
Abstract
Introduction
Polo-like kinase-1 (PLK1) is a crucial driver of cell cycle progression and its down-regulation plays an important checkpoint role in response to DNA damage. Mechanistically, this is mediated by p53 which represses PLK1 expression through chromatin remodelling. Consistent with this model, cultured cells lacking p53 fail to repress PLK1 expression. This study examined PLK1 expression, p53 mutation and clinical outcome in breast cancer.
Methods
Immunohistochemistry was performed using antibodies to PLK1, MDM2 and Ki67 on Tissue Micro-Array (TMA) slides of a cohort of 215 primary breast cancers. The TP53 gene (encoding p53) was sequenced in all tumour samples. Protein expression scored using the "Quickscore" method was compared with clinical and pathological data, including survival.
Results
Staining of PLK1 was observed in 11% of primary breast tumours and was significantly associated with the presence of TP53 mutation (P = 0.0063). Moreover, patients with both PLK1 expression and TP53 mutation showed a significantly worse survival than those with either PLK1 expression or TP53 mutation alone. There was also a close association of elevated PLK1 with triple negative tumours, considered to be poor prognosis breast cancers that generally harbour TP53 mutation. Further association was observed between elevated PLK1 levels and the major p53 negative regulator, MDM2.
Conclusions
The significant association between elevated PLK1 and TP53 mutation in women with breast cancer is consistent with escape from repression of PLK1 expression by mutant p53. Tumours expressing elevated PLK1, but lacking functional p53, may be potential targets for novel anti-PLK1-targeted drugs.
Similar content being viewed by others


Introduction
Breast cancer remains the most common cancer in women in the western world. The genetic and molecular changes underlying the disease are complex. However, understanding the nature of these changes, and their potential for therapeutic exploitation, presents enormous opportunities for individualised approaches to treatment. To explore these possibilities, there is a need to determine whether mechanistic events established in cultured cell systems, which are thought to drive cancer initiation and/or progression, do indeed underlie the development of the disease in the patients. The testing of clinical material, with focus on the presence or absence of key cancer-associated proteins, should help provide such supporting evidence and identify relevant new candidate markers and therapeutic targets.
The p53 tumour suppressor is a short-lived transcription factor that plays a critical role in eliminating tumour cells by coordinating changes in gene expression, leading to cell cycle arrest, senescence or apoptosis [1–3]. p53 regulates the expression of many genes and, accordingly, loss of p53 function during tumour development can have wide-ranging consequences for the pathology of the tumour cells [2, 4]. Most p53 target genes are actually repressed, as opposed to transactivated, by p53 [1], with the outcome that loss of p53 function may lead to dysregulated or even unrestricted levels of oncogenic proteins. Effective transrepression is, therefore, fundamental to p53-mediated tumour suppression, and potentially to clinical outcome. Indeed, mutation of the gene encoding p53 (TP53) is associated with worse survival in breast cancer (for example, see [5]).
The protein kinase PLK1 plays a pivotal role in the maturation of centrosomes, entry into M phase, spindle formation and cytokinesis [6–8]. Ectopic expression of PLK1 in cultured cells is oncogenic [9] and, consistent with this observation, elevated PLK1 levels occur in various human tumour types [10–20], including breast cancers where it is associated with aggressive characteristics, such as vascular invasion, markers of proliferative activity and lack of detectable estrogen receptor [17, 21]. PLK1 is down-regulated by p53 as part of the G2/M cell cycle checkpoint [22–29] and we recently established that this occurs mainly through p53-dependent repression of PLK1 expression [30]. Consistent with this observation, we and others have also shown that p53-null cells are unable to down-regulate PLK1 levels in response to clinically-relevant genotoxic drugs [26, 30]. These data suggest the possibility that tumours lacking functional p53 are likely to have dysregulated PLK1 levels and that these, in turn, may contribute towards the development of malignancy. Recent evidence has also suggested that the viability of stressed cells that lack p53 may become dependent upon PLK1 [26]. Given that PLK1 is widely considered to be a potential therapeutic target, and that several PLK1 inhibitors are currently undergoing clinical trials [7], targeting of cancers lacking p53 with inhibitors of PLK1 could provide an effective tailored therapeutic strategy. However, the relationship between PLK1 levels and the status of the p53 pathway in tumours needs to be established.
In the present study, we confirm that a proportion of breast tumours are positive for PLK1 protein expression as judged by immunohistochemical staining. Strikingly, we find a statistically significant correlation between the detection of PLK1 and the acquisition of mutation(s) in the TP53 gene. Additionally, we find an association between elevated PLK1 levels and increased levels of the major p53 negative regulator, MDM2. These findings support the established in vitro molecular model for PLK1 repression by p53 [30] and suggest that elevated PLK1 levels in tumours arise, at least in part, through the absence of functional p53. To our knowledge, this is the first demonstration of such an association. From a clinical perspective, we find that patients with TP53 mutation and detectable PLK1 show reduced survival and are more likely to have a triple negative genotype. Our data also highlight the possibility that PLK1-targeted therapy may be a useful option for tumours lacking functional p53.
Materials and methods
Patients
The cohort used in this study (TMA24) has been reported previously [31] and comprises 215 unselected pre- and post-menopausal women (aged 28 to 89; median 62 years) with primary, previously untreated breast cancer, who were seen and treated at Tayside University Hospitals, Scotland from 1997 to 2002. Samples were obtained at the time of surgery and only where sufficient cancer material was available. Informed consent was obtained from each patient prior to tissue acquisition and before surgery was carried out. Tumour samples from these patients were constructed on tissue microarray (TMA) slides as described previously following review of tissue blocks by specialist breast pathologists [31]; (the TMAs were constructed from six cores per tumour from across the tumour, not restricted to the edge of the cancer. The values stated are the mean across all six cores). Ethics approval was given by the Tayside Research and Ethics Committee (Ref. 07/S1402/90). The PLK1 analysis was conducted after a median of eight years follow-up during which some patients had recurred or died. Standard adjuvant treatment was used: for those patients with ER-positive tumours tamoxifen or aromatase inhibitors (for post-menopausal women only) and for node-positive patients FEC75 chemotherapy; no Herceptin was used on this cohort.
Western blotting analysis
This was carried out as reported previously [30].
Immunohistochemistry (IHC)
Sections from the TMA block (nominally four microns thick) were cut onto Superfrost® coated glass slides (VWR International Ltd., Dublin, Ireland) and dried for one hour at 60°C before being de-paraffinised in Histoclear (National Diagnostics, Altanta, GA, USA) and then rehydrated through a graded alcohol series. A total of 10 mM Citric acid buffer was used as the standard microwave-based antigen retrieval method. Sections were boiled in a pressure cooker for 10 minutes before being immunostained on a DAKO autostainer (Ely, Cambridgeshire, UK) using Vectastain® ABC kits (Vector Labs, Burlingame, CA, USA) according to the manufacturer's protocol. Briefly, sections were blocked in normal goat serum containing 10% (v/v) from stock avidin solution (Vector Labs) for 20 minutes followed by a 1 h incubation with Polo-like Kinase-1 primary antibody (monoclonal antibody 208G4 from Cell Signaling, Boston, MA, USA) at a dilution of 1 in 25 of the material provided by the supplier), including 10% (v/v) from stock biotin solution (Vector Labs) to reduce non-specific background staining. Sections were then incubated with biotinylated anti-rabbit secondary antibody for 30 minutes followed by Vectastain® Elite ABC reagent for another 30 minutes. Liquid Diaminobenzidine (DAB) (DAKO) was used as a chromogenic agent for five minutes and sections were counterstained with Mayer's haematoxylin. In between each immunostaining step, slides were washed briefly in Tris buffered saline (TBS), pH 7.6. Sections known to stain positively were included in each batch and negative controls were prepared by replacing the primary antibody with TBS buffer. Antibodies specific for other proteins used in the study have been reported elsewhere [31].
TP53 mutation status
The mutation status of the TP53 gene was assessed as described previously in the tissue samples using the Roche p53 Amplichip research test (Roche Molecular Systems, Pleasanton, CA, USA); these data and the detailed methodology are given in reference [32].
Scoring
TMA scoring for PLK1 was carried out by a specialist breast pathologist (CAP) using a Nikon Eclipse E600 light microscope (Amstelveen, The Netherlands), Aperio, Scanscope XT and Spectrum plus (version 9.1, Vista, CA, USA). Scoring was semi quantitative using the Quickscore method [33] for the intensity and proportion of cells stained for the PLK1 antibody. Scores 0 to 3 indicated the intensity (0 = no staining, 1 = light staining, 2 = moderate staining, 3 = strong staining) whilst scores 1 to 6 represented the proportion of staining (1 = 0 to 4%, 2 = 5 to 20%, 3 = 21 to 40%, 4 = 41 to 60%, 5 = 61 to 80%, 6 = 81 to 100%). Multiplication of each score obtained an overall result of 0 to 18. Cores taken from a plug of MCF7 cells were used as a positive control.
Statistics
TMA scoring was analysed to determine appropriate cut-offs for positive PLK1 or negative staining. The frequency of each score was plotted as a histogram and passed through a Dip test to determine the modality of the data. This resulted in a separation between negative (0 to 3) and positive (4 to 18) stained cases (Figure 1).

Histogram of PLK1 staining. Distribution of frequency of patients against immunohistochemical score of PLK1 staining. Dip test statistic = 0.087, alpha = 0.036, P < 0.01, identifying a score of 3 as a cut off.
Data analysis
The scoring data were automatically extracted from the Aperio Digital Pathology system and combined with the pathology and clinical data from the locally developed digital pathology database. This resulting data set was then analysed using an internally developed automated data analysis system, INSPIRE [34]. For this project, INSPIRE was employed to exhaustively test the results of the PLK1 staining against the standard clinical/pathological outcomes and other staining results held within the database using the two-sided Fisher's exact tests and, where appropriate for survival parameters, the Kaplan-Meier method. Results were considered significant at an α level of 5% (P ≤ 0.05), with anything above 10% (P ≤ 0.10) considered not significant. Results falling between these two boundaries (0.05 <P ≤ 0.10) were marked as marginal in the event any warranted further investigation. The data from INSPIRE were then made available to the research team via a Comma Separated Values (CSV) file to allow further manual analysis. Results that were found to be significant were then re-analysed manually with SPSS Statistics (Version 17, IBM Corp., Armonk, NY, USA) to confirm the results found within INSPIRE. Manual analysis, again using SPSS, was also used to selectively combine variables, such as PLK and p53 status.
Survival was calculated from the date of diagnosis until either the last confirmed hospital visit or the date of death. An event was only recorded if there was confirmation of a breast cancer death. All deaths not directly attributable to breast cancer were censored on the date of death. A relapse was defined as clinical, radiological or pathological evidence of disease relapse.
Multivariate analysis
To clarify the results of univariate analysis, PLK1 was tested for significance within a multivariate environment using the Cox regression with Backward Conditional method to select the most relevant data when predicting survival outcome.
Results and discussion
Staining of PLK1 in breast tumour samples
The specificity of the antibody for cell staining was confirmed by Western blotting analysis of cell extracts carried out using the monoclonal antibody, 208G4 (Figure 2). Several different breast cancer-derived cell lines were examined, and were representative of different breast cancer subtypes, p53 wild type/mutant status and the presence or absence of ERα, PR and HER-2. In each case, a single band of expected molecular weight 62 kD (as measured relative to the migration of standards of known molecular weight) was observed corresponding to full length PLK1. These data underpin the specificity of the antibody and validate its application for the immunohistochemical staining of the tumour sections.

Anti-PLK1 antibody 208G4 detects only PLK1 in extracts of several breast cancer-derived cell lines. Cell extracts were prepared by lysing the cells in SDS-sample buffer and the proteins were subsequently resolved by SDS-PAGE. Western blotting analysis reveals a single band of molecular weight 62 kD corresponding to full length PLK1 in each of the cell extracts.
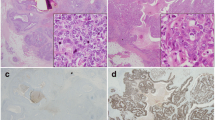
The levels of PLK1 staining in the breast tumour samples varied significantly. A large proportion of samples showed no detectable staining while those that did gave a positive signal (23/215; 11% - see Table 1) varied widely in intensity both from tumour to tumour and, in some cases, within a tumour section, (that is, a small proportion of cells show intense staining as compared with a much lower level of staining in the surrounding cells). Representative examples are shown in Figure 3. These micrographs also indicate that PLK1 staining, where identified, was usually observed in both the nucleus and cytoplasm. Weak or undetectable staining was observed in normal tissue surrounding the tumours. A correlation between PLK1 levels and proliferative activity in cycling cultured cells and in tumours was initially established by Yuan and colleagues [35]. They also noted especially strong staining of mitotic cells, an observation that is consistent with our current knowledge of the cell cycle-dependent regulation of PLK1 expression (that is, only low levels of PLK1 are present in cells in the G1 and S phases as opposed to cells in late G2 and M phase where the levels rise significantly [36–39]). It is, therefore, possible that the variation in staining in the tumour samples could reflect, at least in part, different proliferation rates of the cells. Consistent with this idea we also note that PLK1 was associated with Ki67 expression (a marker for cell proliferation - Table 1). It is also possible that intensely staining cells in any given section are those in G2 or mitosis. Over and above these proposed mechanisms, however, it is likely that cancer-associated changes in the regulation of PLK1 levels additionally contribute to the differences in levels between different tumour samples (see below). Such changes could affect the basal level of expression or the ability to down-regulate PLK1 in response to environmental changes, independently of control mediated by the cell cycle.

Photomicrographs of PLK1 immunohistochemistry from tissue microarray. A and B Grade 3 invasive ductal carcinoma showing a heterogeneous pattern of nuclear and cytoplasmic staining at low (A) and high (B) power. C and D grade 3 invasive ductal carcinoma showing similar staining but in fewer cells at low (C) and high (D) power. The staining is highly specific and confined to the carcinoma cells. There is no staining of vascular, stromal or lymphoid cells.
Relationship of PLK1 expression to TP53status in breast cancer patients
There is a significant association between TP53 mutation status and PLK1 levels (Table 1) such that there is a 3.5-fold greater likelihood of having detectable PLK1 staining if the tumour harbours a mutation(s) in the TP53 gene. We showed recently that PLK1 expression is tightly regulated through p53-mediated transcriptional repression [30]. The association between PLK1 expression and TP53 mutation is, therefore, consistent with our model [30] and suggests that elevated PLK1 levels are likely to occur, at least in part, through the loss of p53 function. We and others have also shown that p53 is protective against apoptosis induced by down-regulation of PLK1 in cultured cells [26, 30, 40]. This observation suggests that tumour cells retaining wild type p53 function are likely to be less susceptible to PLK1 inhibitors and, accordingly, that p53 status is a factor that will have to be taken into account when considering the use of PLK1 inhibitors as part of a "personalised medicine" approach. This is especially significant given that breast cancer patients who have mutant p53 are less responsive to conventional chemotherapies [5]. For these patients, PLK1 inhibitors might be particularly relevant.
Mutation of TP53 can lead not only to inactivation of p53 function but many mutant p53 proteins can interfere with wild type p53 function in a dominant-negative manner in p53 wild type/mutant heterozygotes (reviewed in [41]). Moreover, certain p53 mutants are thought to acquire new functional properties that can influence gene expression levels in a manner that is unaffected by wild type p53. Many of these mutants have been linked to poor prognosis in patients, and studies with animal models indicate that they are likely to promote tumour progression [41, 42]. Our published molecular and cellular analyses suggest that two commonly-occurring tumour-associated "gain-of-function" mutants (R175H and R273H respectively) have no effect on expression from the PLK1 promoter, at least in the context of promoter-reporter (luciferase) assays [30]. The TP53 mutations identified in the present study show no common "hotspots" associated with PLK1 expression (data not shown). As expected, the most common types of the mutations in this cohort were missense mutations, clustered within exon 5 with a smaller group in exon 9 (Table 2). While there were insufficient cases in the cohort to provide unequivocal statistically-supported conclusions, there is a trend for the mutations in exon 5 to be associated with elevated PLK1 levels (in the tumours expressing high PLK1 levels, 6 mutations out of 12 in total are in exon 5 as compared with 11 in exon 5 out of 46 in total for those tumours expressing low or undetectable PLK1: Table 2). Exon 5 encodes amino acids 126 to 186 of p53. Future analyses should test this potential association in a significantly larger cohort. It will also be interesting to determine whether mutation of p53 within this region has any additional influence on PLK1 expression in cultured cells.
An additional issue is that the association between PLK1 expression and TP53 mutational status is not absolute; for example, some tumours show elevated PLK1 levels yet retain wild type TP53 (Table 1). However, p53 function can be lost not only through mutation of the TP53 gene itself but also through mutation, or changes in the expression levels, of proteins that regulate the p53 response. For example, increased expression of the p53 inhibitors, MDM2 (see below) or MDM4, is associated with breast cancer development and is thought to suppress wild type p53 function [41, 42]. It is, therefore, possible that such mechanisms may contribute to permitting PLK1 levels to rise in the tumours where mutation of the TP53 gene is absent. Consistent with this idea we also note a significant association between elevated levels of PLK1 and MDM2 expression (with potential p53 reduction) in our cohort as a whole (Table 1). However, owing to the small size of the subset that has both wild type TP53 and elevated PLK1, we cannot establish any significant association with MDM2 status. It is also possible that the overall association between MDM2 and PLK1 could reflect our previous observation that MDM2 levels may be regulated by PLK1 independently of p53 [30].
Relationship of PLK1 expression to clinical outcome of breast cancer patients
From a clinical perspective, the data show a striking association between elevated PLK1 levels in breast cancer cells and clinical outcome. PLK1 expression was associated almost exclusively with tumour grade, being present predominantly in grade 3 tumours. Moreover, the survival data (Table 1 and Figure 4A) clearly confirm that patients expressing elevated PLK1 expression show significantly reduced survival. These data are consistent with similar findings reported by others linking high levels of PLK1 to aggressive disease and poor outcome [17]. There are no significant associations between PLK1 expression and menopausal status, lymph node status or tumour size (Table 1).

Kaplan-Meier survival analysis. (A) Data correlating PLK1 staining to breast cancer-specific survival in the cohort TMA24. P = 0.02. (B) Stratification of the data to show the influence of p53 status on the association between PLK1 staining and breast cancer-specific survival in the cohort TMA24. P < 0.001. (C) Data correlating PLK1 staining to disease free time in the cohort TMA24. P = 0.072 (D) Stratification of the data to show the influence of p53 status on the association between PLK1 staining and disease free time in the cohort TMA24. P < 0.001. The number of patients at 1-, 5- and 10-year time points are shown below each panel.
By multivariate analysis, in the p53 mutant cancers, only PLK1 (P = 0.015) and the progesterone receptor (P = 0.04) were independently associated with survival. For p53 wild type cancers progesterone receptor (P = 0.041) and size (P = 0.007) were the only independent variables and for all patients (no discrimination by mutation status), progesterone receptor (P < 0.001), tumour grade (P = 0.023), node positivity (P = 0.011) and size (P = 0.031) were independently associated with survival.
There are two additional associations of particular interest and novelty. Firstly, the occurrence of elevated PLK1 levels together with TP53 mutation is associated with poor outcome (Figure 4B), much more so than in patients harbouring TP53 mutation alone. Interestingly, patients with tumours that show elevated PLK1 expression, but retain wild type TP53, have a relatively favourable outcome (Figure 4B). This observation indicates that changes in PLK1 levels may also occur independently of regulation by p53 (that is, through changes to an independent PLK1 regulatory pathway). It also suggests very strongly that p53 is protective against dysregulated PLK1, (which is presumably dysregulated through p53-independent mechanisms), in the context of human disease development. The second interesting feature is the close association of elevated PLK1 with the triple negative phenotype (Table 1). Triple negative tumours tend to have mutation(s) of the TP53 gene and are the most difficult to treat. These data again raise the possibility that tumours of this phenotype, expressing elevated PLK1 and harbouring TP53 mutation, may benefit from the use of novel PLK1 inhibitors [7].
The finding that PLK1 expression is associated almost exclusively with tumour grade (grade 3) could suggest that PLK1 is a passive feature of grade 3 tumours as opposed to a change that may drive tumour development. To address this issue, grade 3 tumours were re-examined and demonstrate clearly that PLK1 status was strongly associated with outcome within the grade 3 tumour subgroup (Figure 5A). This underscores the adverse influence on survival of having both PLK1 over-expression and mutant TP53 status within these high grade cancers (Figure 5B).

Kaplan-Meier survival analysis of patients with Grade 3 tumours. (A) Data correlating PLK1 staining to breast cancer-specific survival. P = 0.004. (B) Stratification of the data to show the influence of p53 status on the association between PLK1 staining and breast cancer-specific survival. P = 0.006. The number of patients at 1-, 5- and 10-year time points are shown below each panel.
Conclusions
Immunohistochemical analysis and DNA sequencing analysis of 215 primary breast tumours have demonstrated a striking association between PLK1 expression and TP53 gene mutation. These observations are consistent with our recently-published molecular model in which wild type functional p53 behaves as an important transcriptional repressor of PLK1. Targeting PLK1 in p53 mutant breast cancer, including poor prognosis, triple negative breast cancer, may offer therapeutic opportunities.
Abbreviations
- CSV:
-
Comma Separated Values
- ERα:
-
estrogen receptor α
- HER-2:
-
human epidermal growth factor receptor-2
- MDM2:
-
murine double minute, clone 2
- MDM4:
-
murine double minute: clone 4
- PLK1:
-
polo-like kinase-1
- PR:
-
progesterone receptor
- TBS:
-
Tris-buffered saline
- TMA:
-
tumour microarray
- TP53 :
-
tumour antigen p53 gene.
References
Riley T, Sontag E, Chen P, Levine A: Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008, 9: 402-412. 10.1038/nrm2395.
Vousden KH, Lane DP: p53 in health and disease. Nat Rev Mol Cell Biol. 2007, 8: 275-283. 10.1038/nrm2147.
Vousden KH, Prives C: Blinded by the light: the growing complexity of p53. Cell. 2009, 137: 413-431. 10.1016/j.cell.2009.04.037.
Goh AM, Coffill CR, Lane DP: The role of mutant p53 in human cancer. J Pathol. 2010, 223: 116-126.
Bonnefoi H, Piccart M, Bogaerts J, Mauriac L, Fumoleau P, Brain E, Petit T, Rouanet P, Jassem J, Blot E, Zaman K, Cufer T, Lortholary A, Lidbrink E, Andre S, Litiere S, Lago LD, Becette V, Cameron DA, Bergh J, Iggo R: TP53 status for prediction of sensitivity to taxane versus non-taxane neoadjuvant chemotherapy in breast cancer (EORTC 10994/BIG 1-00): a randomised phase 3 trial. Lancet Oncol. 2011, 12: 527-539. 10.1016/S1470-2045(11)70094-8.
Archambault V, Glover DM: Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol. 2009, 10: 265-275. 10.1038/nrm2653.
Strebhardt K: Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov. 2010, 9: 643-660. 10.1038/nrd3184.
Strebhardt K, Ullrich A: Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 2006, 6: 321-330. 10.1038/nrc1841.
Smith MR, Wilson ML, Hamanaka R, Chase D, Kung H, Longo DL, Ferris DK: Malignant transformation of mammalian cells initiated by constitutive expression of the polo-like kinase. Biochem Biophys Res Commun. 1997, 234: 397-405. 10.1006/bbrc.1997.6633.
Dietzmann K, Kirches E, von Bossanyi P, Jachau K, Mawrin C: Increased human polo-like kinase-1 expression in gliomas. J Neuro-oncol. 2001, 53: 1-11.
Ito Y, Miyoshi E, Sasaki N, Kakudo K, Yoshida H, Tomoda C, Uruno T, Takamura Y, Miya A, Kobayashi K, Matsuzuka F, Matsuura N, Kuma K, Miyauchi A: Polo-like kinase 1 overexpression is an early event in the progression of papillary carcinoma. Br J Cancer. 2004, 90: 414-418. 10.1038/sj.bjc.6601540.
Knecht R, Elez R, Oechler M, Solbach C, von Ilberg C, Strebhardt K: Prognostic significance of polo-like kinase (PLK) expression in squamous cell carcinomas of the head and neck. Cancer Res. 1999, 59: 2794-2797.
Kneisel L, Strebhardt K, Bernd A, Wolter M, Binder A, Kaufmann R: Expression of polo-like kinase (PLK1) in thin melanomas: a novel marker of metastatic disease. J Cutan Pathol. 2002, 29: 354-358. 10.1034/j.1600-0560.2002.290605.x.
Takahashi T, Sano B, Nagata T, Kato H, Sugiyama Y, Kunieda K, Kimura M, Okano Y, Saji S: Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer Sci. 2003, 94: 148-152. 10.1111/j.1349-7006.2003.tb01411.x.
Tokumitsu Y, Mori M, Tanaka S, Akazawa K, Nakano S, Niho Y: Prognostic significance of polo-like kinase expression in esophageal carcinoma. Int J Oncol. 1999, 15: 687-692.
Weichert W, Denkert C, Schmidt M, Gekeler V, Wolf G, Kobel M, Dietel M, Hauptmann S: Polo-like kinase isoform expression is a prognostic factor in ovarian carcinoma. Br J Cancer. 2004, 90: 815-821. 10.1038/sj.bjc.6601610.
Weichert W, Kristiansen G, Winzer KJ, Schmidt M, Gekeler V, Noske A, Muller BM, Niesporek S, Dietel M, Denkert C: Polo-like kinase isoforms in breast cancer: expression patterns and prognostic implications. Virchows Arch. 2005, 446: 442-450. 10.1007/s00428-005-1212-8.
Weichert W, Schmidt M, Gekeler V, Denkert C, Stephan C, Jung K, Loening S, Dietel M, Kristiansen G: Polo-like kinase 1 is overexpressed in prostate cancer and linked to higher tumor grades. Prostate. 2004, 60: 240-245. 10.1002/pros.20050.
Wolf G, Elez R, Doermer A, Holtrich U, Ackermann H, Stutte HJ, Altmannsberger HM, Rubsamen-Waigmann H, Strebhardt K: Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene. 1997, 14: 543-549. 10.1038/sj.onc.1200862.
Yamada S, Ohira M, Horie H, Ando K, Takayasu H, Suzuki Y, Sugano S, Hirata T, Goto T, Matsunaga T, Hiyama E, Hayashi Y, Ando H, Suita S, Kaneko M, Sasaki F, Hashizume K, Ohnuma N, Nakagawara A: Expression profiling and differential screening between hepatoblastomas and the corresponding normal livers: identification of high expression of the PLK1 oncogene as a poor-prognostic indicator of hepatoblastomas. Oncogene. 2004, 23: 5901-5911. 10.1038/sj.onc.1207782.
Wolf G, Hildenbrand R, Schwar C, Grobholz R, Kaufmann M, Stutte HJ, Strebhardt K, Bleyl U: Polo-like kinase: a novel marker of proliferation: correlation with estrogen-receptor expression in human breast cancer. Pathol Res Pract. 2000, 196: 753-759.
Ando K, Ozaki T, Yamamoto H, Furuya K, Hosoda M, Hayashi S, Fukuzawa M, Nakagawara A: Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J Biol Chem. 2004, 279: 25549-25561. 10.1074/jbc.M314182200.
Incassati A, Patel D, McCance DJ: Induction of tetraploidy through loss of p53 and upregulation of Plk1 by human papillomavirus type-16 E6. Oncogene. 2006, 25: 2444-2451. 10.1038/sj.onc.1209276.
Kho PS, Wang Z, Zhuang L, Li Y, Chew JL, Ng HH, Liu ET, Yu Q: p53-regulated transcriptional program associated with genotoxic stress-induced apoptosis. J Biol Chem. 2004, 279: 21183-21192. 10.1074/jbc.M311912200.
Smits VA, Klompmaker R, Arnaud L, Rijksen G, Nigg EA, Medema RH: Polo-like kinase 1 is a target of the DNA damage checkpoint. Nat Cell Biol. 2000, 2: 672-676. 10.1038/35023629.
Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA, Kinzler KW, Vogelstein B, Papadopoulos N: A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci USA. 2009, 106: 3964-3969. 10.1073/pnas.0813333106.
van Vugt MA, Medema RH: Getting in and out of mitosis with Polo-like kinase-1. Oncogene. 2005, 24: 2844-2859. 10.1038/sj.onc.1208617.
van Vugt MA, Smits VA, Klompmaker R, Medema RH: Inhibition of polo-like kinase-1 by DNA damage occurs in an ATM- or ATR-dependent fashion. J Biol Chem. 2001, 276: 41656-41660. 10.1074/jbc.M101831200.
Zhu H, Chang BD, Uchiumi T, Roninson IB: Identification of promoter elements responsible for transcriptional inhibition of polo-like kinase 1 and topoisomerase IIalpha genes by p21(WAF1/CIP1/SDI1). Cell Cycle. 2002, 1: 59-66.
McKenzie L, King S, Marcar L, Nicol S, Dias SS, Schumm K, Robertson P, Bourdon JC, Perkins N, Fuller-Pace F, Meek DW: p53-dependent repression of polo-like kinase-1 (PLK1). Cell Cycle. 9: 4200-4212.
Hadad SM, Baker L, Quinlan PR, Robertson KE, Bray SE, Thomson G, Kellock D, Jordan LB, Purdie CA, Hardie DG, Fleming S, Thompson AM: Histological evaluation of AMPK signalling in primary breast cancer. BMC Cancer. 2009, 9: 307-10.1186/1471-2407-9-307.
Baker L, Quinlan PR, Patten N, Ashfield A, Birse-Stewart-Bell LJ, McCowan C, Bourdon JC, Purdie CA, Jordan LB, Dewar JA, Wu L, Thompson AM: p53 mutation, deprivation and poor prognosis in primary breast cancer. Br J Cancer. 2010, 102: 719-726. 10.1038/sj.bjc.6605540.
Detre S, Saclani Jotti G, Dowsett M: A "quickscore" method for immunohistochemical semiquantitation: validation for oestrogen receptor in breast carcinomas. J Clin Pathol. 1995, 48: 876-878. 10.1136/jcp.48.9.876.
Quinlan PR, Reed C, Thompson A: INSPIRE: an integrated agent based system for hypothesis generation within cancer datasets. WI-IAT. 2008, 3: 587-590. 2008; IEEE/WIC/ACM International Conference on Web Intelligence and Intelligent Agent Technology: 9-12 December 2008
Yuan J, Horlin A, Hock B, Stutte HJ, Rubsamen-Waigmann H, Strebhardt K: Polo-like kinase, a novel marker for cellular proliferation. Am J Pathol. 1997, 150: 1165-1172.
Golsteyn RM, Schultz SJ, Bartek J, Ziemiecki A, Ried T, Nigg EA: Cell cycle analysis and chromosomal localization of human Plk1, a putative homologue of the mitotic kinases Drosophila polo and Saccharomyces cerevisiae Cdc5. J Cell Sci. 1994, 107: 1509-1517.
Hamanaka R, Smith MR, O'Connor PM, Maloid S, Mihalic K, Spivak JL, Longo DL, Ferris DK: Polo-like kinase is a cell cycle-regulated kinase activated during mitosis. J Biol Chem. 1995, 270: 21086-21091. 10.1074/jbc.270.36.21086.
Lee KS, Yuan YL, Kuriyama R, Erikson RL: Plk is an M-phase-specific protein kinase and interacts with a kinesin-like protein, CHO1/MKLP-1. Mol Cell Biol. 1995, 15: 7143-7151.
Uchiumi T, Longo DL, Ferris DK: Cell cycle regulation of the human polo-like kinase (PLK) promoter. J Biol Chem. 1997, 272: 9166-9174. 10.1074/jbc.272.14.9166.
Yim H, Erikson RL: Polo-like kinase 1 depletion induces DNA damage in early S prior to caspase activation. Mol Cell Biol. 2009, 29: 2609-2621. 10.1128/MCB.01277-08.
Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R, de Graaf P, Francoz S, Gasparini P, Gobbi A, Helin K, Pelicci PG, Jochemsen AG, Marine JC: Amplification of MDMx (or MDM4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol. 2004, 24: 5835-5843. 10.1128/MCB.24.13.5835-5843.2004.
Onel K, Cordon-Cardo C: MDM2 and prognosis. Mol Cancer Res. 2004, 2: 1-8.
Acknowledgements
We are very grateful to Dr. Phil Coates for critical appraisal of the manuscript and helpful feedback. This work was supported by Cancer Research UK (funding to support Dr. Sharon King), Breast Cancer Research Scotland (funding to support Phil Quinlan) and the Breast Cancer Campaign (funding to support activities of the Tayside Tissue Bank).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SIK carried out the experiments and data analysis, interpreted the data and wrote drafts of the manuscript. SB carried out some of the experimental studies and data analysis. PRQ generated the database and carried out the computer-based analysis and statistical application. LJ and CAP undertook all of the pathological analysis and interpretation. AMT conceived and designed the studies, and analysed and interpreted the data. DWM conceived and designed the studies, analysed and interpreted the data, and wrote the final manuscript. All authors read, made suggestions and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
King, S.I., Purdie, C.A., Bray, S.E. et al. Immunohistochemical detection of Polo-like kinase-1 (PLK1) in primary breast cancer is associated with TP53mutation and poor clinical outcome. Breast Cancer Res 14, R40 (2012). https://doi.org/10.1186/bcr3136
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/bcr3136