
Abstract
STAT transcription factors were discovered 10 years ago as mediators of interferon-induced gene expression. They now form an important group, comprising seven members, that are activated by virtually every cytokine and growth factor. Their critical role in development and normal cell signaling has been largely determined through the analysis of transgenic mice lacking individual STAT genes. In addition, cell culture work has further delineated their importance in cellular transformation, apoptosis, differentiation and growth control. This review discusses the specific phenotypes of STAT-deficient animals with a focus on STAT5 and STAT3, as these two STAT molecules are required for normal breast development and involution, respectively, and may play an important role in breast carcinogenesis.
Similar content being viewed by others

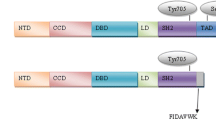

Introduction
Signal transducers and activators of transcription (STAT) proteins are latent transcription factors that become activated by phosphorylation on a single tyrosine (near the carboxy-terminal of the molecule), typically in response to extracellular ligands [1,2]. Virtually every cytokine and growth factor (polypeptide ligands) can cause STAT phosphorylation through either cytokine receptors plus associated Jak kinases or growth factors [eg epidermal growth factor (EGF), platelet-derived growth factor (PDGF), colony-stimulating factor-1] acting through intrinsic receptor tyrosine kinases. This clearly implicates STAT activation in many different biologic events. An active STAT dimer is formed via the reciprocal interactions between the Src homology domain-2 of one monomer and the phosphorylated tyrosine of the other [3]. The dimers accumulate in the nucleus, recognize specific DNA elements, and activate transcription. The STAT proteins are subsequently inactivated by tyrosine dephosphorylation and return to the cytoplasm [4,5].
The relevance of STAT activation to growth control is made apparent in experiments performed in cell lines or animals that lack specific STAT proteins, by the use of antisense molecules to specific STATs or by the use of dominant-negative STAT protein encoding constructs. Of the seven mammalian STATs: STAT2, STAT4, and STAT6 are activated by a small defined group of cytokines; and deficiencies in these STATs, as determined by murine knockout experiments, have been used to demonstrate their restricted roles in interferon-γ signaling and in T cell development (Schindler C, personal communication) [6,7,8,9]. In contrast, STAT1, STAT3, STAT5A, and STAT5B are activated by a large number of different ligands and frequently simultaneously by the same ligand, which raises the question regarding how these STATs participate in particular biologic responses (Table 1).
STAT1 promotes growth inhibition and apoptosis
Interferon-γ activates STAT1 almost exclusively, and mice that lack STAT1 have no innate response to either viral or bacterial pathogens because defense against these pathogens usually requires response to interferon [10,11]. Interestingly, over time STAT1-deficient mice develop spontaneous and chemically induced tumors more readily than wild-type animals, thus defining STAT1 as a 'tumor suppressor' (Schreiber B, personal communication) [12]. The mechanisms responsible for tumor suppression are not completely understood, but are in part due to the lack of tumor surveillance. Work done in tissue culture systems on STAT1-deficient cells [13,14] has demonstrated an important role for STAT1 in ligand-mediated growth arrest and apoptosis. Thus, there is growing evidence that STAT1 activation frequently leads to antiproliferative and proapoptotic events, and may partly explain why the lack of this molecule in vivo leads to increased tumor formation.
STAT5 promotes cell proliferation and is required for mammopoeisis
Two STAT5 genes exist, which are 96% homologous: Stat5A and Stat5B. Both STAT5 molecules are ubiquitously expressed and can be activated by many cytokines (see Table 1), including interleukins, growth hormone, prolactin, EGF, and erythropoietin. In addition, a number of oncogenes such as those that encode Break Point Cluster Region/Abelson (BCR/ABL) tyrosine kinase and human T-cell leukemia virus (HTLV)-1, lead to persistent tyrosine phosphorylation of STAT5 [15,16]. In hematopoietic cells, STAT5 has been demonstrated to play a critical role in regulating apoptosis [17,18,19,20], and in promoting proliferation and cell cycle progression [21,22].
Mammary gland development is defined by the formation of ductal epithelial cells, which requires estrogen and EGF, and of lobular alveolar epithelium, which proliferates in response to prolactin and progesterone [23,24]. STAT5A, which can be activated by EGF and prolactin, is required for mammopoiesis and lactogenesis, as determined by knockout experiments [17,25].
The phosphorylated or activated form of STAT5 is quite low in mammary tissue of virgin animals and during early pregnancy, but rises dramatically after day 14 of pregnancy [26]. Upon weaning the breast involutes, which coincides with a drop in the levels of activated STAT5 [26]. Differentiation of ductal elements occurs in both wild-type and knockout mice. However, the extent or amount of epithelial duct development is decreased in STAT5 knockout animals [17]. Because both prolactin and EGF is required for ductal and alveolar growth, the absolute deficiency of STAT5 (despite the almost nonexistent levels of activated STAT5 in the virgin breast) may reflect the reduced growth or number of epithelial cells. The mammary tissue of STAT5A-deficient animals has a significant paucity of STAT5B protein [25]. STAT5B-deficient animals also have defects in mammopoiesis and lactogenesis (although not nearly as severe as in the STAT5A-deficient animals) [17,27].
In addition to STAT5, EGF, estrogen, and progesterone are factors that are critical to the development and proliferation of mammary tissue. The temporal and physical interaction between STAT5 and these other critical factors has not been well described. However, erbB-2, which can form a heterodimer with other erbB family members, may be essential for morphogenesis of the mammary ducts [28], and erbB-2 as a heterodimer with erB-4 can result in the activation of STAT5 [29]. The progesterone receptor can synergize with activated STAT5 in the induction of transcription from the β-casein gene promoter [30]. These examples suggest that there may be an interplay between these critical molecules and pathways in mammary development.
STAT3 as a regulator of cellular transformation and apoptosis in breast development
STAT3 deficiency leads to early embryonic lethality [31]. STAT3, like STAT5, is ubiquitously expressed in most tissues and is activated by a large number of different ligands, including EGF, PDGF, interleukin-6, ciliary neurotrophic factor, oncostatin M, leukemia inhibitory factor, and granulocyte-macrophage colony-stimulating factor (see Table 1), as well as by a number of oncogenic tyrosine kinases. In normal cells and in animals, ligand-dependent activation of the STATs is a transient process, lasting for several minutes to several hours. In contrast, in many cancerous cell lines and tumors, where growth factor dysregulation is frequently at the heart of cellular transformation, STAT3 protein is persistently tyrosine phosphorylated or activated [32,33]. In studies performed in rodent fibroblasts, STAT3 activation has been demonstrated to be both required and sufficient to mediate cellular transformation. For example, v-src transformation requires activated STAT3, and a constitutively active form of STAT3 (STAT3-C) is sufficient for mediating transformation [34,35,36]. STAT3 activation appears to play an important role in preventing apoptosis in multiple myeloma-derived cell lines [37], squamous cell carcinomas [38], neurons [39,40], and mycosis fungoides tumor cells [41], and mediates cell cycle progression in a pro-B cell line [42].
The standard approach for circumventing embryonic lethality of gene knockouts is the creation of conditional knockouts, whereby only specific tissues lack the gene (protein) of interest [43]. Given that STAT3 deficiency results in death of the embryo, a number of STAT3-conditional knockouts have been generated, demonstrating the importance of STAT3 in preventing interleukin-6-mediated apoptosis of T cells [44], and keratinocytes that lack STAT3 do not migrate properly [45]. These findings, both in vitro and in vivo, suggest that STAT3 activation can prevent apoptosis and promotes proliferative processes, including cellular transformation.
The normal activation pattern of STAT3 in breast development and mammopoiesis reveals prominent levels of activated STAT3 in the virgin breast (presumably in response to EGF), which decrease somewhat during pregnancy and lactation, and subsequently increase during involution [26]. The marked increase in phosphorylated STAT3 during involution coincides with the initial burst of apoptosis.
A recent paper by Chapman et al [46] revealed a requirement for activated STAT3 in the involuting (apoptosing) breast. By crossing a β-lactoglobulin (BLG)-Cre mouse with a STAT3-null/STAT3-flox mouse, mice were generated that had a decrease in the levels of STAT3 in the lactating and involuting breast. The phenotype of the STAT3-deficient mammary gland revealed a delay in involution and apoptosis, suggesting a proapoptotic role for STAT3 in the involuting breast. At the molecular level a concomitant increase in the levels of STAT5 and STAT1 and an increase in the 'proapoptotic' proteins p21, p53 and Bax is observed in the BLG-Cre/Stat3flox/- breast tissue relative to the BLG-Cre/Stat3flox/+ tissue. Given the wealth of evidence demonstrating a proliferative and antiapoptotic role for STAT3 in some cell types, the findings of Chapman et al are somewhat surprising. Nevertheless, there are a few notable examples in which STAT3 activation is clearly responsible for a growth arrest in cultured cells. The first is in a myeloid tumor-derived cell line, M1, in which interleukin-6-mediated STAT3 activation is clearly responsible for inducing G1 arrest of these cells [47,48]. The second is in a macrophage-derived cell line, J774, in which interleukin-10, which activates STAT3, leads to antiproliferation, and expression of an inducibly active STAT3-gyraseB chimera mimics the effects of interleukin-10 [49].
The precise mechanism(s) by which STAT3 activation in the involuting breast leads to apoptosis are not known. Why activated STAT3 has this paradoxical effect as a function of cell type is presumably related to differences in the regulation of STAT3 target genes. For example, activated STAT3 can upregulate expression of the c-myc proto-oncogene [35,50]. It is well known that c-myc dysregulation can lead to both cellular transformation and apoptosis, depending on the cell type and the growth conditions [51]. Regardless of the mechanism, it is clear that STAT3 and STAT5 activation play critical roles in breast tissue development, probably through their ability to influence growth, differentiation, and apoptosis.
The relationship between the molecules responsible for organogenesis and cancer is not known. However, we believe that, in general, normal proteins and signaling pathways become abnormally regulated, which leads to the production of cancer. For example, in the case of breast cancer dysregulation of erbB-2, transforming growth factor-α, and estrogen is associated with the formation of breast cancer in human cells and murine models. STAT3 and STAT5 are clearly involved in breast organogenesis, and there is increasing evidence that the STAT proteins (in particular STAT3, STAT5 and STAT1) are persistently activated in primary breast cancers and breast cancer-derived cell lines (Bromberg J, et al, and Jove R, et al, unpublished observations) [32,52]. It is the persistent nature of STAT activation that is abnormal and is perhaps sufficient to mediate transformation. The potential mechanism(s) responsible for STAT tyrosine phosphorylation in primary breast cancer specimens are many and include the overproduction of the EGF receptor and EGF [38], PDGF receptor, Jak kinases, and c-src [32,53] (Reddy EP, personal communication), and perhaps mutations within the STAT protein itself. In addition, defects in the negative regulators of STAT phosphorylation may also be responsible for the persistence of tyrosine phosphorylated STAT3 in these specimens. These negative regulators include tyrosine phosphatases (ie SHP-2); the suppressor of cytokine signaling (SOC) proteins, which are negative regulators of the Jak kinases [54]; and the protein inhibitor of activated STATs (PIAS) [55], which inhibit activated STATs from binding DNA.
Interestingly, a number of STAT3 and STAT5 target genes (cyclinD1, c-myc [35,50,56] and Bcl-x L [18,35]) are implicated either in breast cancer progression or in prognosis. Overexpression of cyclinD1 in transgenic models leads to hyperproliferation of breast tissue, as well as overt carcinoma [57]. CyclinD1 is overexpressed in preinvasive breast cancer, such as ductal carcinoma in situ, and in invasive breast carcinomas [58]. It has also been suggested that overexpression of cyclin D1 protein is important in the detection of early stages of breast oncogenesis, and may play a crucial role in the development of breast cancer malignancy [58,59,60,61]. C-myc, another Stat3 target gene, when overexpressed correlates with highly proliferative breast tumors [62,63,64]. Bcl-xL, a member of the antiapoptotic bcl-2 family of proteins, may be associated with a prognostically favorable phenotype in breast cancer [65,66,67]. Now, with the development of mice that lack the specific STAT proteins, we can begin to dissect the specific roles of the STATs in breast cancer pathogenesis.
References
Darnell JE: STATs and gene regulation. Science. 1997, 277: 1630-1635. 10.1126/science.277.5332.1630.
Stark GR, Kerrr IM, Williams BR, et al: How cells respond to interferons. Annu Rev Biochem. 1998, 67: 227-264. 10.1146/annurev.biochem.67.1.227.
Chen X, Vinkemeier U, Zhao Y, et al: Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998, 93: 827-839. 10.1016/S0092-8674(00)81443-9.
Haspel RL, Salditt-Georgieff M, Darnell JE: The rapid inactivation of nuclear tyrosine phosphorylated Stat1 depends on a protein tyrosine phosphatase. EMBO J. 1996, 15: 6262-6268.
Haspel RL, Darnell JE: A nuclear protein tyrosine phosphatase is required for the inactivation of Stat1. Proc Natl Acad Sci USA. 1999, 96: 10188-10193. 10.1073/pnas.96.18.10188.
Kaplan MH, Schindler U, Smiley ST, et al: Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996, 4: 313-319.
Thierfelder WE, van Deursen JM, Yamamoto K, et al: Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. 1996, 382: 171-174. 10.1038/382171a0.
Takeda K, Tanaka T, Shi W, et al: Essential role of Stat6 in IL-4 signalling. Nature. 1996, 380: 627-630. 10.1038/380627a0.
Shimoda K, Deursen JV, Sangster MY, et al: Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 1996, 380: 630-633. 10.1038/380630a0.
Meraz MA, White JM, Sheehan KCF, et al: Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996, 84: 431-442. 10.1016/S0092-8674(00)81288-X.
Durbin JE, Hackenmiller R, Simon MC, et al: Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996, 84: 443-450. 10.1016/S0092-8674(00)81289-1.
Kaplan DH, Shankaran V, Dighe AS, et al: Demonstration of an interferon gamma-dependent tumor surveillance system in immuno-competent mice. Proc Natl Acad Sci USA. 1998, 95: 7556-7561. 10.1073/pnas.95.13.7556.
Bromberg JF, Horvath CM, Wen Z, et al: Transcriptionally active Stat1 is required for the antiproliferative effects of both IFN-α and IFN-γ. Proc Natl Acad Sci USA. 1996, 93: 7673-7678. 10.1073/pnas.93.15.7673.
Kumar A, Commane M, Flickinger TW, et al: Defective TNF-alpha-induced apoptosis in STAT1-null cells due to low constitutive levels of caspases. Science. 1997, 278: 1630-1632. 10.1126/science.278.5343.1630.
Shuai K, Halpern J, ten Hoeve J, et al: Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene. 1996, 13: 247-254.
Migone TS, Lin JX, Cereseto A, et al: Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science. 1995, 269: 79-81.
Teglund S, McKay C, Schuetz E, et al: Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998, 93: 841-850. 10.1016/S0092-8674(00)81444-0.
Socolovsky M, Fallon AE, Wang S, et al: Fetal anemia and apoptosis of red cell progenitors in Stat5a-/-5b-/- mice: a direct role for Stat5 in Bcl-X(L) induction. Cell. 1999, 98: 181-191.
Onishi M, Nosaka T, Misawa K, et al: Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol Cell Biol. 1998, 18: 3871-3879.
Nosaka T, Kawashima T, Misawa K, et al: STAT5 as a molecular regulator of proliferation, differentiation and apoptosis in hematopoietic cells. EMBO J. 1999, 18: 4754-4765. 10.1093/emboj/18.17.4754.
Moriggl R, Topham DJ, Teglund S, et al: Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity. 1999, 10: 249-259.
Nieborowska-Skorska M, Wasik M, Slupianek A, et al: Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL is required for leukemogenesis. J Exp Med. 1999, 189: 1229-1242. 10.1084/jem.189.8.1229.
Topper YJ, Freeman CS: Multiple hormone interactions in the developmental biology of the mammary gland. Physiol Rev. 1980, 60: 1049-1056.
Vonderhaar BK: Local effects of EGF, alpha-TGF, and EGF-like growth factors on lobuloalveolar development of the mouse mammary gland in vivo. J Cell Physiol. 1987, 132: 581-584.
Liu X, Robinson GW, Wagner K-U, et al: Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev. 1997, 11: 179-186.
Liu X, Robinson GW, Hennighausen L: Activation of Stat5a and Stat5b by tyrosine phosphorylation is tightly linked to mammary gland differentiation. Mol Endocrinol. 1996, 10: 1496-1506. 10.1210/me.10.12.1496.
Udy GB, Towers RP, Snell RG, et al: Requirement of Stat5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci USA. 1997, 94: 7239-7244. 10.1073/pnas.94.14.7239.
Sebastian J, Richards RG, Walker MP, et al: Activation and function of the epidermal growth factor receptor and erbB-2 during mammary gland morphogenesis. Cell Growth Differ. 1998, 9: 777-785.
Olayioye MA, Beuvink I, Horsch K, et al: ErbB receptor-induced activation of stat transcription factors is mediated by src tyrosine kinases. J Biol Chem. 1999, 274: 17209-17218. 10.1074/jbc.274.24.17209.
Stoecklin E, Wissler M, Schaetzle D, et al: Interactions in the transcriptional regulation exerted by Stat5 and by members of the steroid hormone receptor family. J Steroid Biochem Mol Biol. 1999, 69: 195-204. 10.1016/S0960-0760(99)00052-7.
Takeda K, Noguchi K, Shi W, et al: Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci USA. 1997, 94: 3801-3804. 10.1073/pnas.94.8.3801.
Garcia R, Jove R: Activation of STAT transcription factors in oncogenic tyrosine kinase signaling. J Biomed Sci. 1998, 5: 79-85. 10.1159/000025316.
Weber-Nordt RM, Egen C, Wehinger J, et al: Constitutive activation of STAT proteins in primary lymphoid and myeloid leukemia cells and in Epstein-Barr virus (EBV)-related lymphoma cell lines. Blood. 1996, 88: 809-816.
Bromberg JF, Horvath CM, Besser D, et al: Stat3 activation is required for cellular transformation by v-src . Mol Cell Biol. 1998, 5: 2553-2558.
Bromberg J, Wrzeszczynska M, Devgan G, et al: Stat3 as an Oncogene. Cell. 1999, 98: 295-303.
Turkson J, Bowman T, Garcia R, et al: Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol Cell Biol. 1998, 18: 2545-2552.
Catlett-Falcone R, Landowski TH, Oshiro MM, et al: Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999, 10: 105-115.
Grandis JR, Drenning SD, Chakraborty A, et al: Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor-mediated cell growth in vitro. J Clin Invest. 1998, 102: 1385-1392.
Bonni A, Sun Y, Nadal-Vicens M, et al: Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997, 278: 477-483. 10.1126/science.278.5337.477.
Ihara S, Nakajima K, Fukada T, et al: Dual control of neurite outgrowth by STAT3 and MAP kinase in PC12 cells stimulated with interleukin-6. EMBO J. 1997, 16: 5345-5352. 10.1093/emboj/16.17.5345.
Nielsen M, Kaestel CG, Eriksen KW, et al: Inhibition of constitutively activated Stat3 correlates with altered Bcl-2/Bax expression and induction of apoptosis in mycosis fungoides tumor cells. Leukemia. 1999, 13: 735-738. 10.1038/sj/leu/2401415.
Fukada T, Ohtani T, Yoshida Y, et al: STAT3 orchestrates contradictory signals in cytokine-induced G1 to S cell-cycle transition. EMBO J. 1998, 17: 6670-6677. 10.1093/emboj/17.22.6670.
Kuhn R, Schwenk F, Aguet M, et al: Inducible gene targeting in mice. Science. 1995, 269: 1427-1429.
Takeda K, Kaisho T, Yoshida N, et al: Stat3 activation is responsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell- specific Stat3-deficient mice. J Immunol. 1998, 161: 4652-4660.
Sano S, Itami S, Takeda K, et al: Keratinocyte-specific ablation of stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. EMBO J. 1999, 18: 4657-4668. 10.1093/emboj/18.17.4657.
Chapman RS, Lourenco PC, Tonner E, et al: Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of STAT3. Genes Dev. 1999, 13: 2604-2616. 10.1101/gad.13.19.2604.
Minami M, Inoue M, Wei S, et al: STAT3 activation is a critical step in gp130-mediated terminal differentiation and growth arrest of a myeloid cell line. Proc Natl Acad Sci USA. 1996, 93: 3963-3966. 10.1073/pnas.93.9.3963.
Nakajima K, Yamanaka Y, Nakae K, et al: A centrol role of Stat3 in IL6 induced regulation of growth and differentiation in M1 leukemia cells. EMBO J. 1996, 15: 3651-3658.
O'Farrell AM, Liu Y, Moore KW, et al: IL-10 inhibits macrophage activation and proliferation by distinct signaling mechanisms: evidence for Stat3-dependent and -independent pathways. EMBO J. 1998, 17: 1006-1018. 10.1093/emboj/17.4.1006.
Kiuchi N, Nakajima K, Ichiba M, et al: STAT3 is required for the gp130-mediated full activation of the c-myc gene. J Exp Med. 1999, 189: 63-73. 10.1084/jem.189.1.63.
Cole MD, McMahon SB: The Myc oncoprotein: a critical evaluation of transactivation and target gene regulation. Oncogene. 1999, 18: 2916-2924. 10.1038/sj.onc.1202748.
Watson CJ, Miller WR: Elevated levels of members of the STAT family of transcription factors in breast carcinoma nuclear extracts. Br J Cancer. 1995, 71: 840-844.
Garcia R, Yu CL, Hudnall A, et al: Constitutive activation of Stat3 in fibroblasts transformed by diverse oncoproteins and in breast carcinoma cells. Cell Growth Differ. 1997, 8: 1267-1276.
Naka T, Narazaki M, Hirata M, et al: Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997, 387: 924-929. 10.1038/43219.
Chung CD, Liao J, Liu B, et al: Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997, 278: 1803-1805. 10.1126/science.278.5344.1803.
Matsumura I, Kitamura T, Wakao H, et al: Transcriptional regulation of the cyclin D1 promoter by STAT5: its involvement in cytokine-dependent growth of hematopoietic cells. EMBO J. 1999, 18: 1367-1377. 10.1093/emboj/18.5.1367.
Wang TC, Cardiff RD, Zukerberg L, et al: Mammary hyperplasia and carcinoma in MMTV-cyclin D1 transgenic mice. Nature. 1994, 369: 669-671. 10.1038/369669a0.
Weinstat-Saslow D, Merino MJ, Manrow RE, et al: Overexpression of cyclin D mRNA distinguishes invasive and in situ breast carcinomas from non-malignant lesions. Nature Med. 1995, 1: 1257-1260.
Norton L, Rosen PP, Rosen N: Refining the origins of breast cancer. Nature Med. 1995, 1: 1250-1251.
Gillett CE, Lee AH, Millis RR, et al: Cyclin D1 and associated proteins in mammary ductal carcinoma in situ and atypical ductal hyperplasia. J Pathol. 1998, 184: 396-400. 10.1002/(SICI)1096-9896(199804)184:4<396::AID-PATH1259>3.0.CO;2-G.
Alle KM, Henshall SM, Field AS, et al: Cyclin D1 protein is overexpressed in hyperplasia and intraductal carcinoma of the breast. Clin Cancer Res. 1998, 4: 847-854.
Berns EM, Klijn JG, van Putten WL, et al: c-myc amplification is a better prognostic factor than HER2/neu amplification in primary breast cancer. Cancer Res. 1992, 52: 1107-1113.
Nass SJ, Dickson RB: Epidermal growth factor-dependent cell cycle progression is altered in mammary epithelial cells that overexpress c-myc . Clin Cancer Res. 1998, 4: 1813-1822.
Guerin M, Barrois M, Terrier MJ, et al: Overexpression of either c-myc or c-erbB-2/neu proto-oncogenes in human breast carcinomas: correlation with poor prognosis. Oncogene Res. 1988, 3: 21-31.
Nakopoulou L, Michalopoulou A, Giannopoulou I, et al: bcl-2 protein expression is associated with a prognostically favourable phenotype in breast cancer irrespective of p53 immunostaining. Histopathology. 1999, 34: 310-319. 10.1046/j.1365-2559.1999.00627.x.
Liu JR, Fletcher B, Page C, et al: Bcl-xL is expressed in ovarian carcinoma and modulates chemotherapy-induced apoptosis. Gynecol Oncol. 1998, 70: 398-403. 10.1006/gyno.1998.5125.
Vakkala M, Lahteenmaki K, Raunio H, et al: Apoptosis during breast carcinoma progression. Clin Cancer Res. 1999, 5: 319-324.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bromberg, J. Signal transducers and activators of transcription as regulators of growth, apoptosis and breast development. Breast Cancer Res 2, 86 (2000). https://doi.org/10.1186/bcr38
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/bcr38