
Abstract
Metformin is a widely prescribed anti-diabetic drug and its use is associated with lower cancer incidence. The mechanisms by which metformin attenuates tumorigenesis are not clearly understood. In a paper published in Proceedings of the National Academy of Sciences of the United States of America, Hirsch and colleagues show that metformin interferes with a signaling pathway, mediated by the transcription factor NF-κB, which drives cell transformation and is required for the maintenance of cancer stem cells.
Similar content being viewed by others

Tumors comprise heterogeneous environments that include several distinct cell types [1]. The exact nature of interactions between different cell types within tumors and whether such interactions influence the dynamics of the corresponding cell populations thereby contributing to cancer development are poorly understood issues. Although cultured cancer cells are deprived of the complexity associated with the tumor microenvironment, a different level of heterogeneity persists in that only a discrete, minority fraction of the cancer cell population - referred to as cancer stem cells (CSCs) - has the potential to give rise to tumors when transplanted into animals [2]. CSCs are often resistant to standard therapies and failure to eliminate them has been linked to tumor recurrence [3]. Understanding the molecular mechanisms that underlie CSC identity could therefore be critical in devising more effective cancer therapies.
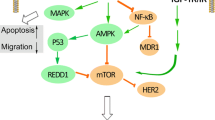
An experimental model that has proven fruitful in elucidating the molecular characteristics of CSCs employs immortalized mammary epithelial cells engineered to express a fusion of the oncogene v-Src (a tyrosine kinase) with the ligand-binding domain of the estrogen receptor (ER). Addition of the synthetic ER ligand tamoxifen induces dimerization of the ER-Src fusion, leading to activation of Src by trans-phosphorylation. Treatment of cells expressing ER-Src with tamoxifen elicits a signaling cascade, referred to as the inflammatory response or inflammatory feedback loop, that is mediated by the transcription factor NF-κB and its downstream target cytokine IL-6 (Figure 1) [4, 5]. Activation of the inflammatory response is essential for Src-induced transformation of mammary epithelial cells, and the cells' ability to form tumors is impeded when this pathway is blocked. Furthermore, Src activation promotes the expansion of the CSC population and CSCs have enhanced activity of the inflammatory pathway compared with non-CSCs. CSCs are therefore likely to exhibit increased dependence on the inflammatory feedback loop, and pharmacological interference with this pathway may limit their tumorigenic potential.

Epithelial cancer cell inflammatory response. Treatment of cells expressing ER-Src with tamoxifen (see text) activates Src and elicits a transcriptional response, mediated by the transcription factor NF-κB, to drive the expression of Lin28, a miRNA binding protein. Lin28 binds to and attenuates the function of let-7 miRNAs, which normally control, among others, the translation of the cytokine IL-6. IL-6 is a potent activator of NF-κB, thereby providing a positive feedback that further amplifies the pathway [4]. In parallel, IL-6 activates another transcription factor, signal transducer and activator of transcription 3 (STAT3), to promote the expression of miR-21 and miR-181b-1, which inhibit the translation of the tumor suppressors PTEN and CYLD, respectively, further enhancing NF-κB activity [5]. Because NF-κB, STAT3 and IL-6 are also players in bona fide inflammatory responses elicited by immune cells, this signaling cascade is known as the inflammatory response or inflammatory feedback loop, alluding to the self-amplifying nature of the pathway.
Metformin is a widely prescribed anti-diabetic drug, and epidemiological studies show that metformin use is associated with lower cancer incidence [6]. Previous work by Hirsch and colleagues showed that metformin selectively kills CSCs over non-CSCs and prolongs tumor remission in mouse xenograft cancer models when combined with chemotherapy agents such as doxorubicin and taxanes [7, 8].
In their latest paper in Proceedings of the National Academy of Sciences of the United States of America, Hirsch and colleagues probed the mechanism of metformin's action and found that metformin treatment inhibits transformation by attenuating the inflammatory feedback loop [9]. In particular, metformin prevented transformation-induced IL-6 expression by inhibiting the translocation of NF-κB to the nucleus. Expression of exogenous Lin28 or treatment with IL-1β (both NF-κB targets) overcomes the anti-transformation effect of metformin. Intriguingly, the inhibitory effect of metformin on inflammatory response components was more pronounced in CSCs than non-CSCs, consistent with the observation that NF-κB nuclear translocation and signal transducer and activator of transcription 3 (STAT3) activity were only inhibited in CSCs. The authors also provided evidence that the sensitivity of other transformed cell lines to metformin is determined by the degree of inflammatory pathway activity (IL-6 levels) that they exhibit.
NF-κB plays a central role in immune-cell-mediated tumor inflammation [10]. The fact that inflamatory pathway activity correlates with metformin sensitivity in xenografts indicates that the effects of metformin are independent of a potential influence of the drug on immune cells. IL-6 from CSCs is sufficient to induce the conversion of non-CSCs to CSCs in a paracrine fashion that establishes a dynamic equilibrium between the two populations [11]. Irrespective of whether metformin can interfere with NF-κB activation in the immune system, IL-6 delivered from immune cells within the tumor is likely to skew the cancer cell population dynamics and this has the potential to impact tumor development [12]. Non-epithelial sources of IL-6 might supersede the role of the epithelial inflammatory response. The influence of metformin on CSC population dynamics in autochthonous tumors remains to be seen.
Another lingering question is how metformin inhibits NF-κB activity. A critical observation by Hirsch and colleagues that may shed some light on this is that metformin only inhibits transformation when administered during an early window of up to 3 hours following Src induction, after which metformin is less effective [9]. This is surprising given that a 5-minute activation of Src suffices to transform cells (albeit with slower kinetics) and NF-κB activity is similar at 1 hour and 4 hours after Src activation [4]. Taken together, these observations suggest that some aspect of metformin function sets the scene to inhibit NF-κB indirectly. Multiple lines of evidence indicate that metformin regulates cellular metabolism, in part, through indirect activation of AMP-activated protein kinase [13], and genetic epistasis experiments are warranted to investigate a potential link between AMP-activated protein kinase and NF-κB in CSCs. Sensitivity to metformin is also influenced by which nutrients are predominantly utilized for anabolic reactions to fuel proliferation [14], so differences in metabolic pathway activity between non-CSCs and CSCs may underlie differential sensitivity to the drug. Regardless of these factors, selective depletion of CSCs promises to pave the way towards more effective therapies [15] and the insights from Hirsch and colleagues bring us a significant step further towards this goal.
Abbreviations
- AMP:
-
adenosine monophosphate
- CSC:
-
cancer stem cell
- ER:
-
estrogen receptor
- IL:
-
interleukin
- miRNA:
-
microRNA
- NF:
-
nuclear factor
- STAT3:
-
signal transducer and activator of transcription 3.
References
Marusyk A, Almendro V, Polyak K: Intra-tumour heterogeneity: a looking glass for cancer?. Nat Rev Cancer. 2012, 12: 323-334.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF: Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003, 100: 3983-3988. 10.1073/pnas.0530291100.
Dean M, Fojo T, Bates S: Tumour stem cells and drug resistance. Nat Rev Cancer. 2005, 5: 275-284. 10.1038/nrc1590.
Iliopoulos D, Hirsch HA, Struhl K: An epigenetic switch involving NF-κB, Lin28, Let-7 microRNA, and IL6 links inflammation to cell transformation. Cell. 2009, 139: 693-706. 10.1016/j.cell.2009.10.014.
Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K: STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell. 2010, 39: 493-506. 10.1016/j.molcel.2010.07.023.
Pollak MN: Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. 2012, 2: 778-790. 10.1158/2159-8290.CD-12-0263.
Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K: Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69: 7507-7511. 10.1158/0008-5472.CAN-09-2994.
Iliopoulos D, Hirsch HA, Struhl K: Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011, 71: 3196-3201. 10.1158/0008-5472.CAN-10-3471.
Hirsch HA, Iliopoulos D, Struhl K: Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc Natl Acad Sci USA. 2013, 110: 972-977. 10.1073/pnas.1221055110.
DiDonato JA, Mercurio F, Karin M: NF-κB and the link between inflammation and cancer. Immunol Rev. 2012, 246: 379-400. 10.1111/j.1600-065X.2012.01099.x.
Iliopoulos D, Hirsch HA, Wang G, Struhl K: Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci USA. 2011, 108: 1397-1402. 10.1073/pnas.1018898108.
Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, Lander ES: Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. 2011, 146: 633-644. 10.1016/j.cell.2011.07.026.
Hardie DG, Ross FA, Hawley SA: AMP-activated protein kinase: a target for drugs both ancient and modern. Chem Biol. 2012, 19: 1222-1236. 10.1016/j.chembiol.2012.08.019.
Javeshghani S, Zakikhani M, Austin S, Bazile M, Blouin MJ, Topisirovic I, St-Pierre J, Pollak MN: Carbon source and myc expression influence the antiproliferative actions of metformin. Cancer Res. 2012, 72: 6257-6267. 10.1158/0008-5472.CAN-12-2907.
Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES: Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009, 138: 645-659. 10.1016/j.cell.2009.06.034.
Acknowledgements
Work in the author's laboratory is supported by the Medical Research Council, UK.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Anastasiou, D. Metformin: a case of divide and conquer. Breast Cancer Res 15, 306 (2013). https://doi.org/10.1186/bcr3387
Published:
DOI: https://doi.org/10.1186/bcr3387