
Abstract
Large-scale collaborative initiatives using next-generation DNA sequencing and other high-throughput technologies have begun to characterize the genomic landscape of breast cancer. These landmark studies have identified infrequent driver mutations that are potential targets for therapeutic intervention with approved or investigational drug treatments, among other important discoveries. Recently, many institutions have launched molecular screening programs that apply high-throughput genomic technologies to patients with advanced solid malignancies, including breast cancer, to inform clinical decision-making. This article provides an overview of the recent molecular insights in breast cancer, including potentially actionable somatic alterations, the technological platforms currently available in a clinical diagnostics setting to detect these alterations, and ongoing institutional or regional molecular screening programs in advanced breast cancer.
Similar content being viewed by others


Introduction
The development of next-generation DNA sequencing (NGS) technology has produced an explosion of research data about point mutations and structural genomic alterations in a wide variety of cancers. The complete genomes of breast cancers and other solid tumors have recently been published [1]. These large-scale initiatives have identified rare genomic alterations that are potential therapeutic targets to guide individualized treatment. The development of personalized medicine has been buoyed by the clinical success of several molecular targeted agents linked to predictive biomarkers such as erlotinib or gefitinib in EGFR mutant non-small cell lung cancer (NSCLC), vemurafenib in BRAF V600E mutant melanoma, and crizotinib in EML4-ALK translocated NSCLC. Sequencing clinical tumor specimens to identify potentially 'druggable' somatic tumor DNA alterations is an emerging paradigm and its application in breast cancer is the focus of this review. Although other high-throughput technologies that quantify RNA expression, such as reverse transcriptase-polymerase chain reaction (PCR) and microarrays, have provided seminal insights into the molecular classification of breast cancer, they have been reviewed previously and will not be discussed [2].
Despite advances in our understanding of the landscape of genomic alterations in breast cancer, molecular diagnostics testing to inform clinical decision-making in breast cancer and other solid malignancies has lagged behind. Few targeted drugs are approved for breast cancer treatment on the basis of a predictive biomarker. Currently, estrogen receptor (ER) expression testing by immunohistochemistry (IHC) and human epidermal growth factor receptor 2 (HER2) overexpression by IHC or gene amplification detected by in situ hybridization are the only routinely used biomarkers to select for molecular targeted treatments. Gene expression signatures, such as Oncotype DX (Genomic Health, Inc., Redwood City, CA, USA) and MammaPrint (Agendia Inc., Irvine, CA, USA), are used to identify patients who have early-stage, ER+ breast cancers that should be treated with adjuvant chemotherapy. ER+ tumors are treated with tamoxifen, aromatase inhibitors (AIs), or other endocrine therapies, whereas HER2+ tumors are treated with trastuzumab, lapatinib, pertuzumab, trastuzumab-DM1, and other HER2-targeted therapies. Testing for certain somatic point mutations, such as KRAS in colorectal cancer (CRC) and BRAF in melanoma, is regularly performed in clinically accredited laboratories. The limitation of this 'single gene, single test' approach, however, is that it fails to identify other potentially relevant aberrations that may impact on clinical decisions [3].
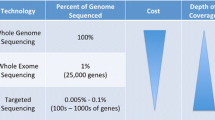
NGS technology refers to methods beyond automated Sanger sequencing that use different techniques to parallelize assays in order to rapidly process thousands to millions of short-read DNA sequences concurrently [4]. These high-throughput, multiplexed assays can identify changes in DNA sequence, gene copy number, structure, or expression. This allows the detection of genetic alterations such as sequence changes in DNA which result from nucleic acid substitutions and insertions or deletions (indels) due to somatic (non-inherited) or germline (inherited) mutations; inherited single-nucleotide polymorphisms (SNPs); structural changes from chromosomal translocations; and copy number variations that involve deleted or amplified DNA segments or genes [5]. These advances have made sequencing entire genomes, exomes (exons in the genome), and transcriptomes (expressed genes) viable. High-throughput technologies are an attractive approach for clinical diagnostic testing because they consolidate single-gene tests and allow deep characterization of targeted regions of the genome enriched in cancer genes that are frequently altered at an affordable cost and in an efficient time frame [6]. This raises the prospect of integrating such technologies at the point of clinical care and addresses the limitations of the 'single gene, single test' paradigm.
Beyond ER and HER2 directed therapies, a variety of promising investigational agents under clinical development are likely to be effective for small subsets of patients whose tumors harbor rare driver mutations. Targeting these genetic alterations is a rational strategy as cells harboring driver mutations may have a survival advantage [7, 8]. The ability to perform multiplexed testing for a range of recurrent molecular alterations provides an opportunity to identify patients with rare driver aberrations who may be candidates for clinical trials with matched targeted therapies [9]. Genomic characterization of tumors with high-throughput technologies is an enrichment strategy that may improve the likelihood of success for testing of new cancer drugs [10, 11].
The true merits of high-throughput genomic screening to promote personalized cancer medicine remain to be established. The traditional paradigm of new drug testing through phase I, II, and III clinical trials is a long and costly process that is further complicated by restricting eligibility to cancers with rare molecular alterations. However, proof-of-concept clinical trials that confirm the value of matching investigational targeted treatments to rare molecular alterations in breast cancer and other malignancies are beginning to emerge [12]. Results from a phase I trial of an alpha-specific PI3-kinase inhibitor, BYL719, in patients with PIK3CA alterations have been presented [13]. Lucitanib (E-3810) is a fibroblast growth factor receptor (FGFR) and vascular endothelial growth factor receptor tyrosine kinase inhibitor that has produced durable responses in patients with FGFR1-amplified tumors, including metastatic breast cancers [14]. Sequential testing of infrequent genomic alterations to identify candidates for clinical trials with matched targeted agents is inefficient, expensive, and wasteful of scarce archived tumor tissue resources [15]. Comprehensive molecular screening programs that provide simultaneous testing of multiple biomarkers early in the course of a patient's disease with access to a broad portfolio of investigational or approved targeted drugs matched to molecular alterations are most likely to advance personalized cancer medicine.
Molecular landscape of breast cancer
Large-scale comprehensive sequencing of breast cancer genomes through initiatives such as The Cancer Genome Atlas (TCGA), the International Cancer Genome Consortium, and the Molecular Taxonomy of Breast Cancer International Consortium [16] reveals the molecular complexity of breast cancer. These studies have several important clinical implications. TP53 (37%) and PIK3CA (36%) are the most frequently mutated genes in breast cancer [1]. Many other genes demonstrate recurrent mutations at lower frequencies (between 1% and 8%), reflecting the broad molecular heterogeneity of breast cancer [1, 17–20]. Certain low-frequency mutations are linked to therapeutic response and others may serve as potential drug targets. Pathways that were thought to be important, such as the PI3-kinase signaling pathway, have been confirmed by frequent mutations or amplifications in its various components (PIK3CA, AKT1, AKT2, AKT3, and INPPB4), especially in ER+ tumors. However, the clinical relevance of particular alterations remains to be established, as demonstrated by the association of PIK3CA mutations with favorable biological characteristics and improved clinical outcome in early-stage ER+ breast cancer [21]. In addition to pathways known to be important in the biology of breast cancer, other novel pathways have been implicated in breast tumors analyzed in large-scale genome projects, such as the stress-induced JUN-kinase pathway via loss-of-function mutations in MAP3K1 and MAP2K4 that may predict sensitivity to chemotherapy in ER+ tumors [1, 19].
Recently, the TCGA project performed exome sequencing for somatic mutations in tumor and matched normal breast tissue in addition to annotating germline variants in a select number of loci with a known predisposition for breast cancer [1]. This project included genome copy number analysis; microRNA, gene, and protein expression by sequencing, mRNA arrays, and reverse-phase protein arrays, in addition to DNA methylation analysis of 507 breast tumors [1]. Distinct mutation patterns were observed in the intrinsic molecular subtypes defined by the prediction analysis of microarrays-50 mRNA expression-based classifier (luminal A and B, HER2-enriched, and basal-like) [22]. TP53 aberrations were observed more frequently in the predominantly ER-HER2-enriched (72%) and basal-like (80%) subtypes, whereas mutually exclusive mutations in MAP3K1, MAP2K4, and GATA3 were seen predominantly in ER+ luminal A and luminal B breast cancers. In silico analysis with the Mutational Significance in Cancer framework discovered 35 genes that were mutated at rates significantly above the background mutation rate, suggesting a pathological role for these implicated genes.
Stephens and colleagues [17] identified nine new cancer genes after exome sequencing of 100 breast tumors, and mutations in seven of these genes were predicted to result in truncated proteins, implying that wild-type versions of these proteins have a tumor-suppressor function. Mutations in tumor-suppressor genes are generally distributed across the gene and do not cluster in hotspots, complicating detection of these aberrations in a clinical setting. The accumulation of certain DNA base substitutions (cytosine at cytosine-thymine dinucleotides) was noted to correlate with age, histological grade, and ER- tumors and thus could be used as an identifying signature [17]. In 104 triple-negative breast cancers, Shah and colleagues [18] used deep re-sequencing technologies and RNA sequencing to demonstrate inter- and intra-tumor variability in somatic mutations and copy number aberrations. Few of the identified mutations were potentially 'druggable', illustrating the challenges of developing new treatments for this aggressive breast cancer subtype.
Ellis and colleagues [19] performed whole-genome sequencing (WGS) (46 cases) and whole-exome sequencing (WES) (31 cases) on 77 post-menopausal ER+ breast cancer samples prior to neoadjuvant treatment with an AI. Among their notable findings were that 18 genes were observed to be mutated above the expected background mutation rate, GATA3 mutations were associated with tumor shrinkage with AI treatment, and tumors with high Ki-67 expression demonstrated a higher frequency of somatic mutations and structural variations that were associated with resistance to AI therapy. In another report of 103 whole-exome and 22 whole-genome sequences of breast tumors from Mexico and Vietnam, Banerji and colleagues [20] described three new aberrations. In ER+ tumors, two loss-of-function mutations in the dimerization partners CBFB transcription factor and RUNX1 may produce resistance to endocrine therapies and have been implicated in the M2 subtype of acute myeloid leukemia. A MAGI3-AKT3 gene fusion was identified in 5 out of 72 basal-like tumor samples and is associated with AKT kinase activation, which may be a potential target for inhibitors of this pathway.
In summary, the major finding from these large-scale projects is that the genomic landscape of breast cancer is very heterogeneous. An aberrant PI3-kinase pathway is significant for both luminal- and basal-type breast cancer. Other pathways important in luminal-type tumors are the P53-MDM2 feedback loop, the stress kinase pathway, and the cell cycle molecules cyclin D1, CDK 4/6, and the retinoblastoma protein. Although somatic alterations, including TP53 mutations, PIK3CA mutations, and ERBB2 amplification, are observed relatively frequently (at least 15%) in breast cancer, many additional potentially 'druggable' genomic alterations occur more infrequently. Classifying low-frequency somatic aberrations as either driver or passenger mutations is a challenge, which is further complicated by the large burden of passenger mutations that can be identified by genome-wide sequencing strategies. Determining the functional significance of low-frequency mutations may be possible through in silico pathway analyses, in addition to functional experiments. Table 1 provides an overview of investigational drugs currently in clinical development with putative molecular predictive biomarkers that could be identified through molecular screening programs.
Molecular screening initiatives
Platforms for clinical testing
Genotyping techniques determine predefined genetic variations at specific locations or 'hotspots' where mutations commonly cluster. Screening panels based on genotyping platforms, such as OncoCarta™ or OncoMap (Sequenom, San Diego, CA, USA) and the SNaPshot™ assay (Applied Biosystems, Foster City, CA, USA), are able to detect known base-pair substitutions and limited indels. This is distinct from direct DNA sequencing technologies, which determine DNA aberrations by establishing the order of nucleic acid pairs and thus can determine translocations, larger indels, or novel base-pair substitutions. The advantage of genotyping or multiplexed PCR-based platforms is that they provide excellent coverage of frequently mutated 'druggable' oncogenes when mutations accumulate in a limited number of DNA sequence regions, such as KRAS (nine bases account for more than 99% of all mutations), BRAF (15 to 18 bases account for more than 90% of all mutations), and PIK3CA (12 to 15 bases account for more than 80% of all mutations) [25, 26]. However, for clinically relevant tumor-suppressor genes, such as TP53, PTEN, BRCA1, or BRCA2, in which mutations are more widely distributed across a much larger DNA coding region, the ability to detect mutations using these platforms is limited to a few selected hotspots. Genotyping systems do not routinely detect gene amplifications, deletions, or translocations that may be clinically relevant.
Sequenom, SNaPshot, and other PCR-based multiplex assays are constrained by bandwidth and throughput. NGS technologies parallelize the sequencing process, producing thousands of DNA sequencing reads at once to allow sequencing of entire exomes or genomes (Table 2). NGS was initially outside the scope of clinical testing and its use was restricted to large research genome centers with access to expensive high-throughput sequencing platforms and high-performance computing to decipher complex NGS data. However, recently developed 'bench-top' NGS instruments, such as the MiSeq (Illumina, Inc., San Diego, CA, USA) and Ion Torrent Personalized Genome Machine (PGM) (Life Technologies, Guilford, CT, USA), have removed the high cost and complexity of genome-scale sequencing. They provide moderate throughput with streamlined library preparation, fast run times, long DNA reads, and relatively automated bioinformatics processing that are well suited to clinical sequencing applications. Both MiSeq and Ion Torrent PGM can sequence a targeted panel of clinically relevant cancer genes (coding regions of 1,000 or more genes with 30× to 50× coverage or approximately 100 genes with at least 1,000× coverage) to identify rare (less than 5% prevalence) mutations and copy number alterations with a rapid turnaround time (of 1 week or less), which can be integrated within clinical workflow. These medium-sequencing throughput systems are unable to do WES or WGS and have limited ability to detect translocations, unless hotspot translocation breakpoints are included to enable detection of such events.
Various sequencing strategies present specific challenges that must be balanced against their perceived and actual benefits (Table 2). WES/WGS will provide the most comprehensive genomic characterization when compared with genotyping or targeted sequencing-based platforms. The clinical utility or actionability of the additional alterations detected by WES/WGS compared with targeted sequencing involving cancer-specific gene panels for treatment selection or matching patients to clinical trials remains to be determined. This is due in part to how actionability is defined, either (a) in a broad context whereby a mutation is actionable if it has a diagnostic, prognostic, or predictive implication or (b) more narrowly to mutations that predict response or resistance to available drugs. In spite of these challenges, NGS is an appealing technology for clinical use because it allows the amalgamation of multiple single-gene tests into a single assay to screen for rare molecular alterations that can be matched to approved or investigational targeted therapies. Such systematic sequencing can deeply characterize a tumor by detecting somatic mutations, SNPs, copy number variations, and translocations. Furthermore, sample heterogeneity does not abrogate the detection of mutations by NGS. The discovery of mutations by programmatic screening of diverse tumor types may enable targeted therapies to be re-purposed into cancers outside that drug's recommended disease sites and thus broaden the population of patients who may benefit from that treatment. As a result, most ongoing molecular screening programs with genotyping or NGS use stored formalin-fixed and paraffin-embedded (FFPE) samples for testing with a targeted cancer or disease site-specific panel, including hotspots, amplicons, or entire exons within genes where mutations are known to occur and may be relevant to treatment selection.
Clinical considerations of molecular screening
Important considerations for molecular screening are tissue selection and the purity of tumor samples. Surgical resection specimens and diagnostic core needle biopsies are routinely FFPE to preserve histology. It is well recognized that DNA and RNA degrades after formalin fixation. The quality of nucleic acids may be particularly poor for FFPE tumor tissues that have been stored for more than 5 years [27]. Stored FFPE tumor tissues are routinely used for biomarker testing of ER and HER2 status; however, this method of tissue fixation may not be appropriate for molecular characterization using high-throughput technologies. Various studies report that NGS can be performed by using FFPE samples [28–30], but whether routinely stored FFPE tumor tissues can be used for clinical NGS testing remains unclear.
Molecular screening using high-throughput technologies is most likely to benefit patients with metastatic breast cancer because these patients have incurable disease that will eventually become refractory to standard treatments. Hence, metastatic relapse or presentation with de novo metastatic disease represents a critical decision point where clinicians could organize somatic DNA sequencing or genotyping of either an archived tissue sample or fresh tumor biopsy of a metastatic lesion. It is increasingly recognized that tumor heterogeneity and clonal evolution may influence biomarker characterization for treatment selection. For example, a discordance of 5% to 15% is observed in ER and HER2 biomarker testing between a primary tumor and a metastatic tumor biopsy obtained from the same patient [31], leading many authors to recommend routine re-biopsy of breast cancer patients at the time of metastatic relapse to guide clinical decision-making [32]. The use of archived primary tumor specimens or fresh metastatic tumor biopsies for high-throughput testing as part of a molecular screening program is an important unresolved question. Furthermore, the genomic diversity within regions of the same tumor (intra-tumor heterogeneity) and between anatomically distinct metastatic tumor sites (inter-tumor heterogeneity) [33] calls into question the adequacy of high-throughput molecular characterization of a single tumor specimen to inform clinical decision-making for all patients.
Notwithstanding these tissue selection issues, there are a number of technical and analytical challenges to routine NGS testing of cancers in a clinical setting. Although the workflow of NGS data alignment and comparison with reference genomes to identify mutations (or 'mutation calling') has improved, NGS is highly dependent on sophisticated computational biology algorithms to identify a region of cancer DNA sequence as wild-type, benign SNP, or a somatic mutation [34]. Parallel sequencing of a patient's germline and tumor tissue may facilitate the identification of somatic alterations; however, this increases the cost and complexity of NGS testing and introduces the potential to identify germline mutations in cancer predisposition genes, such as BRCA1, BRCA2, TP53, and PTEN, that may have additional implications for the patient and family members. There may be variations in the depth of coverage of different regions of the genome with targeted NGS, WES, or WGS. There is no consensus on the minimum threshold requirement for coverage in a clinical setting. Deep coverage of at least 500× to 1,000× may be required to ensure that the required sensitivity and accuracy meet the clinical-grade standard for the detection of somatic alterations in a clinical diagnostic setting. The Next-Generation Sequencing Standardization of Clinical Testing working group recommends that clinically actionable findings be confirmed by independent analysis using an alternative method [35]. This verification step increases the cost of clinical NGS testing and delays the reporting of clinically actionable results.
Targeted genomic and next-generation sequencing using clinical tumor samples
Studies by Thomas and colleagues [3], MacConaill and colleagues [36], and Dias-Santagata and colleagues [37] examined between 250 and 1,000 breast cancer and other tumor specimens for 120 to 400 mutations in 13 to 33 known oncogenes and tumor-suppressor genes by using the Sequenom platforms. These studies identified at least one mutation in 30% to 37% of tumor samples [3, 36, 37].
There is limited information about the clinical impact of high-throughput molecular testing [38, 39]. Sequist and colleagues [40] published their experience at Massachusetts General Hospital with molecular screening of 552 patients with NSCLC by using the multiplex PCR-based SNaPshot assays, which detect around 50 mutations from 14 genes, and fluorescence in situ hybridization (FISH) for ALK translocations. The authors identified at least one mutation in 51% of patients who underwent successful profiling, and directed 78 (22%) of 353 patients with advanced disease to a genotype-matched therapy. Tsimberidou and colleagues [39] successfully performed PCR, FISH, and IHC examining for 11 separate molecular aberrations in 1,144 patients at the MD Anderson Cancer Center. In their cohort, 40% of patients had at least one aberration. They matched each aberration to a targeted treatment when available and demonstrated that patients who received matched targeted therapy (n = 175) had better overall response rates (27% versus 5%; P <0.0001), longer time-to-treatment failure (median 5.2 versus 2.2 months; P <0.0001), and longer overall survival (median 13.4 versus 9.0 months; P = 0.017) compared with patients who did not receive matched treatment (n = 116) [12]. Although these are non-randomized studies, they support the strategy of profiling patients to influence treatment recommendations and demonstrate the potential clinical benefits of tailoring therapy to individual genomic aberrations. These results are in contrast to the findings of Dienstmann and colleagues [41], who profiled 254 patients with chemotherapy-refractory CRC by using the Sequenom platform and did not demonstrate any significant clinical improvement from matched therapy.
More recent molecular profiling initiatives have included broader NGS platforms. Tran and colleagues [42] performed targeted exon sequencing with the NGS PacBio RS platform (Pacific Biosciences, Menlo Park, CA, USA) and genotyping with Sequenom platform on fresh tumor biopsies and archival samples of 50 patients. Nineteen actionable mutations were found in 16 patients, six of whom received matched therapy. Using targeted sequencing of 163 genes on the NGS HiSeq platform (Illumina, Inc.), Wagle and colleagues [43] sequenced 10 patients (with CRC or breast cancer). Each sample was to found to have a clinically meaningful mutation, which included KRAS, BRAF, PIK3CA, or CTNNB1, in addition to common tumor-suppressor genes. WGS and WES were performed for two patients with advanced cancer treated at the University of Michigan by Roychowdhury and colleagues [44]. The patient with CRC was found to have multiple somatic mutations along with CDK8 overexpression and amplification, whereas the patient with melanoma had a somatic mutation in HRAS with a structural rearrangement in CDKN2C. Foundation Medicine Inc. (Cambridge, MA, USA) recently reported that in 169 FFPE samples obtained from patients with metastatic breast cancer that had relapsed after primary surgical resection, 90% of patients had an actionable mutation and that in 124 tumors, one or more actionable mutations that would have been missed by 'hotspot assays' were detected [45]. The company also reported a high concordance rate (>95%) between NGS and IHC/FISH for copy number amplification [46]. Using an NGS assay with a targeted panel of 145 genes, Lipson and colleagues [47] analyzed CRC and NSCLC FFPE samples and found a mutation linked to clinical treatment in 52.5% and 72% of cases (respectively), including two gene fusion aberrations.
Ongoing molecular screening programs
Recognizing that cancer genome sequencing is likely to be integrated into routine clinical decision-making in the near future, many leading cancer research institutions and national cancer agencies have recently launched or are soon to launch broad-scale molecular screening programs for solid tumors, including breast cancer [48]. The majority of institutional programs use a genotyping platform such as Sequenom or SNaPshot and focus their profiling efforts on select tumor types (for example, melanoma, glioblastoma multiforme, breast, lung, colorectal, and ovarian cancer). The size of the gene panel and mutations included vary between each institution; however, in general, the panels include oncogenes linked to targeted drugs that are either approved or in development (Table 3). The Institut Gustav Roussy performs molecular screening as part of their personalized medicine program in several patient subgroups, including metastatic breast cancer (SAFIR01) and potential phase I clinical trial patients (MOlecular Screening for CAncer Treatment and Optimisation, or MOSCATO). Both programs use the Agilent array comparative genome hybridization (Santa Clara, CA, USA) for DNA analysis and the Ion Torrent for mutation analysis. To date, 111 patients underwent a biopsy on MOSCATO; 40% had an actionable mutation and 25 patients received matched therapy [49]. SAFIR01 has screened 423 patients with metastatic breast cancer; of those, 295 samples underwent successful sequencing and 140 patients had a targetable genomic alteration [50]. The majority of these mutations were in the PI3K/AKT pathway.
Beyond these institutional initiatives, nation-wide molecular screening programs have been launched or are in development. Cancer Research United Kingdom has initiated the 'stratified Medicine Programme' across seven cancer research hospitals in the UK to perform molecular profiling for approximately 20 alterations in eight genes by using archival tumor material from 9,000 patients with advanced melanoma, breast, prostate, ovarian, colorectal, and NSCLC over 2 years [51]. The details of the platform that will be used for molecular profiling have not been publicly disclosed. Norway and The Netherlands have initiated similar strategies, using whole-genome and targeted exon sequencing, respectively.
With the advent of commercially available genomic sequencing through for-profit companies like Foundation Medicine, Caris Life Sciences (Irving, TX, USA), and 23andMe (Mountain View, CA, USA), patients and physicians can arrange to have tumor specimens sequenced outside of a clinical trial, or institutional or national screening program. For many patients, the costs of private genomic testing are prohibitive at this point in time. Moreover, the clinical utility of stand-alone genomic testing services is uncertain, as patients are most likely to benefit from genomic testing of their cancers if it is linked to a broader program that provides access to approved or investigational targeted drugs in early-phase clinical testing.
Conclusions
Advances in DNA sequencing technology will revolutionize the concept of personalized breast cancer medicine. Future drug development will likely involve targeted therapies (or combinations of targeted therapies) for molecularly defined subpopulations of patients with breast cancer. The traditional clinical trial paradigm of sequentially screening for individual molecular alterations that determine eligibility for a particular clinical trial with an experimental agent is not well suited to the era of personalized breast cancer medicine. Given the inherent molecular complexity of breast cancer and the growing demand for treatment individualization, a novel 'molecular screening' approach that involves comprehensive genomic characterization early in a patient's disease course that is linked to an experimental therapeutics program with broad access to investigational therapies in early-phase clinical trials is most likely to advance personalized breast cancer medicine. Additional studies are needed to evaluate the feasibility, clinical utility, and cost-effectiveness of molecular screening in breast cancer using high-throughput next-generation sequencing technology.
Abbreviations
- AI:
-
Aromatase inhibitor
- CRC:
-
Colorectal cancer
- ER:
-
Estrogen receptor
- FFPE:
-
Formalin-fixed and paraffin-embedded
- FGFR:
-
Fibroblast growth factor receptor
- FISH:
-
Fluorescence in situ hybridization
- HER2:
-
Human epidermal growth factor receptor 2
- IHC:
-
Immunohistochemistry
- MOSCATO:
-
MOlecular Screening for CAncer Treatment and Optimisation
- NGS:
-
Next-generation DNA sequencing
- NSCLC:
-
Non-small cell lung cancer
- PCR:
-
Polymerase chain reaction
- PGM:
-
Personalized Genome Machine
- SNP:
-
Single-nucleotide polymorphism
- TCGA:
-
The Cancer Genome Atlas
- WES:
-
Whole-exome sequencing
- WGS:
-
Whole-genome sequencing.
References
Cancer Genome Atlas Network: Comprehensive molecular portraits of human breast tumours. Nature. 2012, 490: 61-70. 10.1038/nature11412.
Sotiriou C, Pusztai L: Gene-expression signatures in breast cancer. N Engl J Med. 2009, 360: 790-800. 10.1056/NEJMra0801289.
Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, Wang M, Feng W, Zander T, MacConaill L, Lee JC, Nicoletti R, Hatton C, Goyette M, Girard L, Majmudar K, Ziaugra L, Wong KK, Gabriel S, Beroukhim R, Peyton M, Barretina J, Dutt A, Emery C, Greulich H, Shah K, Sasaki H, Gazdar A, Minna J, Armstrong SA, et al: High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007, 39: 347-351. 10.1038/ng1975.
Metzker ML: Sequencing technologies—the next generation. Nat Rev Genet. 2009, 11: 31-46.
Meyerson M, Gabriel S, Getz G: Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010, 11: 685-696. 10.1038/nrg2841.
Biesecker LG, Burke W, Kohane I, Plon SE, Zimmern R: Next-generation sequencing in the clinic: are we ready?. Nat Rev Genet. 2012, 13: 818-824. 10.1038/nrg3357.
Stratton MR, Campbell PJ, Futreal PA: The cancer genome. Nature. 2009, 458: 719-724. 10.1038/nature07943.
Eifert C, Powers RS: From cancer genomes to oncogenic drivers, tumour dependencies and therapeutic targets. Nat Rev Cancer. 2012, 12: 572-578. 10.1038/nrc3299.
Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O'Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, Bhamra G, Buck G, Choudhury B, Clements J, Cole J, Dicks E, Forbes S, Gray K, Halliday K, Harrison R, Hills K, Hinton J, Jenkinson A, Jones D, et al: Patterns of somatic mutation in human cancer genomes. Nature. 2007, 446: 153-158. 10.1038/nature05610.
Kola I, Landis J: Can the pharmaceutical industry reduce attrition rates?. Nat Rev Drug Discov. 2004, 3: 711-716. 10.1038/nrd1470.
Callaway E: Cancer-gene testing ramps up. Nature. 2010, 467: 766-767. 10.1038/467766a.
Tsimberidou AM, Iskander NG, Hong DS, Wheler JJ, Falchook GS, Fu S, Piha-Paul SA, Naing A, Janku F, Luthra R: Personalized medicine in a phase I clinical trials program: The MD Anderson Cancer Center Initiative. Clin Cancer Res. 2012, 18: 6373-6383. 10.1158/1078-0432.CCR-12-1627.
Juric D, Rodon J, Gonzalez-Angulo AM, Burris HA, Bendell J, Berlin JD, Middleton MR, Bootle D, Boehm M, Schmitt A, Rouyrre N, Quadt C, Baselga J: BYL719, a next generation PI3K alpha specific inhibitor: preliminary safety, PK, and efficacy results from the first-in-human study. Proceedings of the 103rd Annual Meeting of the American Association for Cancer Research. March 31-April 4, 2012. 2012, Chicago, IL: American Association for Cancer Research, Abstract CT-01
Dienstmann R, Andre F, Soria J, Tabernero J, De Braud FGM, Cereda R, Bahleda R, Hollebecque A, Delmonte A, Camboni MG: Significant antitumor activity of E-3810, a novel FGFR and VEGFR inhibitor, in patients with FGFR1 amplified breast cancer. Ann Oncol. 2012, 23: ix116-ix143. 10.1093/annonc/mds393.
Andre F, Delaloge S, Soria JC: Biology-driven phase II trials: what is the optimal model for molecular selection?. J Clin Oncol. 2011, 29: 1236-1238. 10.1200/JCO.2010.31.6877.
Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y: The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012, 486: 346-352.
Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR: The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012, 486: 400-404.
Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, Burleigh A, Yap D, Bernard V, McPherson A, Shumansky K, Crisan A, Giuliany R, Heravi-Moussavi A, Rosner J, Lai D, Birol I, Varhol R, Tam A, Dhalla N, Zeng T, Ma K, Chan SK, et al: The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012, 486: 395-399.
Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC: Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012, 486: 353-360.
Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L: Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012, 486: 405-409. 10.1038/nature11154.
Kalinsky K, Jacks LM, Heguy A, Patil S, Drobnjak M, Bhanot UK, Hedvat CV, Traina TA, Solit D, Gerald W, Moynahan ME: PIK3CA mutation associates with improved outcome in breast cancer. Clin Cancer Res. 2009, 15: 5049-5059. 10.1158/1078-0432.CCR-09-0632.
Parker JS, Mullins M, Cheang MCU, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z: Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009, 27: 1160-1167. 10.1200/JCO.2008.18.1370.
Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, Deleu I, Perez A, Bachelot T, Vittori L, Xu Z, Mukhopadhyay P, Lebwohl D, Hortobagyi GN: Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012, 366: 520-529. 10.1056/NEJMoa1109653.
Kaufman P, Ferrero J, Bourgeois H, Kennecke H, De Boer R, Jacot W, McGreivy J, Suzuki S, Loh E, Robertson J: Abstract S1-4: a randomized, double-blind, placebo-controlled, phase 2 study of AMG 479 with exemestane (E) or fulvestrant (F) in postmenopausal women with hormone-receptor positive (HR+) metastatic (M) or locally advanced (LA) breast cancer (BC). Cancer Res. 2011, 70: S1-S4.
Wellbrock C, Karasarides M, Marais R: The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004, 5: 875-885. 10.1038/nrm1498.
Engelman JA: Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009, 9: 550-562. 10.1038/nrc2664.
Wang F, Wang L, Briggs C, Sicinska E, Gaston SM, Mamon H, Kulke MH, Zamponi R, Loda M, Maher E, Ogino S, Fuchs CS, Li J, Hader C, Makrigiorgos GM: DNA degradation test predicts success in whole-genome amplification from diverse clinical samples. J Mol Diagn. 2007, 9: 441-451. 10.2353/jmoldx.2007.070004.
Adams MD, Veigl ML, Wang Z, Molyneux N, Sun S, Guda K, Yu X, Markowitz SD, Willis J: Global mutational profiling of formalin-fixed human colon cancers from a pathology archive. Mod Pathol. 2012, 25: 1599-1608. 10.1038/modpathol.2012.121.
Hadd AG, Houghton J, Choudhary A, Sah S, Chen L, Marko AC, Sanford T, Buddavarapu K, Krosting J, Garmire L, Wylie D, Shinde R, Beaudenon S, Alexander EK, Mambo E, Adai AT, Latham GJ: Targeted, high-depth, next-generation sequencing of cancer genes in formalin-fixed, paraffin-embedded and fine-needle aspiration tumor specimens. J Mol Diagn. 2013, 15: 234-247. 10.1016/j.jmoldx.2012.11.006.
Wagle N, Berger MF, Davis MJ, Blumenstiel B, Defelice M, Pochanard P, Ducar M, Van Hummelen P, Macconaill LE, Hahn WC, Meyerson M, Gabriel SB, Garraway LA: High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012, 2: 82-93. 10.1158/2159-8290.CD-11-0184.
Amir E, Miller N, Geddie W, Freedman O, Kassam F, Simmons C, Oldfield M, Dranitsaris G, Tomlinson G, Laupacis A, Tannock IF, Clemons M: Prospective study evaluating the impact of tissue confirmation of metastatic disease in patients with breast cancer. J Clin Oncol. 2012, 30: 587-592. 10.1200/JCO.2010.33.5232.
van Diest PJ, Hoefnagel LDC, van der Wall E: Testing for discordance at metastatic relapse of breast cancer matters. J Clin Oncol. 2012, 30: 3031-
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P: Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012, 366: 883-892. 10.1056/NEJMoa1113205.
Ross JS, Cronin M: Whole cancer genome sequencing by next-generation methods. Am J Clin Pathol. 2011, 136: 527-539. 10.1309/AJCPR1SVT1VHUGXW.
Gargis AS, Kalman L, Berry MW, Bick DP, Dimmock DP, Hambuch T, Lu F, Lyon E, Voelkerding KV, Zehnbauer BA: Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat Biotechnol. 2012, 30: 1033-1036. 10.1038/nbt.2403.
MacConaill LE, Campbell CD, Kehoe SM, Bass AJ, Hatton C, Niu L, Davis M, Yao K, Hanna M, Mondal C, Luongo L, Emery CM, Baker AC, Philips J, Goff DJ, Fiorentino M, Rubin MA, Polyak K, Chan J, Wang Y, Fletcher JA, Santagata S, Corso G, Roviello F, Shivdasani R, Kieran MW, Ligon KL, Stiles CD, Hahn WC, Meyerson ML, et al: Profiling critical cancer gene mutations in clinical tumor samples. PLoS One. 2009, 4: e7887-10.1371/journal.pone.0007887.
Dias-Santagata D, Akhavanfard S, David SS, Vernovsky K, Kuhlmann G, Boisvert SL, Stubbs H, McDermott U, Settleman J, Kwak EL, Clark JW, Isakoff SJ, Sequist LV, Engelman JA, Lynch TJ, Haber DA, Louis DN, Ellisen LW, Borger DR, Iafrate AJ: Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO Mol Med. 2010, 2: 146-158. 10.1002/emmm.201000070.
Von Hoff DD, Stephenson JJ, Rosen P, Loesch DM, Borad MJ, Anthony S, Jameson G, Brown S, Cantafio N, Richards DA, Fitch TR, Wasserman E, Fernandez C, Green S, Sutherland W, Bittner M, Alarcon A, Mallery D, Penny R: Pilot study using molecular profiling of patients' tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol. 2010, 28: 4877-4883. 10.1200/JCO.2009.26.5983.
Tsimberidou AM, Iskander NG, Hong DS, Wheler JJ, Fu S, Piha-Paul SA, Naing A, Falchook GS, Janku F, Luthra R, Wen S, Kurzrock R: Personalized medicine in a phase I clinical trials program: The M. D. Anderson Cancer Center Initiative. J Clin Oncol. 2011, 29: abstr CRA2500-
Sequist LV, Heist RS, Shaw AT, Fidias P, Rosovsky R, Temel JS, Lennes IT, Digumarthy S, Waltman BA, Bast E, Tammireddy S, Morrissey L, Muzikansky A, Goldberg SB, Gainor J, Channick CL, Wain JC, Gaissert H, Donahue DM, Muniappan A, Wright C, Willers H, Mathisen DJ, Choi NC, Baselga J, Lynch TJ, Ellisen LW, Mino-Kenudson M, Lanuti M, Borger DR, et al: Implementing multiplexed genotyping of non-small-cell lung cancers into routine clinical practice. Ann Oncol. 2011, 22: 2616-2624. 10.1093/annonc/mdr489.
Dienstmann R, Serpico D, Rodon J, Saura C, Macarulla T, Elez E, Alsina M, Capdevila J, Perez-Garcia J, Sánchez-Ollé G, Aura C, Prudkin L, Landolfi S, Hernández-Losa J, Vivancos A, Tabernero J: Molecular profiling of patients with colorectal cancer and matched targeted therapy in phase I clinical trials. Mol Cancer Ther. 2012, 11: 2062-2071. 10.1158/1535-7163.MCT-12-0290.
Tran B, Brown AM, Bedard PL, Winquist E, Goss GD, Hotte SJ, Welch SA, Hirte HW, Zhang T, Stein LD, Ferretti V, Watt S, Jiao W, Ng K, Ghai S, Shaw P, Petrocelli T, Hudson TJ, Neel BG, Onetto N, Siu LL, McPherson JD, Kamel-Reid S, Dancey JE: Feasibility of real time next generation sequencing of cancer genes linked to drug response: results from a clinical trial. Int J Cancer. 2013, 132: 1547-1555. 10.1002/ijc.27817.
Wagle N, Berger MF, Davis MJ, Blumenstiel B, DeFelice M, Pochanard P, Ducar M, Van Hummelen P, MacConaill LE, Hahn WC: High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012, 2: 82-93. 10.1158/2159-8290.CD-11-0184.
Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X, Kalyana-Sundaram S, Sam L, Balbin OA, Quist MJ, Barrette T, Everett J, Siddiqui J, Kunju LP, Navone N, Araujo JC, Troncoso P, Logothetis CJ, Innis JW, Smith DC, Lao CD, Kim SY, Roberts JS, Gruber SB, Pienta KJ, Talpaz M, Chinnaiyan AM: Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med. 2011, 3: 111ra121-10.1126/scitranslmed.3003161.
Ross JS, Downing S, Yelensky R, Lipson D, Otto G, Palmer GA, Ali SM, Miller VA, Stephens P: Use of the FoundationOne next-generation sequencing (NGS) assay to detect actionable alterations leading to clinical benefit of targeted therapies for relapsed and refractory breast cancer. J Clin Oncol. 2013, 31: abstr 1009-
Lipson D, He J, Yelensky R, Miller V, Sheehan C, Brennan K, Stephens P, Cronin M, Ross J: Next-generation sequencing of FFPE breast cancers demonstrates high concordance with FISH in calling HER2 amplifications and commonly identifies additional clinically relevant genomic alterations. Cancer Res. 2012, 72: 1730-1731. 10.1158/1538-7445.AM2012-1730.
Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, Curran JA, Balasubramanian S, Bloom T, Brennan KW: Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012, 18: 382-384. 10.1038/nm.2673.
Tuma RS: Large-scale genome projects enter the clinic on both sides of the Atlantic. J Natl Cancer Inst. 2011, 103: 1730-1731. 10.1093/jnci/djr503.
Hollebecque A, Massard C, De Baere T, Auger N, Lacroix L, Koubi-Pick V, Vielh P, Lazar V, Bahleda R, Ngo-camus M, Angevin E, Varga A, Deschamps F, Gazzah A, Mazoyer C, Richon C, Vassal G, Eggermont AM, Andre F, Soria JC: Molecular screening for cancer treatment optimization (MOSCATO 01): a prospective molecular triage trial—Interim results. J Clin Oncol. 2013, 31: abstr 2512-
Andre F, Bachelot TD, Campone M, Arnedos M, Dieras V, Lacroix-Triki M, Lazar V, Gentien D, Cohen P, Goncalves A, Lacroix L, Chaffanet M, Dalenc F, Mathieu MC, Bieche I, Olschwang S, Wang Q, Commo F, Jimenez M, Bonnefoi HR: Array CGH and DNA sequencing to personalize targeted treatment of metastatic breast cancer (MBC) patients (pts): a prospective multicentric trial (SAFIR01). J Clin Oncol. 2013, 31: abstr 511-
Aldridge S: 9,000 tumors for stratified medicine. Nat Biotechnol. 2011, 29: 854-854.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
ARH declares that he has no competing interests. PLB has served as a consultant for Novartis (Basel, Switzerland), Roche (Basel, Switzerland), and Sanofi (Paris, France) and has received research support from Bristol-Myers Squibb Company (Princeton, NJ, USA), Genentech (South San Francisco, CA, USA), GlaxoSmithKline (Uxbridge, Middlesex, UK), Roche, Sanofi, and Servier (Neuilly-sur-Seine, France).
Rights and permissions
About this article
Cite this article
Hansen, A.R., Bedard, P.L. Clinical application of high-throughput genomic technologies for treatment selection in breast cancer. Breast Cancer Res 15, R97 (2013). https://doi.org/10.1186/bcr3558
Published:
DOI: https://doi.org/10.1186/bcr3558