
Abstract
Although much feared by clinicians, the ability to produce ketones has allowed humans to withstand prolonged periods of starvation. At such times, ketones can supply up to 50% of basal energy requirements. More interesting, however, is the fact that ketones can provide as much as 70% of the brain's energy needs, more efficiently than glucose. Studies suggest that during times of acute brain injury, cerebral uptake of ketones increases significantly. Researchers have thus attempted to attenuate the effects of cerebral injury by administering ketones exogenously. Hypertonic saline is commonly utilized for management of intracranial hypertension following cerebral injury. A solution containing both hypertonic saline and ketones may prove ideal for managing the dual problems of refractory intracranial hypertension and low cerebral energy levels. The purpose of the present review is to explore the physiology of ketone body utilization by the brain in health and in a variety of neurological conditions, and to discuss the potential for ketone supplementation as a therapeutic option in traumatic brain injury.
Similar content being viewed by others

Introduction
Ketogenesis is the process by which ketone bodies (KB), during times of starvation, are produced via fatty acid metabolism. Although much feared by physicians, mild ketosis can have therapeutic potential in a variety of disparate disease states. The principle ketones include acetoacetate (AcAc), β-hydroxybutyrate (BHB) and ace-tone. In times of starvation and low insulin levels, ketones supply up to 50% of basal energy requirements for most tissues, and up to 70% for the brain. Although glucose is the main metabolic substrate for neurons, ketones are capable of fulfilling the energy requirements of the brain.
The purpose of the present review is to explore the physiology of ketone body utilization by the brain in health and a variety of neurological conditions, and to discuss the potential for ketone supplementation as a therapeutic option in traumatic brain injury (TBI).
Ketones, metabolism and the brain
Ketone metabolism
Ketones are by-products of fat metabolism and are produced in hepatic mitochondria [1]. More specifically, the three KB are: AcAc - if not oxidized to form usable energy, this is the source of the two other KB; acetone - not an energy source, but instead exhaled or excreted as waste; and BHB - not technically a ketone, but reversibly produced from AcAc.
The precursor for the synthesis of ketones is acetyl coenzyme A (acetyl CoA), which is formed by β-oxidation of free fatty acids in the liver. The two important determinants of ketogenesis are the availability of acetyl CoA and the mobilization of fatty acids liberated from white adipose tissue during fasting or catecholaminergic stress [2, 3].
AcAc is the central ketone body. AcAc is reduced to BHB by β-hydroxybutyrate dehydrogenase (BHD) in a NADH-requiring reversible reaction [3]:

The extent of this reaction depends on the state of the cellular pool of NAD+ [4]. When highly reduced, most or all ketones will be in the form of BHB. The ratio of BHB to AcAc reflects the redox state within the mitochondrial matrix [5]. BHB is nonvolatile and is chemically stable. This ketone body's only metabolic fate is interconversion with AcAc. Once synthesized, ketones diffuse into the circulation.
Fate of circulating ketones
Plasma concentrations of circulating KB range from <0.1 mM/l in the postprandial state to as much as 6 mM/l during fasting, with levels reaching 25 mM/l in diabetic ketoacidosis [6–8]. Under normal dietary conditions, ketogenesis from fatty acids is at very low levels. This is attributable to the relatively high blood levels of the ketogenesis-inhibiting hormone insulin, and low blood levels of ketogenesis-promoting hormones, glucagon and cortisol [2]. One of the prerequisites for ketogenesis is therefore low levels of circulating insulin, as seen during starvation and diabetic ketoacidosis
The fate of circulating KB is to be taken up by extra-hepatic tissues, although 10 to 20% may be lost in the urine [8, 9]. KB provide an energy source for several tissues including the brain, skeletal muscle, myocardium and liver, although the heart and kidney have the greatest capacity for utilization [10]. The rate of utilization appears to be a function of the circulating plasma levels [11–13].
Ketones and critical illness
The major illness associated by intensive care physicians with ketosis is diabetic ketoacidosis. Outside diabetic ketoacidosis, ketone measurement is not routinely performed and has been largely used as a research tool to quantify cellular redox status. The arterial ketone body ratio (AKBR) is utilized to reflect the mitochondrial redox status of hepatic cells and may reflect cellular energy status in a number of conditions including hemorrhage, hypoxia, sepsis and hepatic resection [14]. The AKBR (ratio of AcAc to BHB) reflects the cellular NAD+/NADH ratio, which cannot be measured directly and is linked to the phosphorylation potential of the cell (see equation above). In critically ill patients, the entry of acetyl CoA into the tricarboxylic acid cycle is limited. Acetyl CoA consequently combines with another molecule of acetyl CoA to form acetoacetic acid, which in turn is converted to BHB leading to an altered NAD+/NADH ratio and a decreased AKBR [14, 15]. A reduced AKBR is therefore a sign of an inefficient tricarboxylic acid cycle. The AKBR has also been used as an indirect marker of the hepatocellular functional status as the predominant site of metabolism of the ketones is in the liver.
Not surprisingly, metabolic failure at the cellular level is associated with worse clinical outcomes. A low AKBR is associated with poor outcomes in both animal and human models of illness. Yassen and colleagues measured the AKBR in 20 critically ill patients [15]. They demonstrated a progressive decrease in those patients with septic shock who died. In a study of critically ill heart failure patients, Limuro and colleagues noted a survival of only 15% of patients with AKBR <0.7 [16]. Dresing and colleagues found the AKBR to be significantly lower in nonsurvivors than survivors in 30 polytrauma patients (0.6 vs. 1.3; P = 0.001) [17]. Levy and colleagues, however, failed to find a difference in AKBR levels between survivors and nonsurvivors in their cohort of septic patients [18]. In conjunction with the lactate:pyruvate ratio, the AKBR may provide further insight into the cellular redox potential of critically ill patients.
Ketones and the brain
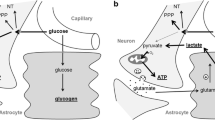
Glucose is the major fuel for the brain in humans on a balanced diet. More recently it was proposed that other substances such as lactate and pyruvate may be utilized by neurons to sustain their activity [19]. Furthermore, during times of starvation, the brain has the capacity to adapt to the use of ketones as its major energy source (and in the metabolic disease diabetes mellitus). In longstanding starvation, ketones can provide 60 to 70% of the energy needs of the brain [20]. The 1.5 kg human brain utilizes 100 to 150 g glucose per day and 20% of the total body oxygen consumption at rest [7]. Protein catabolism can supply somewhere between 17 and 32 g glucose per day, well below the minimum daily cerebral glucose requirements [21]. In fact, to supply sufficient glucose to support brain metabolic requirements from protein alone would lead to death in about 10 days instead of the 57 to 73 days [22]. Moreover in subjects undergoing total starvation for 30 days, the decrease in hunger coincides with the elevation of blood ketones to 7 mM/l. During periods of ongoing starvation, an average-size person produces about 150 g KB per day [23]. One should note, however, that ketones are incapable of maintaining or restoring normal cerebral function in the complete absence of glucose.
Astrocytes may play a significant role in the regulation of brain energy metabolism [24]. Reports suggest that astrocytes can produce KB from fatty acids and leucine and are involved with other pathways of lipid metabolism, such as lipid synthesis and lipoprotein secretion [25–28]. Furthermore, it appears that the production of lactate and KB by astrocytes may increase secondary to neurotransmitters released during enhance synaptic activity [29, 30].
Although the implications of ketone body production by astrocytes are still unclear, the fact that astrocytes outnumber neurons approximately 9:1 and occupy at least 50% of the cerebral volume suggests this constitutes a quantitatively important pathway in the brain [31].
Regulation of cerebral ketone uptake
As previously noted, humans can achieve very high circulating KB concentrations during prolonged fasting, up to 9 mM/l (range 5.8 to 9.7 mM/l) [20]. The plasma concentration of KB is a significant factor affecting the rate of cerebral uptake. The blood-brain barrier (BBB) is relatively impermeable to most hydrophilic substances, such as KB, unless they are transported by a carrier protein. A family of proteins involved in the transport of monocarboxylic acids such as lactic acid and pyruvate across cellular membranes, called monocarboxylic acid transporters (MCT1 and MCT2), are thought to play an integral role.
It is not yet understood how MCTs are regulated. KB are transported at different rates, with the uptake of AcAc being twice that of BHB at a given arterial concentration [12]. Furthermore, prolonged fasting appears to increase the BBB uptake of ketones, with some researchers demonstrating an eightfold increase in the expression of MCT1 [32]. Similarly, Hasselbalch and colleagues reported a 13-fold increase in cerebral uptake of BHB in humans following several days of fasting [33]. In contrast, rapidly increasing plasma ketone concentrations do not lead to such a significant increase in cerebral ketone uptake. Pan and colleagues noted a much smaller increase in cerebral ketone concentrations following a rapid intravenous infusion of BHB as compared with prolonged fasting, implying that MCT upregulation is partly dependent on the length of exposure to increased plasma ketone levels [34, 35].
Once across the BBB, KB enter brain cells to support cellular energy requirements. Studies of the uptake of KB by brain cells suggest entry occurs by two mechanisms: diffusion and a carrier-mediated process. MCTs have been demonstrated on neurons and glial cells although their role here appears less significant as compared with that on the BBB. The impact of mitochondrial enzyme activity involved in ketone metabolism is less clear. Certainly, mitochondrial enzymes can be upregulated by increased levels of BHB, as has been demonstrated following exposure to ketones through starvation or a prolonged high-fat diet [12, 36–39]. In summary, there- fore, a prolonged high blood ketone concentration leads to an increased rate of cerebral ketone uptake and metabolism.
Regional variation in ketone body metabolism
Data for the regional variation in ketone body metabolism are based largely on animal studies. Hawkins and colleagues and Morris noted that KB uptake varies from one brain region to another [40, 41]. Following an intravenous administration of radioactive BHB to adult rats, uptake was greatest in the cerebral cortex (deep layers), in the superior and inferior colliculi, and in regions where the BBB is absent [40, 41]. Areas with a low metabolic rate such as the corpus callosum and other white-matter structures demonstrated greatly reduced uptake. Interestingly, the pattern of KB uptake resembled that of glucose, except in the superficial layers of the cortex where glucose uptake was higher [42]. Similarly, MCT1 expression in the brain may resemble the pattern of KB uptake with some exceptions as MCT1 is found on glia and neurons as well as on BBB epithelial cells and because the pituitary and pineal have no BBB.
Neuroprotective effects of ketones
Neuroprotection is the mechanisms and strategies that, once implemented, may lead to salvage, recovery or regeneration of the nervous system. Ketones may have a neuroprotective effect (see Figure 1). Ketones represent an alternative fuel for both the normal and the injured brain [43].

Potential neuroprotective mechanisms of ketones. Ketones (1) require only three enzymatic steps to enter the tricarboxylic acid (TCA) cycle, (2) reduce the NAD couple, (3) decrease free radical formation, (4) increase production of ATP, (5) increase mitochondrial uncoupling, (6) increase glutathione peroxidase activity, and (7) inhibit pyruvate entry into the TCA cycle. CoA, coenzyme A; GSH, reduced glutathione; GSSG, oxidized glutathione; NAD+, nicotinamide adenine dinucleotide; NADH, nicotinamide adenine dinucleotide hydrogenase; NADP, nicotinamide adenine dinucleotide phosphate; NADPH, nicotinamide adenine dinucleotide phosphate hydrogenase; OHB, hydroxybutyrate; PPP, pentose phosphate pathway; UCP, uncoupling protein. Reprinted with permission from [43].
Firstly, BHB is more energy efficient than glucose and can stimulate mitochondrial biogenesis via the upregulation of genes encoding energy metabolism and mitochondrial enzymes [44]. There is a marked increase in the free energy of ATP hydrolysis, as demonstrated by a 25% increase in hydrolytic work of isolated rat perfused heart preparations when exposed to KB [45]. Also, ketones increase the intermediary metabolites delivered to the citric acid cycle with a 16-fold elevation in acetyl CoA content.
Secondly, ketones protect against glutamate-mediated apoptosis and necrosis through the attenuation of the formation of reactive oxidant species. Ketones can oxidize coenzyme Q, thus decreasing mitochondrial free radical formation. Additionally, the reduction of NAD favors reduction of glutathione, which ultimately favors destruction of hydrogen peroxide by glutathione peroxidase reaction [46, 47].
Thirdly, ketones enhance the conversion of glutamate to GABA with the subsequence enhancement of GABA- mediated inhibition [48, 49]. Cerebral ketone uptake also increases cerebral blood flow [50].
Fourthly, cell death from apoptosis is attenuated by ketone ingestion [48]. Mechanisms include reductions in activation of caspase-3, increases in calbindin and prevention of accumulation of the protein clusterin.
Lastly, cerebral ketone metabolism alters cerebral blood flow [50]. Hasselbalch and colleagues demonstrated a 39% increase in cerebral blood flow following an infusion of sodium BHB.
Current state of research - changes in ketone metabolism during brain injury
Models of injury - animal studies
Adaptive changes to cerebral ketone metabolism during development and starvation are well documented. How-ever, there is evidence that other conditions including TBI, ischemia, hemorrhagic shock and hypoxia induce rapid changes in both vascular and cellular transporters that favor ketone uptake and metabolism. In addition, ketone-metabolizing enzymes demonstrate the ability to change in response to neuropathologic conditions. Collectively, these adaptations suggest that the brain may be more receptive to ketone metabolism under neuropathic conditions.
Researchers have demonstrated an increase in MCT1 and MCT2 expression following cerebral injury [51]. Similarly, ketone-metabolizing enzyme expression also appears to alter. Utilization of BHB in the brain is contingent on its conversion to AcAc by BHD, which is scarce in the adult brain, especially in the basal ganglia. Studies confirm a significant increase in BHD activity following cerebral insult [52].
The brain's ability to utilize exogenous KB is altered. At times of oxidative stress, the ability of the brain to use glucose as a substrate for metabolic activity may become compromised. Certainly, hyperglycemia has been repeatedly shown to be deleterious to the injured brain [53]. During these times, an exogenous supply of ketones may force the brain to shift its reliance from glucose to ketones, thus taking advantage of improved transport and cellular metabolism. This shift can be advantageous in both acute brain injury models (glutamate excitotoxicity, hypoxia/ischemia, TBI) and neurodegenerative conditions (Parkinson's disease, Alzheimer's disease).
Extensive research has been performed using the glutamate-induced injury model, the cerebral hypoxia model and the ischemic injury model. Glutamate neurotoxicity is implicated in a number of neurodegenerative disorders associated with brain ischemia, hypoglycemia and cerebral trauma. During injury, glutamate and aspartate are released through the reverse action of their transport systems or via enhanced exocytotic release. This in turn leads to excitotoxicity and the production of reactive oxidant species. Administration of ketones appears to limit the extent of damage [54–58]. For example, Massieu and colleagues demonstrated that AcAc effectively protects against glutamate neurotoxicity both in vivo and in vitro in rats chronically treated with a glycolysis inhibitor [55].
Hypoxic/ischemic injury leads to a decrease in oxidative metabolism by reducing oxygen availability [59, 60]. Ketone administration ameliorates many of the sequelae of this process, including lactate generation, free radical damage and apoptotic cascade activation [61, 62]. Suzuki and colleagues demonstrated that BHB prolonged the survival time in rat models of hypoxia, anoxia and global ischemia [63]. They further showed that BHB reduced infarct size after middle cerebral artery occlusion, irrespective of whether BHB administration was delayed.
More recently, TBI models have been developed [64, 65]. TBI is associated with impaired cerebral energy production with prolonged glucose metabolic depression and reduction of ATP. Cerebral injury is further exacerbated by free radical production, and activation of poly- ADP-ribose polymerase via DNA damage [66]. As such, under these conditions of impaired glycolytic metabolism, shifting the brain toward ketone metabolism may limit the extent of cerebral injury [63, 67–70] (see Table 1). Much of this work is attributable to Prins and colleagues, who demonstrated that administration of intravenous BHB after TBI in adult rats led to an increase in uptake of BHB and alleviated the decrease in ATP after injury. In a subsequent study they found that a ketogenic diet was effective in reducing the cortical contusion volume in a rat model of TBI [64, 65].
Neurodegenerative conditions may also benefit from ketone administration as there is evidence of altered cerebral metabolism. Studies of Parkinson's disease and Alzheimer's disease confirm the encouraging effects of ketones. Mechanisms are complex but include decreasing free radical production and providing acetyl CoA for energy production [45, 52, 71–74]. Other conditions that may be responsive to ketones include amyotrophic lateral sclerosis and certain brain tumors.
Models of injury - human research
Mild ketosis is induced in humans by either fasting or the consumption of a high-fat, low-carbohydrate diet. Most studies in humans have delivered ketones via the enteral route [75]. In adults, achieving blood levels of 5 to 7 mM/l total KB requires the production or intake of about 150 g per day [23]. This level, however, is subject to the influences of glucose, insulin, cortisol and catecholamines. To achieve a therapeutic response in refractory epilepsy, it has been suggested that the level of blood ketone should be above 4 mM/l [76].
Salts of KB can be produced. Several authors (see below) have administered sodium AcAc or sodium BHB successfully with few side effects. A new formulation called KTX 0101 consists of a combination of NaCl and BHB, although relevant concentrations are not described [77]. The main issues related to intravenous administration of ketone solutions include volume loading, sodium loading and the potential for significant metabolic alkalosis. These issues will depend on the relevant concentrations of anions and cations in the mixture and on the osmolality.
There is a paucity of research examining the effects of ketone administration in humans. Interpretation of results is further complicated by variable routes of administration, concentrations and age variability of research subjects. For instance, research on the effects of intraenous ketone preparations are confined almost exclusively to adults, whereas dietary studies include a large number of children. As far as we are aware, only two clinical studies examining the neuroprotective effects of KB in humans have been undertaken. The first by Ritter and colleagues examined the effects of a ketogenic diet on biochemical markers of metabolism in head-injury patients but did not include clinical outcomes [78]. The second study, reported by Smith and colleagues, has never been published but the authors reported on the use of a sodium BHB solution for neuroprotection during cardiac surgery [77]. The bulk of the research has taken place using the rodent model.
Some general statements, however, can be made. Studies have demonstrated that ketones can be given enterally or intravenously with minimal side effects; that utilization of KB increases with rising blood concentrations, with a plateau at approximately 128 g/24 hours; that hyperketotic states are associated with improved glucose control; that increased plasma levels lead to an increase in cerebral uptake; and that ketogenic diets may lead to improved cognitive function in patients with chronic dementia (see Table 2). Well-designed outcome studies, however, are lacking [34, 35, 77–85].
Adverse effects of ketones
There are minimal data on the adverse effects of ketone administration, much of which are based on chronic intake via the enteral route rather than the parenteral route. In the acute setting, effects of parenteral formulations on the pH, sodium level, lipids and glucose are likely to dominate; for example, intravenous formulations would theoretically be alkalinizing, depending on the ketone concentration. This was confirmed by Hiraide and colleagues, who noted a significant increase in pH and sodium concentrations following the intravenous administration of a 20% solution of sodium BHB to severe trauma patients [81]. Also of potential concern is the reduction in glucose cerebral metabolism and the increase in cerebral blood flow demonstrated by Hasselbalch and colleagues during administration of intravenous BHB [50]. The long-term consequences of these changes are not yet known.
Some side effects are predictable following enteral administration, such as dehydration and hypoglycemia. Other side effects are less common and present following prolonged use, including growth retardation, obesity, nutrient deficiency, decreased bone density, hepatic failure, and immune dysfunction. Freeman and colleagues reported a significant rise in mean blood cholesterol to over 250 mg/dl following prolonged intake a of ketogenic diet [86]. This effect in turn may be atherogenic, leading to lipid deposition in blood vessels. In addition there are reports of dilated cardiomyopathy in patients on the ketogenic diet, possibly as a consequence of the toxic effects of elevated plasma free fatty acids. Finally, an increased incidence in nephrolithiasis in patients on the ketogenic diet as well as increases in serum uric acid secondary have been reported [66, 87].
Potential clinical applications for ketone solutions - traumatic brain injury
TBI is a major cause of mortality and morbidity in young adults [88]. Cerebral edema and subsequent intracranial hypertension is an important factor influencing patient outcome, and, despite considerable research, has proven difficult to manage. Over the past 30 years, osmotherapy has become an important tool in the management of intracranial hypertension after TBI [88]. Various substances, including urea, glycerol, sorbitol and mannitol, have been utilized [89, 90]. More recently, hypertonic saline (HTS) has proven useful in controlling intracranial pressure (ICP).
HTS has been extensively studied in animal and human models of TBI [91–94]. These experiments suggest that small-volume resuscitation with HTS solution may be beneficial for increasing cerebral perfusion pressure and cerebral blood flow and for decreasing ICP while maintaining hemodynamic stability following TBI. Animal studies comparing HTS and mannitol suggest that HTS is more effective in reducing ICP and has a longer duration of action [95, 96]. In humans, HTS appears effective for reducing ICP in cases of refractory intracranial hyper- tension following TBI. Several studies have confirmed a significant decrease in ICP correlated with an increasing serum osmolality [97–99].
In the past, researchers have combined hypertonic sodium with a variety of anions including acetate and lactate [100, 101]. Acetate, a source of hydrogen ion acceptors, is an alternate source of bicarbonate by metabolic conversion in the liver. Acetate is a myocardial depressant, however, and can lead to hypotension in susceptible individuals. Hypertonic sodium/lactate solutions have also been investigated. Whilst HTS/lactate is generally considered safe and may provide cerebral nutrition, there are limited animal data suggesting that fluids containing lactate may be associated with significant toxic effects [102]. Further research is required to clarify these issues.
There is therefore a need for a solution able to address two fundamental problems related to TBI; namely, refractory intracerebral hypertension and low cerebral energy levels. This requirement could theoretically be accomplished by incorporating ketones into a hypertonic solution such as HTS. There are, however, a number of challenges associated with the development of new parenteral solutions.
Firstly, the impact of the constituents on individual metabolism needs to be evaluated. The hypertonicity induced by 3% NaCl is well documented and desirable. The target sodium level is not known, however, and various authors have reported levels as high as 185 mmol/l. Secondly, although the long-term metabolic effects of ketone administration are described, little information exists on the acute changes induced by intravenous infusions. For example, the entry of ketones into cerebrospinal fluid is partly concentration dependent and therefore a certain serum level (possibly above 4 mM/l) is required for maximum utilization. What then is the effect of high ketone concentration on blood pH? Such a solution would be expected to be alkalinizing, but this will be altered by the rate of metabolism. Thirdly, which ketone should be incorporated, AcAc or BHB? Both ketones have been administered to humans in the past without ill effects. Fourthly, should cerebrospinal fluid or plasma levels be utilized to guide therapy? Lastly, ketones are expensive to produce, so can a commercially viable solution be developed?
Conclusion
There is both theoretical and clinical evidence that administering ketones to patients with cerebral injury may provide significant benefit. Furthermore, combining ketones with HTS may provide the added advantage of ICP control with improved cerebral metabolism. There are, however, a number of outstanding questions requiring further consideration. These questions include dosing, timing, and the route and duration of administration. Further research is necessary to clarify these issues and to determine whether an HTS ketone solution could be employed in the management of acute brain injury.
Abbreviations
- AcAc:
-
acetoacetate
- acetyl CoA:
-
coenzyme A
- AKBR:
-
arterial ketone body ratio
- BBB:
-
blood-brain barrier
- BHB:
-
β-hydroxybutyrate
- BHD:
-
β-hydroxybutyrate dehydrogenase
- GABA:
-
γ-aminobutyric acid
- HTS:
-
hypertonic saline
- ICP:
-
intracranial pressure
- KB:
-
ketone bodies
- MCT:
-
monocarboxylic acid transporter
- NAD+:
-
nicotinamide adenine dinucleotide
- NADH:
-
nicotinamide adenine dinucleotide hydrogenase
- TBI:
-
traumatic brain injury.
References
Fukao T, Lopaschuk GD, Mitchell GA: Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot Essent Fatty Acids 2004, 70: 243-251. 10.1016/j.plefa.2003.11.001
DeFronzo R, Ferrannini E: Regulation of intermediary metabolism during fasting and feeding. In Endocrinology. Edited by: DeGroot L, Jameson J. Philadelphia: W.B. Saunders; 2001:737-755.
Veech RL: The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids 2004, 70: 309-319. 10.1016/j.plefa.2003.09.007
Williamson DH, Lund P, Krebs HA: The redox state of free nicotinamideadenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J 1967, 103: 514-527.
Veech RL, Eggleston LV, Krebs HA: The redox state of free nicotinamideadenine dinucleotide phosphate in the cytoplasm of rat liver. Biochem J 1969, 115: 609-619.
Bonnefont JP, Specola NB, Vassault A, Lombes A, Ogier H, de Klerk JB, Munnich A, Coude M, Paturneau-Jouas M, Saudubray JM: The fasting test in paediatrics: application to the diagnosis of pathological hypo- and hyperketotic states. Eur J Pediatr 1990, 150: 80-85. 10.1007/BF02072043
Cahill GF, Herrera MG, Morgan AP, Soeldner JS, Steinke J, Levy PL, Reichard GA, Kipnis DM: Hormone-fuel interrelationships during fasting. J Clin Invest 1966, 45: 1751-1769. 10.1172/JCI105481
Owen OE, Felig P, Morgan AP, Wahren J, Cahill GF Jr: Liver and kidney metabolism during prolonged starvation. J Clin Invest 1969, 48: 574-583. 10.1172/JCI106016
Garber AJ, Menzel PH, Boden G, Owen OE: Hepatic ketogenesis and gluconeogenesis in humans. J Clin Invest 1974, 54: 981-989. 10.1172/JCI107839
Williamson DH: Ketone body production and metabolism in the fetus and newborn. In Neonatal Physiology. Edited by: Polin R, Fox W. Philadelphia: W.B. Saunders; 1992:330-340.
Balasse EO, Fery F: Ketone body production and disposal: effects of fasting, diabetes, and exercise. Diabetes Metab Rev 1989, 5: 247-270. 10.1002/dmr.5610050304
Hawkins RA: Uptake of ketone bodies by rat brain in vivo. Biochem J 1971, 121: 17P.
Williamson DH, Bates MW, Page MA, Krebs HA: Activities of enzymes involved in acetoacetate utilization in adult mammalian tissues. Biochem J 1971, 121: 41-47.
Singer M: Mitochondrial function in sepsis: acute phase versus multiple organ failure. Crit Care Med 2007, 35: S441-S448. 10.1097/01.CCM.0000278049.48333.78
Yassen KA, Galley HF, Lee A, Webster NR: Mitochondrial redox state in the critically ill. Br J Anaesth 1999, 83: 325-327.
Limuro Y, Yamamoto M, Inoue S, Kohno H, Matsumoto Y: Superoxide production by liver macrophages in septic shock model - relation to arterial ketone body ratio. Eur Surg Res 1992, 24: 363-371. 10.1159/000129229
Dresing K, Armstrong VW, Leip CL, Streit F, Burchardi H, Sturmer KM, Oellerich M: Real-time assessment of hepatic function is related to clinical outcome in critically ill patients after polytrauma. Clin Biochem 2007, 40: 1194-1200. 10.1016/j.clinbiochem.2007.06.013
Levy B, Sadoune LO, Gelot AM, Bollaert PE, Nabet P, Larcan A: Evolution of lactate/pyruvate and arterial ketone body ratios in the early course of catecholamine-treated septic shock. Crit Care Med 2000, 28: 114-119. 10.1097/00003246-200001000-00019
Pellerin L: How astrocytes feed hungry neurons. Mol Neurobiol 2005, 32: 59-72. 10.1385/MN:32:1:059
Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF Jr: Brain metabolism during fasting. J Clin Invest 1967, 46: 1589-1595. 10.1172/JCI105650
Casazza JP, Felver ME, Veech RL: The metabolism of acetone in rat. J Biol Chem 1984, 259: 231-236.
VanItallie TB, Nufert TH: Ketones: metabolism's ugly duckling. Nutr Rev 2003, 61: 327-341. 10.1301/nr.2003.oct.327-341
Reichard GA, Owen OE, Haff AC, Paul P, Bortz WM: Ketone-body production and oxidation in fasting obese humans. J Clin Invest 1974, 53: 508-515. 10.1172/JCI107584
Guzman M, Blazquez C: Ketone body synthesis in the brain: possible neuroprotective effects. Prostaglandins Leukot Essent Fatty Acids 2004, 70: 287-292. 10.1016/j.plefa.2003.05.001
Auestad N, Korsak RA, Morrow JW, Edmond J: Fatty acid oxidation and ketogenesis by astrocytes in primary culture. J Neurochem 1991, 56: 1376-1386. 10.1111/j.1471-4159.1991.tb11435.x
Bixel MG, Hamprecht B: Generation of ketone bodies from leucine by cultured astroglial cells. J Neurochem 1995, 65: 2450-2461. 10.1046/j.1471-4159.1995.65062450.x
Ladu MJ, Reardon C, Van Eldik L, Fagan AM, Bu G, Holtzman D, Getz GS: Lipoproteins in the central nervous system. Ann N Y Acad Sci 2000, 903: 167-175. 10.1111/j.1749-6632.2000.tb06365.x
Lopes-Cardozo M, Larsson OM, Schousboe A: Acetoacetate and glucose as lipid precursors and energy substrates in primary cultures of astrocytes and neurons from mouse cerebral cortex. J Neurochem 1986, 46: 773-778. 10.1111/j.1471-4159.1986.tb13039.x
Guzman M, Blazquez C: Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol Metab 2001, 12: 169-173. 10.1016/S1043-2760(00)00370-2
Pellerin L, Pellegri G, Martin JL, Magistretti PJ: Expression of monocarboxylate transporter mRNAs in mouse brain: support for a distinct role of lactate as an energy substrate for the neonatal vs. adult brain. Proc Natl Acad Sci USA 1998, 95: 3990-3995. 10.1073/pnas.95.7.3990
Pope A: Neurolgia: quantitative aspects. In Dynamic Properties of Glial Cells: An Interdisciplinary Approach to their Study in the Central and Peripheral Nervous System. Edited by: Schoff eniels E, Tower F. New York: Pergamon Press; 1978:13-20.
Leino RL, Gerhart DZ, Duelli R, Enerson BE, Drewes LR: Diet-induced ketosis increases monocarboxylate transporter (MCT1) levels in rat brain. Neurochem Int 2001, 38: 519-527. 10.1016/S0197-0186(00)00102-9
Hasselbalch SG, Knudsen GM, Jakobsen J, Hageman LP, Holm S, Paulson OB: Brain metabolism during short-term starvation in humans. J Cereb Blood Flow Metab 1994, 14: 125-131.
Pan JW, Rothman TL, Behar KL, Stein DT, Hetherington HP: Human brain beta-hydroxybutyrate and lactate increase in fasting-induced ketosis. J Cereb Blood Flow Metab 2000, 20: 1502-1507. 10.1097/00004647-200010000-00012
Pan JW, Telang FW, Lee JH, de Graaf RA, Rothman DL, Stein DT, Hetherington HP: Measurement of beta-hydroxybutyrate in acute hyperketonemia in human brain. J Neurochem 2001, 79: 539-544. 10.1046/j.1471-4159.2001.00575.x
Kante A, Cherkaoui Malki M, Coquard C, Latruffe N: Metabolic control of the expression of mitochondrial D-beta-hydroxybutyrate dehydrogenase, a ketone body converting enzyme. Biochim Biophys Acta 1990, 1033: 291-297.
Smith AL, Satterthwaite HS, Sokoloff L: Induction of brain D(-)-betahydroxybytrate dehydrogenase activity by fasting. Science 1969, 163: 79-81. 10.1126/science.163.3862.79
Gjedde A, Crone C: Induction processes in blood-brain transfer of ketone Bodies during starvation. Am J Physiol 1975, 229: 1165-1169.
Moore TJ, Lione AP, Sugden MC, Regen DM: Beta-hydroxybutyrate transport in rat brain: developmental and dietary modulations. Am J Physiol 1976, 230: 619-630.
Hawkins RA, Mans AM, Davis DW: Regional ketone body utilization by rat brain in starvation and diabetes. Am J Physiol 1986, 250: E169-E178.
Morris AA: Cerebral ketone body metabolism. J Inherit Metab Dis 2005, 28: 109-121. 10.1007/s10545-005-5518-0
Hawkins RA, Biebuyck JF: Ketone bodies are selectively used by individual brain regions. Science 1979, 205: 325-327. 10.1126/science.451608
Prins ML: Cerebral metabolic adaptation and ketone metabolism after brain injury. J Cereb Blood Flow Metab 2008, 28: 1-16. 10.1038/sj.jcbfm.9600543
Veech RL, Chance B, Kashiwaya Y, Lardy HA, Cahill GF Jr: Ketone bodies, potential therapeutic uses. IUBMB Life 2001, 51: 241-247. 10.1080/152165401753311780
Kashiwaya Y, Sato K, Tsuchiya N, Thomas S, Fell DA, Veech RL, Passonneau JV: Control of glucose utilization in working perfused rat heart. J Biol Chem 1994, 269: 25502-25514.
Krebs HA, Veech RL: Equilibrium relations between pyridine nucleotides and adenine nucleotides and their roles in the regulation of metabolic processes. Adv Enzyme Regul 1969, 7: 397-413. 10.1016/0065-2571(69)90030-2
Ziegler DR, Ribeiro LC, Hagenn M, Siqueira IR, Araujo E, Torres IL, Gottfried C, Netto CA, Goncalves CA: Ketogenic diet increases glutathione peroxidase activity in rat hippocampus. Neurochem Res 2003, 28: 1793-1797. 10.1023/A:1026107405399
Gasior M, Rogawski MA, Hartman AL: Neuroprotective and diseasemodifying effects of the ketogenic diet. Behav Pharmacol 2006, 17: 431-439. 10.1097/00008877-200609000-00009
Yudkoff M, Daikhin Y, Nissim I, Lazarow A: Ketogenic diet, amino acid metabolism, and seizure control. J Neurosci Res 2001, 66: 931-940. 10.1002/jnr.10083
Hasselbalch SG, Madsen PL, Hageman LP, Olsen KS, Justesen N, Holm S, Paulson OB: Changes in cerebral blood flow and carbohydrate metabolism during acute hyperketonemia. Am J Physiol 1996, 270: E746-E751.
Prins ML, Giza CC: Induction of monocarboxylate transporter 2 expression and ketone transport following traumatic brain injury in juvenile and adult rats. Dev Neurosci 2006, 28: 447-456. 10.1159/000094170
Tieu K, Perier C, Caspersen C, Teismann P, Wu DC, Yan SD, Naini A, Vila M, Jackson-Lewis V, Ramasamy R, Przedborski S: D-β-Hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J Clin Invest 2003, 112: 892-901.
Salim A, Hadjizacharia P, Dubose J, Brown C, Inaba K, Chan LS, Margulies D: Persistent hyperglycemia in severe traumatic brain injury: an independent predictor of outcome. Am Surg 2009, 75: 25-29.
Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM: Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 2007, 145: 256-264. 10.1016/j.neuroscience.2006.11.065
Massieu L, Haces ML, Montiel T, Hernandez-Fonseca K: Acetoacetate protects hippocampal neurons against glutamate-mediated neuronal damage during glycolysis inhibition. Neuroscience 2003, 120: 365-378. 10.1016/S0306-4522(03)00266-5
Mejia-Toiber J, Montiel T, Massieu L: D-β-Hydroxybutyrate prevents glutamate-mediated lipoperoxidation and neuronal damage elicited during glycolysis inhibition in vivo. Neurochem Res 2006, 31: 1399-1408. 10.1007/s11064-006-9189-5
Noh HS, Kim YS, Kim YH, Han JY, Park CH, Kang AK, Shin HS, Kang SS, Cho GJ, Choi WS: Ketogenic diet protects the hippocampus from kainic acid toxicity by inhibiting the dissociation of bad from 14-3-3. J Neurosci Res 2006, 84: 1829-1836. 10.1002/jnr.21057
Noh HS, Kim YS, Lee HP, Chung KM, Kim DW, Kang SS, Cho GJ, Choi WS: The protective effect of a ketogenic diet on kainic acid-induced hippocampal cell death in the male ICR mice. Epilepsy Res 2003, 53: 119-128. 10.1016/S0920-1211(02)00262-0
Masuda R, Monahan JW, Kashiwaya Y: D-β-Hydroxybutyrate is neuroprotective against hypoxia in serum-free hippocampal primary cultures. J Neurosci Res 2005, 80: 501-509. 10.1002/jnr.20464
Puchowicz MA, Emancipator DS, Xu K, Magness DL, Ndubuizu OI, Lust WD, LaManna JC: Adaptation to chronic hypoxia during diet-induced ketosis. Adv Exp Med Biol 2005, 566: 51-57. full_text
Hu ZG, Wang HD, Jin W, Yin HX: Ketogenic diet reduces cytochrome c release and cellular apoptosis following traumatic brain injury in juvenile rats. Ann Clin Lab Sci 2009, 39: 76-83.
Hu ZG, Wang HD, Qiao L, Yan W, Tan QF, Yin HX: The protective effect of the ketogenic diet on traumatic brain injury-induced cell death in juvenile rats. Brain Injury 2009, 23: 459-465. 10.1080/02699050902788469
Suzuki M, Suzuki M, Kitamura Y, Mori S, Sato K, Dohi S, Sato T, Matsuura A, Hiraide A: Beta-hydroxybutyrate, a cerebral function improving agent, protects rat brain against ischemic damage caused by permanent and transient focal cerebral ischemia. Jpn J Pharmacol 2002, 89: 36-43. 10.1254/jjp.89.36
Prins ML, Fujima LS, Hovda DA: Age-dependent reduction of cortical contusion volume by ketones after traumatic brain injury. J Neurosci Res 2005, 82: 413-420. 10.1002/jnr.20633
Prins ML, Lee SM, Fujima LS, Hovda DA: Increased cerebral uptake and oxidation of exogenous betaHB improves ATP following traumatic brain injury in adult rats. J Neurochem 2004, 90: 666-672. 10.1111/j.1471-4159.2004.02542.x
Hall ED, Andrus PK, Yonkers PA: Brain hydroxyl radical generation in acute experimental head injury. J Neurochem 1993, 60: 588-594. 10.1111/j.1471-4159.1993.tb03189.x
Biros MH, Nordness R: Eff ects of chemical pretreatment on posttraumatic cortical edema in the rat. Am J Emerg Med 1996, 14: 27-32. 10.1016/S0735-6757(96)90008-X
Go KG, Prenen GH, Korf J: Protective effect of fasting upon cerebralhypoxic-ischemic injury. Metab Brain Dis 1988, 3: 257-263. 10.1007/BF00999535
Marie C, Bralet AM, Gueldry S, Bralet J: Fasting prior to transient cerebral ischemia reduces delayed neuronal necrosis. Metab Brain Dis 1990, 5: 65-75. 10.1007/BF01001047
Suzuki M, Suzuki M, Sato K, Dohi S, Sato T, Matsuura A, Hiraide A: Effect of beta-hydroxybutyrate, a cerebral function improving agent, on cerebral hypoxia, anoxia and ischemia in mice and rats. Jpn J Pharmacol 2001, 87: 143-150. 10.1254/jjp.87.143
Imamura K, Takeshima T, Kashiwaya Y, Nakaso K, Nakashima K: D-β- Hydroxybutyrate protects dopaminergic SH-SY5Y cells in a rotenone model of Parkinson's disease. J Neurosci Res 2006, 84: 1376-1384. 10.1002/jnr.21021
Kashiwaya Y, Takeshima T, Mori N, Nakashima K, Clarke K, Veech RL: D-β- Hydroxybutyrate protects neurons in models of Alzheimer's and Parkinson's disease. Proc Natl Acad Sci USA 2000, 97: 5440-5444. 10.1073/pnas.97.10.5440
Kweon GR, Marks JD, Krencik R, Leung EH, Schumacker PT, Hyland K, Kang UJ: Distinct mechanisms of neurodegeneration induced by chronic complex I inhibition in dopaminergic and non-dopaminergic cells. J Biol Chem 2004, 279: 51783-51792. 10.1074/jbc.M407336200
Van der Auwera I, Wera S, Van Leuven F, Henderson ST: A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer's disease. Nutr Metab (Lond) 2005, 2: 28. 10.1186/1743-7075-2-28
Van Hove JL, Grunewald S, Jaeken J, Demaerel P, Declercq PE, Bourdoux P, Niezen-Koning K, Deanfeld JE, Leonard JV: D, L-3-Hydroxybutyrate treatment of multiple acyl-CoA dehydrogenase defi ciency (MADD). Lancet 2003, 361: 1433-1435. 10.1016/S0140-6736(03)13105-4
Gilbert DL, Pyzik PL, Freeman JM: The ketogenic diet: seizure control correlates better with serum beta-hydroxybutyrate than with urine ketones. J Child Neurol 2000, 15: 787-790. 10.1177/088307380001501203
Smith SL, Heal DJ, Martin KF: KTX 0101: a potential metabolic approach to cytoprotection in major surgery and neurological disorders. CNS Drug Rev 2005, 11: 113-140. 10.1111/j.1527-3458.2005.tb00265.x
Ritter AM, Robertson CS, Goodman JC, Contant CF, Grossman RG: Evaluation of a carbohydrate-free diet for patients with severe head injury. J Neurotrauma 1996, 13: 473-485. 10.1089/neu.1996.13.473
Blomqvist G, Alvarsson M, Grill V, Von Heijne G, Ingvar M, Thorell JO, Stone-Elander S, Widen L, Ekberg K: Effect of acute hyperketonemia on the cerebral uptake of ketone bodies in nondiabetic subjects and IDDM patients. Am J Physiol Endocrinol Metab 2002, 283: E20-E28.
Dashti HM, Mathew TC, Khadada M, Al-Mousawi M, Talib H, Asfar SK, Behbahani AI, Al-Zaid NS: Beneficial effects of ketogenic diet in obese diabetic subjects. Mol Cell Biochem 2007, 302: 249-256. 10.1007/s11010-007-9448-z
Hiraide A, Katayama M, Sugimoto H, Yoshioka T, Sugimoto T: Effect of 3-hydroxybutyrate on posttraumatic metabolism in man. Surgery 1991, 109: 176-181.
Miles JM, Nissen SL, Rizza RA, Gerich JE, Haymond MW: Failure of infused beta-hydroxybutyrate to decrease proteolysis in man. Diabetes 1983, 32: 197-205. 10.2337/diabetes.32.3.197
Owen OE, Reichard GA, Markus H, Boden G, Mozzoli MA, Shuman CR: Rapid intravenous sodium acetoacetate infusion in man. Metabolic and kinetic responses. J Clin Invest 1973, 52: 2606-2616. 10.1172/JCI107453
Reger MA, Henderson ST, Hale C, Cholerton B, Baker LD, Watson GS, Hyde K, Chapman D, Craft S: Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol Aging 2004, 25: 311-314. 10.1016/S0197-4580(03)00087-3
Hasselbalch SG, Knudsen GM, Jakobsen J, Hageman LP, Holm S, Paulson OB: Blood-brain barrier permeability of glucose and ketone bodies during short-term starvation in humans. Am J Physiol 1995, 268: E1161-E1166.
Freeman JM, Vining EP, Pillas DJ, Pyzik PL, Casey JC, Kelly LM: The efficacy of the ketogenic diet - 1998: a prospective evaluation of intervention in 150 children. Pediatrics 1998, 102: 1358-1363. 10.1542/peds.102.6.1358
Kielb S, Koo HP, Bloom DA, Faerber GJ: Nephrolithiasis associated with the ketogenic diet. J Urol 2000, 164: 464-466. 10.1016/S0022-5347(05)67400-9
The Brain Trauma Foundation. The American Association of Neurological Surgeons. The Joint Section on Neurotrauma and Critical Care. Initial management J Neurotrauma 2000, 17: 463-469. 10.1089/neu.2000.17.463
Bhardwaj A, Ulatowski JA: Cerebral edema: hypertonic saline solutions. Curr Treat Options Neurol 1999, 1: 179-188. 10.1007/s11940-999-0002-z
Harukuni I, Kirsch JR, Bhardwaj A: Cerebral resuscitation: role of osmotherapy. J Anesth 2002, 16: 229-237. 10.1007/s005400200030
Dubick MA, Wade CE: A review of the efficacy and safety of 7.5% NaCl/6% dextran 70 in experimental animals and in humans. J Trauma 1994, 36: 323-330. 10.1097/00005373-199403000-00007
Eilig I, Rachinsky M, Artru AA, Alonchin A, Kapuler V, Tarnapolski A, Shapira Y: The effect of treatment with albumin, hetastarch, or hypertonic saline on neurological status and brain edema in a rat model of closed head trauma combined with uncontrolled hemorrhage and concurrent resuscitation in rats. Anesth Analg 2001, 92: 669-675. 10.1097/00000539-200103000-00023
Gunnar W, Jonasson O, Merlotti G, Stone J, Barrett J: Head injury and hemorrhagic shock: studies of the blood brain barrier and intracranial pressure after resuscitation with normal saline solution, 3% saline solution, and dextran-40. Surgery 1988, 103: 398-407.
Shackford SR: Effect of small-volume resuscitation on intracranial pressure and related cerebral variables. J Trauma 1997, 42: S48-S53. 10.1097/00005373-199705001-00009
Berger S, Schurer L, Hartl R, Deisbock T, Dautermann C, Murr R, Messmer K, Baethmann A: 7.2% NaCl/10% dextran 60 versus 20% mannitol for treatment of intracranial hypertension. Acta Neurochir Suppl (Wien) 1994, 60: 494-498.
Mirski AM, Denchev ID, Schnitzer SM, Hanley FD: Comparison between hypertonic saline and mannitol in the reduction of elevated intracranial pressure in a rodent model of acute cerebral injury. J Neurosurg Anesthesiol 2000, 12: 334-344. 10.1097/00008506-200010000-00006
Cooper DJ, Myles PS, McDermott FT, Murray LJ, Laidlaw J, Cooper G, Tremayne AB, Bernard SS, Ponsford J: Prehospital hypertonic saline resuscitation of patients with hypotension and severe traumatic brain injury: a randomized controlled trial. JAMA 2004, 291: 1350-1357. 10.1001/jama.291.11.1350
Vassar MJ, Fischer RP, O'Brien PE, Bachulis BL, Chambers JA, Hoyt DB, Holcroft JW: A multicenter trial for resuscitation of injured patients with 7.5% sodium chloride. The effect of added dextran 70. The Multicenter Group for the Study of Hypertonic Saline in Trauma Patients. Arch Surg 1993, 128: 1003-1011. discussion 1011-1013
Vassar MJ, Perry CA, Holcroft JW: Prehospital resuscitation of hypotensive trauma patients with 7.5% NaCl versus 7.5% NaCl with added dextran: a controlled trial. J Trauma 1993, 34: 622-632. discussion 632-633 10.1097/00005373-199305000-00003
Qureshi AI, Suarez JI, Bhardwaj A: Malignant cerebral edema in patients with hypertensive intracerebral hemorrhage associated with hypertonic saline infusion: a rebound phenomenon? J Neurosurg Anesthesiol 1998, 10: 188-192. 10.1097/00008506-199807000-00010
Qureshi AI, Suarez JI, Bhardwaj A, Mirski M, Schnitzer MS, Hanley DF, Ulatowski JA: Use of hypertonic (3%) saline/acetate infusion in the treatment of cerebral edema: effect on intracranial pressure and lateral displacement of the brain. Crit Care Med 1998, 26: 440-446. 10.1097/00003246-199803000-00011
Chan L, Slater J, Hasbargen J, Herndon DN, Veech RL, Wolf S: Neurocardiac toxicity of racemic D, L-lactate fluids. Integr Physiol Behav Sci 1994, 29: 383-394. 10.1007/BF02691358
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
White, H., Venkatesh, B. Clinical review: Ketones and brain injury. Crit Care 15, 219 (2011). https://doi.org/10.1186/cc10020
Published:
DOI: https://doi.org/10.1186/cc10020