical ventilation with high tidal volume induces inflammation in patients without lung disease. Crit Care 2010, 14: R39. 10.1186/cc8919
Determann RM, Royakkers A, Wolthuis EK, Vlaar AP, Choi G, Paulus F, Hofstra JJ, de Graaff MJ, Korevaar JC, Schultz MJ: Ventilation with lower tidal volumes as compared with conventional tidal volumes for patients without acute lung injury: a preventive randomized controlled trial. Crit Care 2010, 14: R1. 10.1186/cc8230
Dreyfuss D, Saumon G: Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med 1998, 157: 294-323.
Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network N Engl J Med 2000, 342: 1301-1308. 10.1056/NEJM200005043421801
Phoenix SI, Paravastu S, Columb M, Vincent JL, Nirmalan M: Does a higher positive end expiratory pressure decrease mortality in acute respiratory distress syndrome? A systematic review and meta-analysis. Anesthesiology 2009, 110: 1098-1105. 10.1097/ALN.0b013e31819fae06
Zambon M, Vincent JL: Mortality rates for patients with acute lung injury/ARDS have decreased over time. Chest 2008, 133: 1120-1127. 10.1378/chest.07-2134
Vassilakopoulos T, Petrof BJ: Ventilator-induced diaphragmatic dysfunction. Am J Respir Crit Care Med 2004, 169: 336-341. 10.1164/rccm.200304-489CP
Bernard N, Matecki S, Py G, Lopez S, Mercier J, Capdevila X: Effects of prolonged mechanical ventilation on respiratory muscle ultrastructure and mitochondrial respiration in rabbits. Intensive Care Med 2003, 29: 111-118.
Capdevila X, Lopez S, Bernard N, Rabischong E, Ramonatxo M, Martinazzo G, Prefaut C: Effects of controlled mechanical ventilation on respiratory muscle contractile properties in rabbits. Intensive Care Med 2003, 29: 103-110.
Hermans G, Agten A, Testelmans D, Decramer M, Gayan-Ramirez G: Increased duration of mechanical ventilation is associated with decreased diaphragmatic force: a prospective observational study. Crit Care 2010, 14: R127. 10.1186/cc9094
Hussain SN, Mofarrahi M, Sigala I, Kim HC, Vassilakopoulos T, Maltais F, Bellenis I, Chaturvedi R, Gottfried SB, Metrakos P, Danialou G, Matecki S, Jaber S, Petrof BJ, Goldberg P: Mechanical ventilation-induced diaphragm disuse in humans triggers autophagy. Am J Respir Crit Care Med 2010, 182: 1377-1386. 10.1164/rccm.201002-0234OC
Jaber S, Petrof BJ, Jung B, Chanques G, Berthet JP, Rabuel C, Bouyabrine H, Courouble P, Koechlin-Ramonatxo C, Sebbane M, Similowski T, Scheuermann V, Mebazaa A, Capdevila X, Mornet D, Mercier J, Lacampagne A, Philips A, Matecki S: Rapidly progressive diaphragmatic weakness and injury during mechanical ventilation in humans. Am J Respir Crit Care Med 2010, 183: 364-371. 10.1164/rccm.201004-0670OC
Levine S, Biswas C, Dierov J, Barsotti R, Shrager JB, Nguyen T, Sonnad S, Kucharchzuk JC, Kaiser LR, Singhal S, Budak MT: Increased proteolysis, myosin depletion and atrophic AKT-FOXO signaling in human diaphragm disuse. Am J Respir Crit Care Med 2010, 183: 483-490. 10.1164/rccm.200910-1487OC
Levine S, Nguyen T, Taylor N, Friscia ME, Budak MT, Rothenberg P, Zhu J, Sachdeva R, Sonnad S, Kaiser LR, Rubinstein NA, Powers SK, Shrager JB: Rapid disuse atrophy of diaphragm fibers in mechanically ventilated humans. N Engl J Med 2008, 358: 1327-1335. 10.1056/NEJMoa070447
Radell PJ, Remahl S, Nichols DG, Eriksson LI: Effects of prolonged mechanical ventilation and inactivity on piglet diaphragm function. Intensive Care Med 2002, 28: 358-364. 10.1007/s00134-002-1207-8
Anzueto A, Peters JI, Tobin MJ, de los Santos R, Seidenfeld JJ, Moore G, Cox WJ, Coalson JJ: Effects of prolonged controlled mechanical ventilation on diaphragmatic function in healthy adult baboons. Crit Care Med 1997, 25: 1187-1190. 10.1097/00003246-199707000-00021
Futier E, Constantin JM, Combaret L, Mosoni L, Roszyk L, Sapin V, Attaix D, Jung B, Jaber S, Bazin JE: Pressure support ventilation attenuates ventilator-induced protein modifications in the diaphragm. Crit Care 2008, 12: R116. 10.1186/cc7010
Gayan-Ramirez G, de Paepe K, Cadot P, Decramer M: Detrimental effects of short-term mechanical ventilation on diaphragm function and IGF-I mRNA in rats. Intensive Care Med 2003, 29: 825-833.
Jaber S, Sebbane M, Koechlin C, Hayot M, Capdevila X, Eledjam JJ, Prefaut C, Ramonatxo M, Matecki S: Effects of short vs. prolonged mechanical ventilation on antioxidant systems in piglet diaphragm. Intensive Care Med 2005, 31: 1427-1433. 10.1007/s00134-005-2694-1
Jung B, Constantin JM, Rossel N, Le Goff C, Sebbane M, Coisel Y, Chanques G, Futier E, Hugon G, Capdevila X, Petrof B, Matecki S, Jaber S: Adaptive support ventilation prevents ventilator-induced diaphragmatic dysfunction in piglet: an in vivo and in vitro study. Anesthesiology 2010, 112: 1435-1443. 10.1097/ALN.0b013e3181d7b036
Le Bourdelles G, Viires N, Boczkowski J, Seta N, Pavlovic D, Aubieri M: Effects of mechanical ventilation on diaphragmatic contractile properties in rats. Am J Respir Crit Care Med 1994, 149: 1539-1544.
Powers SK, Shanely RA, Coombes JS, Koesterer TJ, McKenzie M, Van Gammeren D, Cicale M, Dodd SL: Mechanical ventilation results in progressive contractile dysfunction in the diaphragm. J Appl Physiol 2002, 92: 1851-1858.
Sassoon CS, Caiozzo VJ, Manka A, Sieck GC: Altered diaphragm contractile properties with controlled mechanical ventilation. J Appl Physiol 2002, 92: 2585-2595.
Yang L, Luo J, Bourdon J, Lin MC, Gottfried SB, Petrof BJ: Controlled mechanical ventilation leads to remodeling of the rat diaphragm. Am J Respir Crit Care Med 2002, 166: 1135-1140. 10.1164/rccm.2202020
McClung JM, Van Gammeren D, Whidden MA, Falk DJ, Kavazis AN, Hudson MB, Gayan-Ramirez G, Decramer M, DeRuisseau KC, Powers SK: Apocynin attenuates diaphragm oxidative stress and protease activation during prolonged mechanical ventilation. Crit Care Med 2009, 37: 1373-1379. 10.1097/CCM.0b013e31819cef63
Shanely RA, Zergeroglu MA, Lennon SL, Sugiura T, Yimlamai T, Enns D, Belcastro A, Powers SK: Mechanical ventilation-induced diaphragmatic atrophy is associated with oxidative injury and increased proteolytic activity. Am J Respir Crit Care Med 2002, 166: 1369-1374. 10.1164/rccm.200202-088OC
DeRuisseau KC, Kavazis AN, Deering MA, Falk DJ, Van Gammeren D, Yimlamai T, Ordway GA, Powers SK: Mechanical ventilation induces alterations of the ubiquitin-proteasome pathway in the diaphragm. J Appl Physiol 2005, 98: 1314-1321. 10.1152/japplphysiol.00993.2004
Maes K, Testelmans D, Powers S, Decramer M, Gayan-Ramirez G: Leupeptin inhibits ventilator-induced diaphragm dysfunction in rats. Am J Respir Crit Care Med 2007, 175: 1134-1138. 10.1164/rccm.200609-1342OC
McClung JM, Kavazis AN, DeRuisseau KC, Falk DJ, Deering MA, Lee Y, Sugiura T, Powers SK: Caspase-3 regulation of diaphragm myonuclear domain during mechanical ventilation-induced atrophy. Am J Respir Crit Care Med 2007, 175: 150-159. 10.1164/rccm.200601-142OC
Zhu E, Sassoon CS, Nelson R, Pham HT, Zhu L, Baker MJ, Caiozzo VJ: Early effects of mechanical ventilation on isotonic contractile properties and MAF-box gene expression in the diaphragm. J Appl Physiol 2005, 99: 747-756. 10.1152/japplphysiol.00126.2005
Radell P, Edstrom L, Stibler H, Eriksson LI, Ansved T: Changes in diaphragm structure following prolonged mechanical ventilation in piglets. Acta Anaesthesiol Scand 2004, 48: 430-437. 10.1111/j.1399-6576.2004.00352.x
Zergeroglu MA, McKenzie MJ, Shanely RA, Van Gammeren D, DeRuisseau KC, Powers SK: Mechanical ventilation-induced oxidative stress in the diaphragm. J Appl Physiol 2003, 95: 1116-1124.
Belcastro AN, Shewchuk LD, Raj DA: Exercise-induced muscle injury: a calpain hypothesis. Mol Cell Biochem 1998, 179: 135-145. 10.1023/A:1006816123601
Du J, Wang X, Miereles C, Bailey JL, Debigare R, Zheng B, Price SR, Mitch WE: Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J Clin Invest 2004, 113: 115-123.
Jackman RW, Kandarian SC: The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol 2004, 287: C834-843. 10.1152/ajpcell.00579.2003
ATS/ERS Statement on respiratory muscle testing Am J Respir Crit Care Med 2002, 166: 518-624. 10.1164/rccm.166.4.518
Boles JM, Bion J, Connors A, Herridge M, Marsh B, Melot C, Pearl R, Silverman H, Stanchina M, Vieillard-Baron A, Welte T: Weaning from mechanical ventilation. Eur Respir J 2007, 29: 1033-1056. 10.1183/09031936.00010206
Similowski T, Fleury B, Launois S, Cathala HP, Bouche P, Derenne JP: Cervical magnetic stimulation: a new painless method for bilateral phrenic nerve stimulation in conscious humans. J Appl Physiol 1989, 67: 1311-1318.
Laghi F, Cattapan SE, Jubran A, Parthasarathy S, Warshawsky P, Choi YS, Tobin MJ: Is weaning failure caused by low-frequency fatigue of the diaphragm? Am J Respir Crit Care Med 2003, 167: 120-127. 10.1164/rccm.200210-1246OC
Watson AC, Hughes PD, Louise Harris M, Hart N, Ware RJ, Wendon J, Green M, Moxham J: Measurement of twitch transdiaphragmatic, esophageal, and endotracheal tube pressure with bilateral anterolateral magnetic phrenic nerve stimulation in patients in the intensive care unit. Crit Care Med 2001, 29: 1325-1331. 10.1097/00003246-200107000-00005
Cattapan SE, Laghi F, Tobin MJ: Can diaphragmatic contractility be assessed by airway twitch pressure in mechanically ventilated patients? Thorax 2003, 58: 58-62. 10.1136/thorax.58.1.58
Knisely AS, Leal SM, Singer DB: Abnormalities of diaphragmatic muscle in neonates with ventilated lungs. J Pediatr 1988, 113: 1074-1077. 10.1016/S0022-3476(88)80585-7
Gayan-Ramirez G, Testelmans D, Maes K, Racz GZ, Cadot P, Zador E, Wuytack F, Decramer M: Intermittent spontaneous breathing protects the rat diaphragm from mechanical ventilation effects. Crit Care Med 2005, 33: 2804-2809. 10.1097/01.CCM.0000191250.32988.A3
Sassoon CS, Zhu E, Caiozzo VJ: Assist-control mechanical ventilation attenuates ventilator-induced diaphragmatic dysfunction. Am J Respir Crit Care Med 2004, 170: 626-632. 10.1164/rccm.200401-042OC
Gerovasili V, Stefanidis K, Vitzilaios K, Karatzanos E, Politis P, Koroneos A, Chatzimichail A, Routsi C, Roussos C, Nanas S: Electrical muscle stimulation preserves the muscle mass of critically ill patients: a randomized study. Crit Care 2009, 13: R161. 10.1186/cc8123
Gerovasili V, Tripodaki E, Karatzanos E, Pitsolis T, Markaki V, Zervakis D, Routsi C, Roussos C, Nanas S: Short-term systemic effect of electrical muscle stimulation in critically ill patients. Chest 2009, 136: 1249-1256. 10.1378/chest.08-2888
Gayan-Ramirez GN, Decramer ML: Diaphragm antioxidant system in controlled mechanical ventilation in piglets: short term vs. prolonged mechanical ventilation response. Intensive Care Med 2005, 31: 1303-1305. 10.1007/s00134-005-2693-2
Wang KK: Calpain and caspase: can you tell the difference? Trends Neurosci 2000, 23: 20-26. 10.1016/S0166-2236(99)01479-4
Dobrowolny G, Aucello M, Rizzuto E, Beccafico S, Mammucari C, Boncompagni S, Belia S, Wannenes F, Nicoletti C, Del Prete Z, Rosenthal N, Molinaro M, Protasi F, Fanò G, Sandri M, Musarò A: Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab 2008, 8: 425-436. 10.1016/j.cmet.2008.09.002
Nathens AB, Neff MJ, Jurkovich GJ, Klotz P, Farver K, Ruzinski JT, Radella F, Garcia I, Maier RV: Randomized, prospective trial of antioxidant supplementation in critically ill surgical patients. Ann Surg 2002, 236: 814-822. 10.1097/00000658-200212000-00014
Chen M, Won DJ, Krajewski S, Gottlieb RA: Calpain and mitochondria in ischemia/reperfusion injury. J Biol Chem 2002, 277: 29181-29186. 10.1074/jbc.M204951200
Jaber S, Petrof BJ, Jung B, Chanques G, Berthet JP, Rabuel C, Bouyabrine H, Courouble P, Koechlin-Ramonatxo C, Sebbane M, Similowski T, Scheuermann V, Mebazaa A, Capdevila X, Mornet D, Mercier J, Lacampagne A, Philips A, Matecki S: Rapidly progressive diaphragmatic weakness and injury during mechanical ventilation in humans. Am J Respir Crit Care Med 2011, 183: 364-371. 10.1164/rccm.201004-0670OC
Whidden MA, McClung JM, Falk DJ, Hudson MB, Smuder AJ, Nelson WB, Powers SK: Xanthine oxidase contributes to mechanical ventilation-induced diaphragmatic oxidative stress and contractile dysfunction. J Appl Physiol 2009, 106: 385-394. 10.1152/japplphysiol.91106.2008
Scarabelli TM, Stephanou A, Pasini E, Comini L, Raddino R, Knight RA, Latchman DS: Different signaling pathways induce apoptosis in endothelial cells and cardiac myocytes during ischemia/reperfusion injury. Circ Res 2002, 90: 745-748. 10.1161/01.RES.0000015224.07870.9A
Vassilakopoulos T, Zakynthinos S, Roussos C: The tension-time index and the frequency/tidal volume ratio are the major pathophysiologic determinants of weaning failure and success. Am J Respir Crit Care Med 1998, 158: 378-385.
Maes K, Testelmans D, Cadot P, Deruisseau K, Powers SK, Decramer M, Gayan-Ramirez G: Effects of acute administration of corticosteroids during mechanical ventilation on rat diaphragm. Am J Respir Crit Care Med 2008, 178: 1219-1226. 10.1164/rccm.200702-296OC
Dekhuijzen PN, Decramer M: Steroid-induced myopathy and its significance to respiratory disease: a known disease rediscovered. Eur Respir J 1992, 5: 997-1003.
Testelmans D, Maes K, Wouters P, Gosselin N, Deruisseau K, Powers S, Sciot R, Decramer M, Gayan-Ramirez G: Rocuronium exacerbates mechanical ventilation-induced diaphragm dysfunction in rats. Crit Care Med 2006, 34: 3018-3023.
Testelmans D, Maes K, Wouters P, Powers SK, Decramer M, Gayan-Ramirez G: Infusions of rocuronium and cisatracurium exert different effects on rat diaphragm function. Intensive Care Med 2007, 33: 872-879. 10.1007/s00134-007-0584-4
Papazian L, Forel JM, Gacouin A, Penot-Ragon C, Perrin G, Loundou A, Jaber S, Arnal JM, Perez D, Seghboyan JM, Constantin JM, Courant P, Lefrant JY, Guérin C, Prat G, Morange S, Roch A, ACURASYS Study Investigators: Neuromuscular blockers in early acute respiratory distress syndrome. N Engl J Med 2010, 363: 1107-1116. 10.1056/NEJMoa1005372
Ottenheijm CA, Heunks LM, Dekhuijzen PN: Diaphragm muscle fiber dysfunction in chronic obstructive pulmonary disease: toward a pathophysiological concept. Am J Respir Crit Care Med 2007, 175: 1233-1240. 10.1164/rccm.200701-020PP
Testelmans D, Crul T, Maes K, Agten A, Crombach M, Decramer M, Gayan-Ramirez G: Atrophy and hypertrophy signalling in the diaphragm of patients with COPD. Eur Respir J 2010, 35: 549-556. 10.1183/09031936.00091108
Ebihara S, Hussain SN, Danialou G, Cho WK, Gottfried SB, Petrof BJ: Mechanical ventilation protects against diaphragm injury in sepsis: interaction of oxidative and mechanical stresses. Am J Respir Crit Care Med 2002, 165: 221-228.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Jaber, S., Jung, B., Matecki, S. et al. Clinical review: Ventilator-induced diaphragmatic dysfunction - human studies confirm animal model findings!. Crit Care 15, 206 (2011). https://doi.org/10.1186/cc10023
Published:
DOI: https://doi.org/10.1186/cc10023