
Abstract
Introduction
HER2 gene amplification and protein overexpression (HER2+) define a clinically challenging subgroup of breast cancer with variable prognosis and response to therapy. Although gene expression profiling has identified an ERBB2 molecular subtype of breast cancer, it is clear that HER2+ tumors reside in all molecular subtypes and represent a genomically and biologically heterogeneous group, needed to be further characterized in large sample sets.
Methods
Genome-wide DNA copy number profiling, using bacterial artificial chromosome (BAC) array comparative genomic hybridization (aCGH), and global gene expression profiling were performed on 200 and 87 HER2+ tumors, respectively. Genomic Identification of Significant Targets in Cancer (GISTIC) was used to identify significant copy number alterations (CNAs) in HER2+ tumors, which were related to a set of 554 non-HER2 amplified (HER2-) breast tumors. High-resolution oligonucleotide aCGH was used to delineate the 17q12-q21 region in high detail.
Results
The HER2-amplicon was narrowed to an 85.92 kbp region including the TCAP, PNMT, PERLD1, HER2, C17orf37 and GRB7 genes, and higher HER2 copy numbers indicated worse prognosis. In 31% of HER2+ tumors the amplicon extended to TOP2A, defining a subgroup of HER2+ breast cancer associated with estrogen receptor-positive status and with a trend of better survival than HER2+ breast cancers with deleted (18%) or neutral TOP2A (51%). HER2+ tumors were clearly distinguished from HER2- tumors by the presence of recurrent high-level amplifications and firestorm patterns on chromosome 17q. While there was no significant difference between HER2+ and HER2- tumors regarding the incidence of other recurrent high-level amplifications, differences in the co-amplification pattern were observed, as shown by the almost mutually exclusive occurrence of 8p12, 11q13 and 20q13 amplification in HER2+ tumors. GISTIC analysis identified 117 significant CNAs across all autosomes. Supervised analyses revealed: (1) significant CNAs separating HER2+ tumors stratified by clinical variables, and (2) CNAs separating HER2+ from HER2- tumors.
Conclusions
We have performed a comprehensive survey of CNAs in HER2+ breast tumors, pinpointing significant genomic alterations including both known and potentially novel therapeutic targets. Our analysis sheds further light on the genomically complex and heterogeneous nature of HER2+ tumors in relation to other subgroups of breast cancer.
Similar content being viewed by others


Introduction
Gene amplification is a frequent mechanism of oncogene activation in breast cancer (BC) [1]. Amplification and overexpression of the HER2 (HER2/neu, ERBB2) oncogene on chromosome 17q12 occur in 15-25% of invasive BC [2]. HER2-amplified (HER2+) tumors define a clinically important BC subgroup, generally associated with poor prognosis [2, 3]. Strategies to therapeutically target the HER2 protein by monoclonal antibodies (for example, trastuzumab) or tyrosine kinase inhibitors (for example, lapatinib) have been successful [4–7]. As these drugs are most effective in HER2+ BC, considerable efforts have been devoted to accurate assessment of HER2 status, currently performed by immunohistochemistry (IHC) and/or in situ hybridization [8]. However, despite the success of targeted treatment, many HER2+ cases fail to respond or develop resistance over time.
It is evident that the HER2-amplicon has a variable structure, comprising other genes in the 17q12-q21 region that may contribute to tumor progression and treatment effect in HER2+ BC. One of these genes is topoisomerase IIα (TOP2A), located 700 kb telomeric of HER2, that may be either co-amplified, unaffected or deleted in HER2+ tumors [9]. TOP2A status has been reported to significantly influence the response to anthracycline-based therapy [10–12] although conflicting results exist [13, 14]. Furthermore, it is evident that HER2+ tumors constitute a biologically heterogeneous subgroup of BC. Global gene expression profiling defines an ERBB2 molecular subtype of BC that predominantly consists of estrogen receptor (ER) negative HER2+ tumors (HER2+/ER-) [15, 16], while HER2+/ER+ tumors are more heterogeneously classified. In addition, we recently used gene expression profiling to characterize three distinct subgroups of HER2+ tumors, and to create a HER2-derived prognostic gene signature with strong correlation to outcome for patients with HER2+ disease [17].
Genomic profiling using array comparative genomic hybridization (aCGH) analysis has revealed frequent complex copy number alterations (CNAs) on chromosome 17q, often including high-level amplifications, in HER2+ BC [18–20]. Moreover, although HER2+ tumors share other commonly gained or lost regions with non-HER2 amplified (HER2-) tumors, the genomic profiles of HER2+ tumors are more often heterogeneous and complex in nature [18–20]. We designed a study to comprehensively investigate CNA patterns in 200 HER2+ tumors using high-density bacterial artificial chromosome (BAC) aCGH in concert with custom-designed high-density zoom-in aCGH [21]. We provide evidence of considerable genomic heterogeneity in HER2+ BC, and further delineate the boundaries of the 17q12-q21 amplicon. In addition, matched global gene expression profiles were available for 87 tumors allowing correlation of CNAs to mRNA expression levels for identification of putative target genes.
Materials and methods
Patients and tumor material
Freshly frozen HER2+ BC tissue (n = 188) was obtained from the Southern Sweden Breast Cancer Group's tissue bank at the Department of Oncology, Lund University Hospital and from Department of Pathology, Reykjavik University Hospital. Additionally, 12 formalin-fixed paraffin embedded (FFPE) tumors were obtained from the Department of Pathology, Lund University Hospital. Confirmatory IHC and/or fluorescence in situ hybridization (FISH) data were available in 69 of 200 HER2+ tumors [17, 22]. Patient and tumor characteristics are summarized in Table 1 and described in detail in Additional file 1. The study was approved by the regional Ethical Committee in Lund (reg. no. LU240-01 and 2009/658), waiving the requirement for informed consent for the study, and the Icelandic Data Protection Committee and the National Bioethics Committee of Iceland. For Icelandic patients written informed consent was obtained according to the national guidelines.
aCGH analysis
BAC microarrays were produced by the SCIBLU Genomics Center [23] in three array formats, 32K (Gene expression Omnibus, GEO, GPL4723), 33K (GPL7247) and 38K (GPL9077) all mapped to the UCSC Human Genome browser build 17 [24]. DNA from fresh frozen and FFPE tumor tissue was extracted as described (Additional file 2). Array printing, labeling, hybridization, scanning and image analysis were performed as previously described [25]. Technical replicate experiments were performed for 15 tumors. Copy number estimates (log2ratios) for each array were normalized [26] and replicated samples were merged after normalization. Breakpoint analysis was performed using circular binary segmentation (CBS) with α = 0.01 [27]. Only segments ≥4 BAC probes were used in further analyses. Following segmentation, array platforms were combined into a common array design. CNAs were detected using sample adaptive thresholds from 250 kb smoothed data [26] (Additional file 1). Threshold for amplification was set to segmented log2ratio ≥0.5, and for high-level amplification to segmented log2ratio ≥1 for HER2+ tumors. Recurrent high-level amplifications were defined as single peaks computed from the shortest region of amplification overlap occurring in >2% of tumors. HER2/TOP2A co-amplification was defined as segmented log2ratio ≥0.5 for HER2 and TOP2A. Co-amplification percentages were calculated as the number of tumors with co-amplification divided by the lowest number of the individual amplifications, if not stated otherwise. Pericentromeric BAC probes on the p- and q-arm of chromosome 17 were identified as the three probes closest to the chromosome 17 centromer (CEP17) (Additional file 2). CEP17 amplification was defined as the average segmented log2ratio ≥0.5 of either the probes on the p- or q-arm. BAC aCGH data are available through GEO [28] as [GEO:GSE21259].
Zoom-in aCGH analysis
Custom-designed 60-mer oligonucleotide zoom-in aCGH arrays with an average probe-to-probe spacing of 100 bp in the 17q12-q21 region were designed using the Agilent eArray ver. 5.3 software (Agilent Technologies, Santa Clara, CA) as described (Additional file 2) and performed on 20 tumors (Additional file 1). Microarrays were processed as described [21]. Breakpoint analysis was performed using CBS (α = 0.01). Agilent probes were mapped to the UCSC build 18 [24]. Thresholds for amplification and high-level amplification were set similarly as for BAC aCGH data.
Identification of significant copy number alterations and fraction of the genome altered
Genomic Identification of Significant Targets in Cancer (GISTIC) [29] was used to identify significant CNAs in the 200 tumors (Additional file 2). GISTIC regions with q-value < 0.25 were identified as significant. Student's t-test performed on average log2ratios for GISTIC regions were used to identify regions associated with different clinical variables, such as ER status, lymph node (LN) status, histological grade (grade 3 vs. 1 and 2), DNA ploidy (diploid vs. aneuploid), tumor size (≤20 mm vs. >20 mm) and patient age (<50 years vs. ≥50 years) for HER2+ tumors. A false discovery rate-adjusted P-value < 0.05 was considered significant (Additional file 2). Genomic coordinates for GISTIC regions are mapped to the UCSC Human Genome browser build 17 [24]. A firestorm-like amplification pattern [30] was defined as at least three high-level amplifications larger than three BAC probes located on the same chromosomal arm, separated by non-amplified segments, and with a maximum inter-peak distance <50% of the chromosome arm length. The fraction of the genome altered (FGA) was calculated as previously described [17].
External aCGH data sets for comparison
CNAs and amplification frequencies in HER2+ tumors were compared to an assembled HER2- reference BC data set (n = 554) comprising four BC aCGH data sets, Chin et al. [31], Fridlyand et al. [32], Adelaide et al. [33], and Jönsson et al. (submitted) (Table 1 and Additional file 3). Data sets were processed individually (Additional file 2), transformed to a common 100 kb probe set as described [34], and merged. Clinical follow-up information was available for the Chin, Fridlyand, and Jönsson data sets. Gene expression subtype classification [15] was available for the Chin, Adelaide, and Jönsson data sets (Additional file 2). Threshold for amplification was set to segmented log2ratio ≥0.5, and for high-level amplification to segmented log2ratio >0.8 for HER2- tumors in the reference data set.
Gene expression analysis
Gene expression profiles for 87 HER2+ tumors were available as either oligonucleotide data (n = 58, Jönsson et al. submitted) or cDNA data (n = 29) [22] part of larger BC data sets. Data sets were individually processed and classified according to different gene signatures (Additional file 2).
Correlation of gene expression data with genomic aberrations
Gene expression data were compared to GISTIC aCGH log2ratios using Pearson correlation as described [25]. A correlation cut-off representing P = 0.05 obtained from 10,000 permutations of aCGH sample labels was used to identify significantly correlated genes in GISTIC regions. Global correlation analysis using genes mapped to individual BAC probes was performed similarly, with two modifications; segmented log2ratios were used for individual BAC probes and 1,000 permutations were performed for P-value estimation.
Survival analysis
Overall survival (OS), univariate and multivariate regression analyses were performed in R [35] using the Survival package. Survival curves were compared using Kaplan-Meier estimates and the log-rank test. The full follow-up time was used for log-rank tests and regression analyses if not specified otherwise. In multivariate analysis stratified tumor size and LN status were included as covariates. Tick marks in Kaplan-Meier plots indicate censored follow-up.
Results
Extent and patterns of 17q12-q21 amplification in HER2+ breast cancer
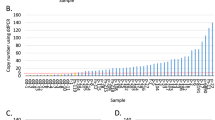
The 17q12-q21 amplification pattern was analyzed in 200 HER2+ tumors using BAC aCGH. The ability of the BAC aCGH platform to accurately estimate HER2 copy numbers was confirmed by parallel FISH analysis (Additional file 2) in 13 FFPE HER2-amplified tumors, showing a good correlation between the techniques (Figure S1A in Additional file 4). The smallest region of amplification overlap (SRO) for the HER2 amplicon was 248 kbp (chr17:34979166-35227087, hg17 build), involving 10 RefSeq genes (Figure 1A). The most frequently up-regulated genes (gene expression log2ratio ≥1) in this SRO were HER2 (92% of samples) followed by GRB7 (85%), C17orf37 (79%), PERLD1 (72%), PPP1R1B (63%) and STARD3 (62%), while gene expression data were unavailable for NEUROD2, TCAP, PNMT and ZNFN1A3. The HER2 SRO was further delineated by zoom-in aCGH to 85.92 kbp (chr17:35074472-35160391, hg18 build) including TCAP, PNMT, PERLD1, HER2, C17orf37 and GRB7. Thus, this analysis excluded STARD3, since three tumors showed an amplicon breakpoint within the gene, while one tumor had an amplicon starting immediately telomeric of STARD3 (Figures 1B and 1C).

Extent and pattern of the HER2 -amplicon on chromosome 17q12-q21 in HER2+ BC. (A) Frequency of amplification across 200 HER2-amplified tumors analyzed by BAC aCGH. Frequency estimates correspond to number of tumors with segmented log2ratio >0.5 for respective BAC probe and are displayed at respective BAC probe's center position (red circles). Shortest region of amplification overlap (SRO) was defined from involved BAC probes genomic start and stop position and is marked with a light gray background. Genomic position of eight BAC probes mapping to the SRO is displayed together with their center position (black circle). (B) Close-up of chr17:35000001-35200000 (hg18 build) for tumor TAX577717 analyzed by zoom-in oligonucleotide aCGH. (C) Close-up of chr17:35000001-35867695 (hg18 build) for tumor TAX577700 analyzed by zoom-in aCGH.
HER2/TOP2A co-amplification was observed in 61 tumors (31%) with an SRO of approximately 1,050 kbp (chr17:34873000-35921000, hg17 build) using BAC aCGH, and narrowed to 783.64 kbp (chr17:35067680-35851322, hg18 build) using zoom-in aCGH (Figure 1C). TOP2A mRNA levels were significantly higher in HER2/TOP2A co-amplified cases as compared to non-amplified HER2+ tumors (P = 3 × 10-5 and 7 × 10-8, respectively for TOP2A loss and TOP2A normal cases, t-test) in agreement with Arriola et al. [20], as well as compared to HER2- tumors, classified according to gene expression subtypes (for example, P = 0.0002 for HER2+/TOP2A+ vs. HER2- basal-like tumors, t-test) (Figure S1B in Additional file 4). Intriguingly, HER2+/TOP2A+ tumors showed significantly lower S-phase fractions and lower correlation to a gene expression grade signature [36] than HER2+/TOP2A- tumors (P = 0.004 and 0.001, respectively, t-test) (Figure S1B in Additional file 4). Additionally, HER2/TOP2A co-amplification was associated with ER+ status, as also observed by others [19, 37], as well as patient age ≥50 years (P = 0.008 and 0.03, respectively, Fisher's exact test), but not to other clinical variables. Loss of TOP2A was found in 36 (18%) HER2+ tumors, while the remaining 51% had neither loss nor amplification of TOP2A. Moreover, none of 554 HER2- tumors had focal TOP2A amplification.
Extent and patterns of significant CNAs on chromosome 17 in HER2+ breast cancer
Chromosome 17q has been reported to frequently harbor complex CNAs in HER2+ BC, often involving other high-level amplifications together with the 17q12 locus [18–20]. GISTIC analysis was used to identify and delineate 17 significant regions (10 gains, including the HER2 amplicon, and 7 losses) on chromosome 17 in the 200 HER2+ tumors (Figure 2, Additional file 5). Recurrent high-level amplifications were observed in 12 of 17 regions, of which amplifications on 17q11.2, 17q12 (centromeric of HER2), 17q12 (HER2), 17q21.33 and 17q23.2 (centromeric) were more prevalent in HER2+ compared to HER2- tumors. While 41% of all HER2+ tumors contained ≥1 other recurrent 17q amplicon besides HER2, no recurrent amplifications were identified on 17p. Several genes in the 17 GISTIC regions showed significant correlation between mRNA expression and copy number levels, including genes and miRNAs implicated in BC oncogenesis like RPS6KB1 [38], PPM1D [18] and mir-21 [39] on 17q23.2 (Additional file 5). MYST2, proposed as the candidate oncogene in the 17q21.33 region [40], was amplified in 20% of the tumors, however, located centromeric of the 17q21.33 GISTIC region. Besides the association of amplification on 17q11.2 with ER+ tumor status (P = 0.009, Fisher's exact test), none of the other 17q amplicons were significantly correlated with ER status, LN status, tumor size, histological grade or patient age, possibly due to the small sample numbers for individual amplicons.

Extent, frequency and patterns of CNAs on chromosome 17 in HER2+ BC. Regions of loss are shown in green, normal in black, gain in dark red, amplification in red, and high-level amplification in white for each sample (row). Frequency of gain (red) and loss (green) across all 200 tumors are shown for chromosome 17. Read boxes, above the cytoband bar, indicate GISTIC regions of gain and green boxes GISTIC regions of loss. GISTIC regions with recurrent amplifications that are more frequent in HER2+ BC compared to HER2- BC are named. Vertical purple line corresponds to centromer limit.
Amplification of centromeric regions on chromosome 17 (CEP17 amplification) was observed in 22 (11%) tumors using the closest pericentromeric BAC probes. CEP17 amplification based on copy number status for BAC probes on either 17p11.1 or 17q11.1 was found in 37 (19%) HER2+ tumors. By comparison, only four (1%) HER2- tumors in the Jönsson et al. data set showed CEP17 amplification based on either pericentromeric BAC probes, or average copy number of BAC probes on either 17p11.1 or 17q11.1. Zoom-in aCGH analysis further delineated the pattern of amplification in the centromeric region, identifying amplification of a region including the WSB1 gene (chr17:22558232-22751802, hg18) as the most frequent in tumors with CEP17 amplification.
Recurrent amplifications and firestorm-like amplification patterns in HER2+ breast cancer
Excluding chromosome 17, recurrent high-level amplifications were observed with varying frequencies on chromosomes 1, 5, 6, 8, 11, 19 and 20 in 90 (45%) of HER2+ tumors (Additional file 6). Interestingly, there was no significant difference in their overall prevalence in HER2+ and HER2- tumors (P > 0.05, Bonferroni adjusted Fisher's exact test). Identified amplifications included several known BC amplicons and oncogenes, for example, 8p12 (FGFR1, LSM1, RAB11F1P1, PPAPDC1B), 8q24.21 (MYC), 11q13.3 (CCND1) and 20q13.2 (ZNF217). Co-occurrence of amplicons was also common as 30%, 9% and 8% of the 90 tumors had two, three or more than three recurrent amplifications, respectively. A number of chromosome regions were frequently co-amplified: amplifications on 8q (one with another), most regions on 17q (with each other), 1q32.1-q32.2 with 8q24.21, and 20q13.2 with 20q13.32 (Figure 3A). Interestingly, while high-level amplifications of 8p12, 11q13.3 and 20q13.2 were mutually exclusive in the HER2+ tumors, these co-amplifications were not uncommon in HER2- tumors (Figure 3). Furthermore, co-occurrence of high-level amplifications at 8p12 with 8q24.21, and 20q13.2 with 8q24.21 were also rare in HER2+ tumors compared to HER2- tumors (Figure 3). High-level amplifications of other putative oncogenes, for example, PIK3CA, ESR1, EGFR, KIT, MDM2 and MYB, were rare in HER2+ tumors (≤1%). Amplifications on 11q13.3, 11q13.5 and 19q13.42 were associated with ER+ tumor status (P = 0.002, 0.06, 0.03 respectively, Fisher's exact test), corroborating previously reported association of 11q13.3 to HER2+/ER+ disease [19]. No recurrent amplification was associated with LN status, tumor size or patient age.

Pattern of co-occurrence of recurrent amplifications in HER2+ and HER2- BC. (A) Fraction of co-amplification of recurrent amplifications in HER2+ BC excluding the 17q12 HER2 locus. For each amplification (vertical axis), the fraction of samples with a co-amplification (horizontal axis) is indicated in each box. Only co-amplifications occurring in ≥2 tumors with fractions ≥0.2 are displayed. For example, 20% of tumors with 17q24.2 amplification also have 1q21.2 amplification, while 40% of tumors with 1q21.2 amplifications also show amplification at 17q24.2 indicating that the number of 1q21.2 amplified tumors is lower than the number of tumors with 17q24.2 amplification. (B) The fraction of co-amplification of recurrent amplifications in A in HER2- breast tumors. Only co-amplifications occurring in ≥3 tumors with fractions ≥0.2 are displayed. Fractions are calculated similarly as in A.
A firestorm-like amplification pattern (firestorms) has been defined as multiple closely spaced high-level amplifications limited to single chromosome arms [30]. In total, 115 firestorms were observed in 88 (44%) HER2+ tumors. Firestorms observed on p-arms (n = 14) were predominantly located on 1p (36%), 6p (21%) and 12p (14%), while firestorms observed on q-arms (n = 101) were predominantly located on 17q (57%), 8q (16%), 20q (5%), 6q (4%), 12q (3%), and 1q (3%). By comparison, a total of 39 firestorms were observed in 30 (10%) HER2- tumors in the Jönsson et al. data set analyzed similarly on the same BAC aCGH platform. Firestorms in these HER2- tumors were predominantly located on 1q, 6q, 8q, 11q, 12p, 12q, and 17q (>1 firestorm). Amplification peaks in observed firestorms were rarely recurrent across HER2+ tumors, except for a few peaks on 17q that were observed in multiple tumors. Prevalence of firestorms was correlated with LN+ status and DNA aneuploidy in HER2+ tumors (P = 0.02 and 0.009, respectively, Fisher's exact test), but not to ER status or tumor size.
Comparison of DNA copy number alterations in HER2+ and HER2- breast cancer
HER2+ tumors revealed considerable genomic heterogeneity with the most frequent CNAs (>30% of tumors) being gain on 1q, 5p, 6p, 8q, 9q, 11q, 12p, 12q, 16p, 17q, 19p, 19q, 20p, 20q and 21q, and loss on 1p, 3p, 8p, 9p, 11q, 16q, 17p, 17q and 18q (Figure 4A). GISTIC analysis identified 117 regions (58 gains, 59 losses), located across all autosomes including numerous candidate genes and miRNAs (Figure 4A, Additional file 5).

Significant CNAs in HER2+ BC in relation to molecular subtypes. (A) Frequency of gain (red) and loss (green) in 200 HER2+ tumors. Blue regions indicate significant CNAs identified by GISTIC analysis. (B) GISTIC regions differing HER2+ tumors from HER2- tumors classified according to the molecular subtypes as basal-like, luminal A, luminal B and normal-like (P < 0.05, Bonferroni adjusted Fisher's exact test). Each box represents a GISTIC region, red indicates more frequent gain, and green indicates more frequent loss. (C) GISTIC regions differing HER2+/ER- tumors from HER2- tumors classified as basal-like. Regions identified by Bonferroni adjusted Fisher's exact test (P < 0.05). Each box represents a GISTIC region, red indicates more frequent gain, and green indicates more frequent loss. (D) GISTIC regions associated with ER status and DNA ploidy in HER2+ BC. Regions identified by Student's t-test with FDR-adjusted P < 0.05. Each box represents a GISTIC region, red indicates more frequent gain, and green indicates more frequent loss.
Several GISTIC regions showed significant differences in frequency when HER2+ tumors were compared to HER2- tumors classified into gene expression subtypes (Table 1, Figure 4B, Additional file 7). Basal-like tumors were characterized by more frequent losses on 2q37.1, 4p, 4q, 5q, 9q, 10q, 11p15.5 and 14q, and gain of 1p22.1, 3q26.32, 10p13 and 12p13.31 compared to HER2+ tumors. Luminal A tumors were characterized by more frequent losses on 16q, while other GISTIC regions were less frequently altered compared to HER2+ tumors. Luminal B tumors were characterized by more frequent losses on 6q, 11q, 16q and 22q compared to HER2+ tumors, while normal-like tumors were characterized by an overall lower CNA frequency.
The average fraction of the genome altered (FGA), representing the percentage of BAC clones subjected to gain or loss for each sample, for HER2+ tumors was 0.34, equally divided between gains (0.18) and losses (0.16). HER2+/ER- tumors showed significantly lower FGA than HER2-/ER- tumors (P = 9 × 10-11, t-test), while HER2+/ER+ tumors were not different from HER2-/ER+ tumors (P = 0.11). A similar comparison of HER2+ tumors to HER2- tumors of various gene expression subtypes showed significantly higher FGA in basal-like (P < 2 × 10-16) and luminal B (P = 0.0001) tumors, but lower FGA in luminal A (P = 0.0003) and normal-like tumors (P = 0.04) compared to HER2+ tumors.
Differences between HER2+ and basal-like breast cancer
Since most ER- tumors are found in the ERBB2 and basal-like gene expression subtypes [15, 41], we performed separate comparisons of HER2+/ER- tumors vs. HER2- basal-like tumors, and ERBB2 classified tumors vs. HER2- tumors classified according to gene expression subtypes. Comparison of HER2+/ER- tumors vs. basal-like tumors resulted in similar findings as for all HER2+ tumors vs. basal-like tumors (Figure 4C). Moreover, comparison of ERBB2 classified HER2+ tumors (n = 51) to basal-like tumors identified similar regions as in the comparison of HER2+/ER- tumors (Additional file 8). We were not able to confirm findings that loss of 15q14-q21 and 9p21.3 separate basal-like tumors from HER2+ tumors [18]. In addition, HER2+ tumors displayed differences in mRNA expression of two different ER gene expression modules [42, 43] compared to HER2- tumors classified according to the gene expression subtypes in the Jönsson et al. data set (Additional file 9). Notably, basal-like tumors displayed considerably lower expression of the ER gene expression modules compared to HER2+/ER- tumors, while the difference between HER2+/ER- and HER2+/ER+ tumors was less pronounced.
DNA copy number alterations in subgroups of HER2+ breast cancer
Highly similar CNA frequencies were observed when HER2+ tumors were stratified by ER status, with apparent differences being limited to more frequent loss of 1p, 11q and 16q and gain of 11q13 in HER2+/ER+ tumors, and loss of 5q in HER2+/ER- tumors (Additional file 10). Supervised analysis of subgroups of HER2+ tumors defined by clinical or tumor biomarkers identified 11 GISTIC regions significantly associated with ER status or DNA ploidy (Figure 4D). Several GISTIC regions (for example, +1p31.3, - 5q14.3, +11q13.3, - 16q23.3) separated ER+ from ER- tumors in HER2+ BC, but their effect was evident also in HER2- tumors (P = 0.06, 5 × 10-23, 0.0002, 0.0003 respectively in HER2- tumors, Bonferroni adjusted t-test). No GISTIC regions separated HER2+ tumors stratified by patient age, LN status, tumor size, or histological grade 1 or 2 vs. 3. Differences in FGA were observed for HER2+ tumors stratified by LN status (P = 0.02, t-test) and DNA ploidy (P = 1 × 10-8), but not by ER status, tumor size, histological grade or patient age.
Of the 200 HER2+ tumors analyzed by BAC aCGH, 87 had concurrent gene expression data and were classified according to the gene expression subtypes [15] (Table 1). Notably, 24% of tumors classified to the ERBB2 subtype were ER-positive. The individually small subtype groups prevented individual pair-wise comparisons with the ERBB2 subtype. However, no significant GISTIC regions, and no significant difference in FGA were found in a pair-wise comparison of the tumors in the ERBB2 subtype (n = 51) vs. tumors in remaining expression subtypes combined (n = 30). Strikingly, 83% of HER2+/ER- cases with available gene expression data were classified into either the basal-like or ERBB2 subtype, while only 30% of HER2+/ER+ tumors were classified to any of the two luminal subtypes.
Associations of histopathological and genomic characteristics with overall survival
LN status, DNA ploidy and tumor size were independent significant variables for OS in 176 patients with primary HER2+ BC, while ER status, patient age and histological grade were not associated with OS (Table 2). HER2+ cases classified to the ERBB2 gene expression subtype have been reported to show a tendency for poorer relapse-free survival compared to HER2+ cases classified to the other subtypes [44]. However, we were not able to verify such a prognostic association using OS as an endpoint in the current study, for either all five subtypes separately, or the ERBB2 subtype vs. remaining four subtypes combined (data not shown).
There was a trend towards a different outcome for patients stratified according to TOP2A status (Figure 5A), and stratification by HER2 copy number estimates showed that patients with the highest HER2 copy number estimates had significantly worse OS compared to tumors with the lowest estimates (Figures 5B and 5C). However, the difference in OS for the latter case is at least partly explained by that cases in the group with the highest HER2 copy numbers were more frequently DNA aneuploid (P = 0.1, Fisher's exact test) and displayed higher FGA values (P = 0.04, t-test) compared to cases in the group with the lowest copy numbers. No association with outcome was seen for the presence of recurrent amplifications or firestorms in patients with primary HER2+ BC. This lack of association remained significant also when stratifying patients with DNA aneuploid or diploid HER2+ tumors for presence of recurrent amplifications or firestorms respectively (data not shown). In contrast, patients with HER2- tumors displaying a firestorm-like amplification pattern in the Jönsson et al. data showed significantly worse OS (log-rank P = 0.0008) supported by multivariate analysis (n = 250, P = 0.002, HR = 2.3, 95% confidence interval (CI) = 1.4 to 3.8). A tendency for worse OS was observed for HER2+ cases with high vs. low FGA (log-rank P = 0.08) especially for HER2+/ER+ tumors (Table 2). The association was however weakened considerably when FGA was stratified for DNA aneuploidy (log-rank P = 0.52 for all tumors and P = 0.65 for HER2+/ER+ tumors), while DNA aneuploidy still added prognostic information when stratified for FGA (log-rank P = 0.02 for all tumors, P = 0.07 for HER2+/ER+ tumors). Low sample numbers hampered the investigation of individual recurrent amplifications, but 10 GISTIC regions showed moderate association to OS (log-rank P < 0.1) when comparing gain or loss vs. normal copy number in HER2+ cases (Figure 5D). Three of these regions (-3p.13, +5q35.2 and +8p12) were also associated with OS in the combined HER2- reference data set (log-rank P = 0.004, 0.002 and 0.003, respectively) adding either independent or near independent prognostic information in multivariate analysis (P = 0.08, 0.007 and 0.06, respectively) (Figure 5E). Of these regions, 8p12 has previously been identified as an indicator of poor breast cancer prognosis [31]. The association with outcome for the 5q35.2 GISTIC region was stronger for HER2-/ER- tumors compared to HER2-/ER+ tumors (log-rank P = 0.006 and 0.07 respectively).

Association of OS with TOP2A-status, HER2 copy number levels, and GISTIC regions in HER2+ and HER2- tumors. (A) OS in primary HER2+ tumors stratified by TOP2A-status. (B) OS in primary HER2+ tumors stratified by the 25th (HER2+ CN low) and 75th percentile (HER2+ CN high) of the mean HER2 segmented log2ratio. (C) OS in primary HER2+ tumors stratified by the 15th (HER2+ CN low) and 85th percentile (HER2+ CN high) of the mean HER2 log2ratio. (D) GISTIC regions showing association with OS (log-rank P < 0.1) in the 176 primary HER2+ tumors. The vertical axis represents -log10(p) for log-rank, univariate and multivariate analysis. Tumor size and LN status were included as covariates in multivariate analyses beside GISTIC regions. Dashed lines indicate P = 0.1, 0.05 and 0.01. GISTIC regions are ordered according to their genomic position. (E) Association to OS for GISTIC regions in D in HER2- tumors. The vertical axis represents -log10(p) for log-rank, univariate and multivariate analysis. Tumor size and LN status were included as covariates in multivariate analyses besides GISTIC regions. Dashed lines indicate P = 0.1, 0.05 and 0.01. GISTIC regions are ordered according to their genomic position.
Correlation of gene expression data with genomic aberrations
Gene expression data were compared with genomic aberrations in 87 HER2+ tumors in order to identify genes affected by gene dosage. First, genes in the 117 GISTIC regions (n = 1,750) were matched to available gene expression data (n = 1,078) and correlated across tumor samples as previously described [25]. This approach identified 460 significantly correlated genes, 284 and 176 in GISTIC regions of gain and loss, respectively (Additional file 5). Second, analysis was re-performed without the restriction to genes in GISTIC regions. Of 10,162 matched genes, 5,853 genes (58%) showed a standard deviation >0.5 in mRNA expression, and 2,242 (38%) of these genes showed significant correlation between expression and copy numbers (P < 0.05 adjusted for multiple testing) (Additional file 11). In summary, correlation of mRNA and copy number identified numerous breast cancer tumor suppressor genes and oncogenes, such as STAT5A, SNIP, ZNF217, TOP2A, BCL2, PAK1, PER3, CCND1, NME1, PPAPDC1B, LSM1, IL17RB and FGFR1, to be significantly correlated in HER2+ tumors. In addition, genes previously reported to be significantly correlated in HER2+/TOP2A co-amplified cases (for example, CASC3, CDC6, RARA and SMARCE1) [20] were also identified in this study (Additional file 11).
Discussion
We have characterized a large set of HER2+ BC in comparison to HER2- BC using a combination of molecular techniques to delineate the HER2-amplicon in high-detail, and to pinpoint, on a genome-wide scale, critical regions of focal amplifications, gains and losses that may be important for tumor development and reflect the heterogeneity of HER2+ BC.
The HER2-amplicon
By using a custom-designed zoom-in aCGH platform we delineated the shortest region of overlapping amplification for the HER2-amplicon to an 85.92 kbp region including six genes. The identified region is considerably smaller than previously reported [19, 20, 45], mainly due to the larger number of tumors and the extreme probe density provided by the zoom-in aCGH platform. While the role of some amplicon genes may be less relevant for breast cancer development, the function of GRB7 is intriguing. GRB7 showed strong correlation between mRNA transcript levels and copy number status in this study, and increased mRNA expression has been shown to correlate with protein overexpression in breast cancer cell lines [46, 47]. As an SH2-containing adapter protein GRB7 can interact with phosphorylated HER2 and mediate aspects of cell migration through binding with focal adhesion kinase [48, 49]. Furthermore, GRB7 has been pinpointed as one of the top-ranked genes in a HER2-derived prognostic gene signature [17], arguing that it is not merely a silent passenger of the amplicon. However, we found no case of focal GRB7 amplification in HER2- cases suggesting that its activation is linked to the selected advantage conferred by HER2 activation.
CEP17 amplification in HER2+ breast cancer
Accurate evaluation of HER2 status is important for identification of patients that would benefit from HER2 targeted therapy. FISH analysis for determination of the ratio of HER2 copy number to chromosomes 17 copy number, represented by a centromeric chromosome 17 FISH probe (HER2/CEP17 ratio), has been suggested as the current golden standard [50–52]. However, concerns have been raised, based on aCGH and MLPA studies, whether CEP17 copy number status accurately reflects true chromosome 17 copy number and polysomy (≥3 copies of the entire chromosome 17) [53–55]. We found that polysomic chromosome 17, using the definition suggested by Marchio et al. [53], was a rare event in HER2+ breast tumors (1.5% of tumors). While CEP17 amplification was a rare event in HER2- tumors, it was observed in a much higher frequency in HER2+ tumors, consistent with recent reports [14]. Thus, our data support the notion that abnormal CEP17 copy numbers more likely stem from CNAs on chromosome 17q, and that CEP17 correction may for certain cases be misleading for interpretation of HER2 status [53, 54].
TOP2A aberrations in HER2+ breast cancer
The variable structure of the HER2 amplicon frequently involves additional genes telomeric of HER2 [20, 56], for instance TOP2A encoding a protein target of anthracyclines [57]. It has become increasingly evident that TOP2A alterations rarely occur in HER2- breast tumors (reviewed in [9]) in line with our finding that no HER2- tumor showed focal amplification of TOP2A. In this study, TOP2A amplification or deletion was observed in 31% and 18% of HER2+ tumors, respectively, concordant with previous reports (reviewed in [9]). Consistent with previous reports [56], we found in tumors analyzed by high-resolution zoom-in aCGH that co-amplification of HER2 and TOP2A was not separated by chromosomal regions with normal or deleted copy numbers, and that discordance between HER2 and TOP2A copy numbers existed in co-amplified cases. The strong correlation between TOP2A copy number and expression level (Figure S1B in Additional file 4, Additional file 11) and the abrupt breaks in the HER2/TOP2A amplicon telomeric of TOP2A in several tumors (for example, Figure 1C), suggest a selective retention of TOP2A activation in the development of some tumors through, for instance, breakage-fusion-bridge cycles [56]. TOP2A alterations clearly have a potential role in tumor progression and treatment response, and have been linked to better disease-free survival for patients with HER2+ disease treated with anthracyclines [11, 37]. However, TOP2A protein expression has been reported to correlate more with cellular proliferation than gene amplification [58], in line with our findings of elevated TOP2A mRNA levels in highly proliferative HER2- basal-like and luminal B tumors for which no focal TOP2A amplification was observed (Figure S1B in Additional file 4). Moreover, it has recently been suggested that alterations in the centromeric region of chromosome 17 is a more powerful predictor of response to anthracycline-based treatment than alterations in either HER2 or TOP2A [14]. Taken together, the finding of a trend of better OS for HER2+/TOP2A+ tumors in the present study is difficult to interpret, as we had no specific treatment information available for patients in this study. Clearly, the complex relationship of individual genes in the 17q12-q21 region, as well as other genomic alterations on chromosome 17q, to breast cancer development and treatment efficacy warrants further investigation.
Recurrent amplifications and firestorm patterns in HER2+ breast cancer
Recurrent high-level amplifications and firestorms were frequent in HER2+ breast tumors as also observed by others [18–20]. In line with previous reports, HER2+ tumors were firmly associated with an amplifier/firestorm-like genomic pattern, as firestorms or recurrent amplifications, excluding the HER2-amplicon, were observed in 70% of all HER2+ tumors [18, 30, 31]. However, only a few recurrent amplifications on chromosome 17q were more common in HER2+ than in HER2- tumors. In contrast to recent reports [30, 31], presence of firestorms or recurrent high-level amplifications were not associated with clinical outcome for patients with HER2+ breast tumors. Instead, DNA aneuploidy was a stronger indicator of poor prognosis, especially for HER2+/ER-positive tumors, in line with previous reports for breast cancer irrespective of HER2 status [59, 60]. Although firestorms were significantly more frequent in DNA aneuploid HER2+ tumors, 25% of DNA diploid HER2+ cases displayed firestorms suggesting that occurrence of gross chromosomal alterations and amplifier patterns may be unrelated mechanisms of genomic instability.
The majority of identified recurrent amplifications in this study have previously been reported in both HER2+ and HER2- breast tumors, although with different frequencies and co-amplification patterns [18, 31, 32, 38]. A few discrepancies exist in comparison to recent aCGH studies on HER2+ tumors, for example, we did not observe recurrent (>2%) high-level amplifications on chromosome 7p, 14q and 18q [18, 20]. Interestingly, while combinations of high-level amplifications of 8p12, 11q13.3 and 20q13.2 were not uncommon in HER2- tumors, they were mutually exclusive in HER2+ tumors. When the threshold for high-level amplification was lowered to that of amplification, co-occurrence of these regions was still rare in HER2+ tumors compared to HER2- tumors (data not shown). Individual amplification of 8p12, 11q13.3, or 20q13.2 was associated with worse OS in HER2- tumors (log-rank P ≤ 0.02), but not in HER2+ tumors. Furthermore, co-amplification of 11q13.3 with 8p12 and 11q13.3 with 20q13.2 remained significantly associated with outcome in HER2- tumors despite fewer cases, while co-amplification of 8p12 with 20q13.2 showed only a trend (log-rank P = 0.10). Presumably, these amplifications activate cellular pathways that drive tumor progression synergistically in HER2- tumors, whereas having more than one of these amplifications provides no advantage in HER2+ tumors.
Significant CNAs in HER2+ breast cancer
The molecular subtypes of breast cancer [15] have been associated with distinct CNAs and genomic patterns [18, 31, 38, 61] that may contribute to their transcriptional profiles and biological phenotypes [62]. The overall pattern of CNAs in HER2+ breast tumors observed in this study corroborates earlier findings [18, 19]. However, a few discordances exist, mainly corresponding to more frequent gain of 16p and less frequent aberrations on chromosome 7 in the current study. Comparison of CNAs in HER2+ tumors to HER2- tumors revealed differences in FGA and frequency of several GISTIC regions (Figure 4, Additional files 5, 7, 8). On the other hand, some of the most recurrent CNAs in HER2+ tumors irrespective of ER status, including +1q, +8q, -8p, and -17p, were also commonly observed in HER2- tumors indicating their importance in breast cancer. Several of the regions discriminating HER2+ from HER2- tumors have previously been reported as specific for basal-like, luminal A and B tumors respectively [31–33, 40, 61, 63, 64]. However, certain discriminatory regions are explained by the ER status of the 200 HER2+ tumors and the gene expression subtypes. For instance, HER2- luminal A and B tumors displayed more frequent loss of 16q than HER2+ tumors as a whole (Figure 4B). However, when HER2+/ER+ tumors were compared to luminal A and B tumors, loss of 16q was no longer significant, reflecting the strong association of specific CNAs with ER status (data not shown). Furthermore, our data, supported by subtype classifications of independent breast cancer data sets, indicate that the ERBB2 subgroup, although dominated by HER2+/ER- tumors, contains a sizeable fraction of HER2+/ER+ tumors.
The finding of GISTIC regions stratifying both HER2+ and HER2- tumors based on ER status is in contrast to a recent smaller aCGH study on HER2+ breast tumors [19]. Interestingly, although HER2+/ER- tumors harbor a pattern of CNAs similar to HER2-/ER- and basal-like tumors, the frequencies of these aberrations are significantly lower in HER2+/ER- tumors as well as ERBB2 subtype classified HER2+ tumors, in agreement with Marchio et al. [19] (Figure 4C, Additional files 5 and 8). HER2+/ER- tumors have been linked by gene expression analysis to an apocrine/steroid response-positive subgroup of ER-negative BC characterized by overexpression of genes related to steroid estrogen response [65, 66]. Moreover, there is increasing support for crosstalk between the HER2 and ER-signaling pathways (reviewed by [67, 68]). Consistent with these observations and recent reports [42, 69], HER2+ tumors in the current study showed intermediate expression of two ER gene expression modules, which were significantly less expressed in HER2- basal-like tumors. Further substantiating the difference between HER2+/ER- and HER2- basal-like tumors we did not find elevated frequencies of CNAs characteristic of HER2- basal-like BC in HER2+ tumors with high correlation to the basal-like gene expression centroid (data not shown).
In summary, HER2+ tumors display a wide range of frequently complex CNAs including firestorms and recurrent amplifications. However, with the exception of a limited number of CNAs primarily located on chromosome 17q, the vast majority of CNAs does not appear specifically associated with HER2+ tumors per se, as revealed when compared to other breast cancer subgroups. These findings underline the genomically complex and heterogeneous nature of HER2+ breast cancer in relation to other subgroups of breast cancer.
Conclusions
We have conducted a comprehensive survey of copy number alterations in HER2+ breast tumors using a combination of aCGH and gene expression analysis, pinpointing significant genomic alterations including both known and potentially novel therapeutic targets. Our analysis sheds further light on the genetically complex and heterogeneous nature of HER2+ tumors in relation to other breast cancer subgroups.
Abbreviations
- aCGH:
-
array-based CGH
- BAC:
-
Bacterial artificial chromosome
- BC:
-
breast cancer
- CBS:
-
circular binary segmentation
- CEP17:
-
chromosome 17 centromere
- CNA:
-
copy number alteration
- ER:
-
estrogen receptor
- FFPE:
-
formalin fixed paraffin embedded
- FGA:
-
fraction of the genome altered
- FISH:
-
fluorescence in situ hybridization
- GISTIC:
-
Genomic Identification of Significant Targets in Cancer
- IHC:
-
immunohistochemistry
- LN:
-
lymph node
- OS:
-
overall survival
- SRO:
-
shortest region of overlap.
References
Jonkers J, Berns A: Oncogene addiction: sometimes a temporary slavery. Cancer Cell. 2004, 6: 535-538.
Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL: Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987, 235: 177-182. 10.1126/science.3798106.
Paik S, Hazan R, Fisher ER, Sass RE, Fisher B, Redmond C, Schlessinger J, Lippman ME, King CR: Pathologic findings from the National Surgical Adjuvant Breast and Bowel Project: prognostic significance of erbB-2 protein overexpression in primary breast cancer. J Clin Oncol. 1990, 8: 103-112.
Smith I, Procter M, Gelber RD, Guillaume S, Feyereislova A, Dowsett M, Goldhirsch A, Untch M, Mariani G, Baselga J, Kaufmann M, Cameron D, Bell R, Bergh J, Coleman R, Wardley A, Harbeck N, Lopez RI, Mallmann P, Gelmon K, Wilcken N, Wist E, Sanchez Rovira P, Piccart-Gebhart MJ: 2-year follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer: a randomised controlled trial. Lancet. 2007, 369: 29-36. 10.1016/S0140-6736(07)60028-2.
Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE, Davidson NE, Tan-Chiu E, Martino S, Paik S, Kaufman PA, Swain SM, Pisansky TM, Fehrenbacher L, Kutteh LA, Vogel VG, Visscher DW, Yothers G, Jenkins RB, Brown AM, Dakhil SR, Mamounas EP, Lingle WL, Klein PM, Ingle JN, Wolmark N: Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005, 353: 1673-1684. 10.1056/NEJMoa052122.
Baselga J, Tripathy D, Mendelsohn J, Baughman S, Benz CC, Dantis L, Sklarin NT, Seidman AD, Hudis CA, Moore J, Rosen PP, Twaddell T, Henderson IC, Norton L: Phase II study of weekly intravenous trastuzumab (Herceptin) in patients with HER2/neu-overexpressing metastatic breast cancer. Semin Oncol. 1999, 26: 78-83.
Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello-Gruszfeld A, Crown J, Chan A, Kaufman B, Skarlos D, Campone M, Davidson N, Berger M, Oliva C, Rubin SD, Stein S, Cameron D: Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006, 355: 2733-2743. 10.1056/NEJMoa064320.
Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, Dowsett M, Fitzgibbons PL, Hanna WM, Langer A, McShane LM, Paik S, Pegram MD, Perez EA, Press MF, Rhodes A, Sturgeon C, Taube SE, Tubbs R, Vance GH, Vijver van de M, Wheeler TM, Hayes DF: American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol. 2007, 25: 118-145. 10.1200/JCO.2006.09.2775.
Slamon DJ, Press MF: Alterations in the TOP2A and HER2 genes: association with adjuvant anthracycline sensitivity in human breast cancers. J Natl Cancer Inst. 2009, 101: 615-618. 10.1093/jnci/djp092.
O'Malley FP, Chia S, Tu D, Shepherd LE, Levine MN, Bramwell VH, Andrulis IL, Pritchard KI: Topoisomerase II alpha and responsiveness of breast cancer to adjuvant chemotherapy. J Natl Cancer Inst. 2009, 101: 644-650. 10.1093/jnci/djp067.
Tanner M, Isola J, Wiklund T, Erikstein B, Kellokumpu-Lehtinen P, Malmstrom P, Wilking N, Nilsson J, Bergh J: Topoisomerase IIalpha gene amplification predicts favorable treatment response to tailored and dose-escalated anthracycline-based adjuvant chemotherapy in HER-2/neu-amplified breast cancer: Scandinavian Breast Group Trial 9401. J Clin Oncol. 2006, 24: 2428-2436. 10.1200/JCO.2005.02.9264.
Knoop AS, Knudsen H, Balslev E, Rasmussen BB, Overgaard J, Nielsen KV, Schonau A, Gunnarsdottir K, Olsen KE, Mouridsen H, Ejlertsen B: retrospective analysis of topoisomerase IIa amplifications and deletions as predictive markers in primary breast cancer patients randomly assigned to cyclophosphamide, methotrexate, and fluorouracil or cyclophosphamide, epirubicin, and fluorouracil: Danish Breast Cancer Cooperative Group. J Clin Oncol. 2005, 23: 7483-7490. 10.1200/JCO.2005.11.007.
Tubbs R, Barlow WE, Budd GT, Swain E, Porter P, Gown A, Yeh IT, Sledge G, Shapiro C, Ingle J, Haskell C, Albain KS, Livingston R, Hayes DF: Outcome of patients with early-stage breast cancer treated with doxorubicin-based adjuvant chemotherapy as a function of HER2 and TOP2A status. J Clin Oncol. 2009, 27: 3881-3886. 10.1200/JCO.2008.20.1566.
Bartlett JM, Munro AF, Dunn JA, McConkey C, Jordan S, Twelves CJ, Cameron DA, Thomas J, Campbell FM, Rea DW, Provenzano E, Caldas C, Pharoah P, Hiller L, Earl H, Poole CJ: Predictive markers of anthracycline benefit: a prospectively planned analysis of the UK National Epirubicin Adjuvant Trial (NEAT/BR9601). Lancet Oncol. 11: 266-274. 10.1016/S1470-2045(10)70006-1.
Perou CM, Sorlie T, Eisen MB, Rijn van de M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, Zhu SX, Lonning PE, Borresen-Dale AL, Brown PO, Botstein D: Molecular portraits of human breast tumours. Nature. 2000, 406: 747-752. 10.1038/35021093.
Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, Demeter J, Perou CM, Lonning PE, Brown PO, Borresen-Dale AL, Botstein D: Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003, 100: 8418-8423. 10.1073/pnas.0932692100.
Staaf J, Ringner M, Vallon-Christersson J, Jonsson G, Bendahl PO, Holm K, Arason A, Gunnarsson H, Hegardt C, Agnarsson BA, Luts L, Grabau D, Ferno M, Malmstrom P, Johannsson O, Loman N, Barkardottir R, Borg A: Identification of subtypes in HER2-positive breast cancer reveals a gene signature prognostic of outcome. J Clin Oncol.
Natrajan R, Lambros MB, Rodriguez-Pinilla SM, Moreno-Bueno G, Tan DS, Marchio C, Vatcheva R, Rayter S, Mahler-Araujo B, Fulford LG, Hungermann D, Mackay A, Grigoriadis A, Fenwick K, Tamber N, Hardisson D, Tutt A, Palacios J, Lord CJ, Buerger H, Ashworth A, Reis-Filho JS: Tiling Path Genomic Profiling of Grade 3 Invasive Ductal Breast Cancers. Clin Cancer Res. 2009, 15: 2711-2722. 10.1158/1078-0432.CCR-08-1878.
Marchio C, Natrajan R, Shiu K, Lambros M, Rodriguez-Pinilla S, Tan D, Lord C, Hungermann D, Fenwick K, Tamber N, Mackay A, Palacios J, Sapino A, Buerger H, Ashworth A, Reis-Filho J: The genomic profile of HER2-amplified breast cancers: the influence of ER status. J Pathol. 2008, 216: 399-407. 10.1002/path.2423.
Arriola E, Marchio C, Tan DS, Drury SC, Lambros MB, Natrajan R, Rodriguez-Pinilla SM, Mackay A, Tamber N, Fenwick K, Jones C, Dowsett M, Ashworth A, Reis-Filho JS: Genomic analysis of the HER2/TOP2A amplicon in breast cancer and breast cancer cell lines. Lab Invest. 2008, 88: 491-503. 10.1038/labinvest.2008.19.
Staaf J, Torngren T, Rambech E, Johansson U, Persson C, Sellberg G, Tellhed L, Nilbert M, Borg A: Detection and precise mapping of germline rearrangements in BRCA1, BRCA2, MSH2, and MLH1 using zoom-in array comparative genomic hybridization (aCGH). Hum Mutat. 2008, 29: 555-564. 10.1002/humu.20678.
Honeth G, Bendahl PO, Ringner M, Saal LH, Gruvberger-Saal SK, Lovgren K, Grabau D, Ferno M, Borg A, Hegardt C: The CD44+/CD24- phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008, 10: R53-10.1186/bcr2108.
SCIBLU Genomics, Lund University, Sweden. [http://www.lth.se/sciblu]
UCSC Genome Browser. [http://genome.ucsc.edu/]
Jonsson G, Staaf J, Olsson E, Heidenblad M, Vallon-Christersson J, Osoegawa K, de Jong P, Oredsson S, Ringner M, Hoglund M, Borg A: High-resolution genomic profiles of breast cancer cell lines assessed by tiling BAC array comparative genomic hybridization. Genes Chromosomes Cancer. 2007, 46: 543-558. 10.1002/gcc.20438.
Staaf J, Jonsson G, Ringner M, Vallon-Christersson J: Normalization of array-CGH data: influence of copy number imbalances. BMC Genomics. 2007, 8: 382-10.1186/1471-2164-8-382.
Venkatraman ES, Olshen AB: A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007, 23: 657-663. 10.1093/bioinformatics/btl646.
Omnibus GE: [http://www.ncbi.nlm.nih.gov/geo/]
Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, Vivanco I, Lee JC, Huang JH, Alexander S, Du J, Kau T, Thomas RK, Shah K, Soto H, Perner S, Prensner J, Debiasi RM, Demichelis F, Hatton C, Rubin MA, Garraway LA, Nelson SF, Liau L, Mischel PS, Cloughesy TF, Meyerson M, Golub TA, Lander ES, Mellinghoff IK, et al: Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci USA. 2007, 104: 20007-20012. 10.1073/pnas.0710052104.
Hicks J, Krasnitz A, Lakshmi B, Navin NE, Riggs M, Leibu E, Esposito D, Alexander J, Troge J, Grubor V, Yoon S, Wigler M, Ye K, Borresen-Dale AL, Naume B, Schlicting E, Norton L, Hagerstrom T, Skoog L, Auer G, Maner S, Lundin P, Zetterberg A: Novel patterns of genome rearrangement and their association with survival in breast cancer. Genome Res. 2006, 16: 1465-1479. 10.1101/gr.5460106.
Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, Lapuk A, Neve RM, Qian Z, Ryder T, Chen F, Feiler H, Tokuyasu T, Kingsley C, Dairkee S, Meng Z, Chew K, Pinkel D, Jain A, Ljung BM, Esserman L, Albertson DG, Waldman FM, Gray JW: Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006, 10: 529-541. 10.1016/j.ccr.2006.10.009.
Fridlyand J, Snijders AM, Ylstra B, Li H, Olshen A, Segraves R, Dairkee S, Tokuyasu T, Ljung BM, Jain AN, McLennan J, Ziegler J, Chin K, Devries S, Feiler H, Gray JW, Waldman F, Pinkel D, Albertson DG: Breast tumor copy number aberration phenotypes and genomic instability. BMC Cancer. 2006, 6: 96-10.1186/1471-2407-6-96.
Adelaide J, Finetti P, Bekhouche I, Repellini L, Geneix J, Sircoulomb F, Charafe-Jauffret E, Cervera N, Desplans J, Parzy D, Schoenmakers E, Viens P, Jacquemier J, Birnbaum D, Bertucci F, Chaffanet M: Integrated profiling of basal and luminal breast cancers. Cancer Res. 2007, 67: 11565-11575. 10.1158/0008-5472.CAN-07-2536.
Gunnarsson R, Staaf J, Jansson M, Ottesen AM, Goransson H, Liljedahl U, Ralfkiaer U, Mansouri M, Buhl AM, Smedby KE, Hjalgrim H, Syvanen AC, Borg A, Isaksson A, Jurlander J, Juliusson G, Rosenquist R: Screening for copy-number alterations and loss of heterozygosity in chronic lymphocytic leukemia-A comparative study of four differently designed, high resolution microarray platforms. Genes Chromosomes Cancer. 2008
The R Project for Statistical Computing. [http://www.r-project.org]
Ivshina AV, George J, Senko O, Mow B, Putti TC, Smeds J, Lindahl T, Pawitan Y, Hall P, Nordgren H, Wong JE, Liu ET, Bergh J, Kuznetsov VA, Miller LD: Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res. 2006, 66: 10292-10301. 10.1158/0008-5472.CAN-05-4414.
Arriola E, Rodriguez-Pinilla SM, Lambros MB, Jones RL, James M, Savage K, Smith IE, Dowsett M, Reis-Filho JS: Topoisomerase II alpha amplification may predict benefit from adjuvant anthracyclines in HER2 positive early breast cancer. Breast Cancer Res Treat. 2007, 106: 181-189. 10.1007/s10549-006-9492-5.
Haverty PM, Fridlyand J, Li L, Getz G, Beroukhim R, Lohr S, Wu TD, Cavet G, Zhang Z, Chant J: High-resolution genomic and expression analyses of copy number alterations in breast tumors. Genes Chromosomes Cancer. 2008, 47: 530-542. 10.1002/gcc.20558.
Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY: miR-21-mediated tumor growth. Oncogene. 2007, 26: 2799-2803. 10.1038/sj.onc.1210083.
Hu X, Stern HM, Ge L, O'Brien C, Haydu L, Honchell CD, Haverty PM, Peters BA, Wu TD, Amler LC, Chant J, Stokoe D, Lackner MR, Cavet G: Genetic alterations and oncogenic pathways associated with breast cancer subtypes. Mol Cancer Res. 2009, 7: 511-522. 10.1158/1541-7786.MCR-08-0107.
Hu Z, Fan C, Oh DS, Marron JS, He X, Qaqish BF, Livasy C, Carey LA, Reynolds E, Dressler L, Nobel A, Parker J, Ewend MG, Sawyer LR, Wu J, Liu Y, Nanda R, Tretiakova M, Ruiz Orrico A, Dreher D, Palazzo JP, Perreard L, Nelson E, Mone M, Hansen H, Mullins M, Quackenbush JF, Ellis MJ, Olopade OI, Bernard PS, et al: The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics. 2006, 7: 96-10.1186/1471-2164-7-96.
Desmedt C, Haibe-Kains B, Wirapati P, Buyse M, Larsimont D, Bontempi G, Delorenzi M, Piccart M, Sotiriou C: Biological processes associated with breast cancer clinical outcome depend on the molecular subtypes. Clin Cancer Res. 2008, 14: 5158-5165. 10.1158/1078-0432.CCR-07-4756.
Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J, Sahin A, Agarwal R, Joy C, Liu W, Stivers D, Baggerly K, Carey M, Lluch A, Monteagudo C, He X, Weigman V, Fan C, Palazzo J, Hortobagyi GN, Nolden LK, Wang NJ, Valero V, Gray JW, Perou CM, Mills GB: Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009, 69: 4116-4124. 10.1158/0008-5472.CAN-08-3441.
Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, Quackenbush JF, Stijleman IJ, Palazzo J, Marron JS, Nobel AB, Mardis E, Nielsen TO, Ellis MJ, Perou CM, Bernard PS: Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009, 27: 1160-1167. 10.1200/JCO.2008.18.1370.
Kauraniemi P, Kuukasjarvi T, Sauter G, Kallioniemi A: Amplification of a 280-kilobase core region at the ERBB2 locus leads to activation of two hypothetical proteins in breast cancer. Am J Pathol. 2003, 163: 1979-1984.
Bai T, Luoh SW: GRB-7 facilitates HER-2/Neu-mediated signal transduction and tumor formation. Carcinogenesis. 2008, 29: 473-479. 10.1093/carcin/bgm221.
Kao J, Pollack JR: RNA interference-based functional dissection of the 17q12 amplicon in breast cancer reveals contribution of coamplified genes. Genes Chromosomes Cancer. 2006, 45: 761-769. 10.1002/gcc.20339.
Han DC, Guan JL: Association of focal adhesion kinase with Grb7 and its role in cell migration. J Biol Chem. 1999, 274: 24425-24430. 10.1074/jbc.274.34.24425.
Janes PW, Lackmann M, Church WB, Sanderson GM, Sutherland RL, Daly RJ: Structural determinants of the interaction between the erbB2 receptor and the Src homology 2 domain of Grb7. J Biol Chem. 1997, 272: 8490-8497. 10.1074/jbc.272.13.8490.
Bempt Vanden I, Van Loo P, Drijkoningen M, Neven P, Smeets A, Christiaens MR, Paridaens R, De Wolf-Peeters C: Polysomy 17 in breast cancer: clinicopathologic significance and impact on HER-2 testing. J Clin Oncol. 2008, 26: 4869-4874. 10.1200/JCO.2007.13.4296.
Downs-Kelly E, Yoder BJ, Stoler M, Tubbs RR, Skacel M, Grogan T, Roche P, Hicks DG: The influence of polysomy 17 on HER2 gene and protein expression in adenocarcinoma of the breast: a fluorescent in situ hybridization, immunohistochemical, and isotopic mRNA in situ hybridization study. Am J Surg Pathol. 2005, 29: 1221-1227. 10.1097/01.pas.0000165528.78945.95.
Dal Lago L, Durbecq V, Desmedt C, Salgado R, Verjat T, Lespagnard L, Ma Y, Veys I, Di Leo A, Sotiriou C, Piccart M, Larsimont D: Correction for chromosome-17 is critical for the determination of true Her-2/neu gene amplification status in breast cancer. Mol Cancer Ther. 2006, 5: 2572-2579. 10.1158/1535-7163.MCT-06-0129.
Marchio C, Lambros MB, Gugliotta P, Di Cantogno LV, Botta C, Pasini B, Tan DS, Mackay A, Fenwick K, Tamber N, Bussolati G, Ashworth A, Reis-Filho JS, Sapino A: Does chromosome 17 centromere copy number predict polysomy in breast cancer? A fluorescence in situ hybridization and microarray-based CGH analysis. J Pathol. 2009, 219: 16-24. 10.1002/path.2574.
Moelans CB, de Weger RA, van Diest PJ: Absence of chromosome 17 polysomy in breast cancer: analysis by CEP17 chromogenic in situ hybridization and multiplex ligation-dependent probe amplification. Breast Cancer Res Treat. 2009
Yeh IT, Martin MA, Robetorye RS, Bolla AR, McCaskill C, Shah RK, Gorre ME, Mohammed MS, Gunn SR: Clinical validation of an array CGH test for HER2 status in breast cancer reveals that polysomy 17 is a rare event. Mod Pathol. 2009, 22: 1169-1175. 10.1038/modpathol.2009.78.
Jacobson KK, Morrison LE, Henderson BT, Blondin BA, Wilber KA, Legator MS, O'Hare A, Van Stedum SC, Proffitt JH, Seelig SA, Coon JS: Gene copy mapping of the ERBB2/TOP2A region in breast cancer. Genes Chromosomes Cancer. 2004, 40: 19-31. 10.1002/gcc.20019.
Wang JC: DNA topoisomerases. Annu Rev Biochem. 1996, 65: 635-692. 10.1146/annurev.bi.65.070196.003223.
Mueller RE, Parkes RK, Andrulis I, O'Malley FP: Amplification of the TOP2A gene does not predict high levels of topoisomerase II alpha protein in human breast tumor samples. Genes Chromosomes Cancer. 2004, 39: 288-297. 10.1002/gcc.20008.
Ewers SB, Attewell R, Baldetorp B, Borg A, Ferno M, Langstrom E, Killander D: Prognostic significance of flow cytometric DNA analysis and estrogen receptor content in breast carcinomas--a 10 year survival study. Breast Cancer Res Treat. 1992, 24: 115-126. 10.1007/BF01961244.
Rennstam K, Ahlstedt-Soini M, Baldetorp B, Bendahl PO, Borg A, Karhu R, Tanner M, Tirkkonen M, Isola J: Patterns of chromosomal imbalances defines subgroups of breast cancer with distinct clinical features and prognosis. A study of 305 tumors by comparative genomic hybridization. Cancer Res. 2003, 63: 8861-8868.
Bergamaschi A, Kim YH, Wang P, Sorlie T, Hernandez-Boussard T, Lonning PE, Tibshirani R, Borresen-Dale AL, Pollack JR: Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes Chromosomes Cancer. 2006, 45: 1033-1040. 10.1002/gcc.20366.
Natrajan R, Weigelt B, Mackay A, Geyer FC, Grigoriadis A, Tan DS, Jones C, Lord CJ, Vatcheva R, Rodriguez-Pinilla SM, Palacios J, Ashworth A, Reis-Filho JS: An integrative genomic and transcriptomic analysis reveals molecular pathways and networks regulated by copy number aberrations in basal-like, HER2 and luminal cancers. Breast Cancer Res Treat. 2010, 121: 575-589. 10.1007/s10549-009-0501-3.
Chin SF, Teschendorff AE, Marioni JC, Wang Y, Barbosa-Morais NL, Thorne NP, Costa JL, Pinder SE, Wiel van de MA, Green AR, Ellis IO, Porter PL, Tavare S, Brenton JD, Ylstra B, Caldas C: High-resolution aCGH and expression profiling identifies a novel genomic subtype of ER negative breast cancer. Genome Biol. 2007, 8: R215-10.1186/gb-2007-8-10-r215.
Melchor L, Honrado E, Huang J, Alvarez S, Naylor TL, Garcia MJ, Osorio A, Blesa D, Stratton MR, Weber BL, Cigudosa JC, Rahman N, Nathanson KL, Benitez J: Estrogen receptor status could modulate the genomic pattern in familial and sporadic breast cancer. Clin Cancer Res. 2007, 13: 7305-7313. 10.1158/1078-0432.CCR-07-0711.
Teschendorff AE, Miremadi A, Pinder SE, Ellis IO, Caldas C: An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol. 2007, 8: R157-10.1186/gb-2007-8-8-r157.
Farmer P, Bonnefoi H, Becette V, Tubiana-Hulin M, Fumoleau P, Larsimont D, Macgrogan G, Bergh J, Cameron D, Goldstein D, Duss S, Nicoulaz AL, Brisken C, Fiche M, Delorenzi M, Iggo R: Identification of molecular apocrine breast tumours by microarray analysis. Oncogene. 2005, 24: 4660-4671. 10.1038/sj.onc.1208561.
Bender LM, Nahta R: Her2 cross talk and therapeutic resistance in breast cancer. Front Biosci. 2008, 13: 3906-3912. 10.2741/2978.
Osborne CK, Shou J, Massarweh S, Schiff R: Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin Cancer Res. 2005, 11: 865s-870s.
Wirapati P, Sotiriou C, Kunkel S, Farmer P, Pradervand S, Haibe-Kains B, Desmedt C, Ignatiadis M, Sengstag T, Schutz F, Goldstein DR, Piccart M, Delorenzi M: Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res. 2008, 10: R65-10.1186/bcr2124.
Acknowledgements
The authors would like to acknowledge Professor Bo Baldetorp at the Department of Oncology, Lund University, Sweden for support with flow cytometry data. Financial support for this study was provided by the Swedish Cancer Society, the Knut & Alice Wallenberg Foundation, the Foundation for Strategic Research through the Lund Centre for Translational Cancer Research (CREATE Health), the Mrs. Berta Kamprad Foundation, the Gunnar Nilsson Cancer Foundation, the Swedish Research Council, the Lund University Hospital Research Funds, the American Cancer Society, the Gustav V:s Jubilee Foundation, the IngaBritt and Arne Lundberg Foundation, The Icelandic Centre for Research, the Research Fund of Landspitali-University Hospital, and a donation in memory of Marianne Nygren. The SCIBLU Genomics Center is supported by governmental funding of clinical research within the national health services (ALF) and by Lund University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
NL has received honoraria from Roche and AstraZeneca. The other authors declare that they have no competing interests.
Authors' contributions
ÅB and JS conceived of the study. JS, GJ, AA and HG performed array CGH experiments. DG performed FISH and IHC experiments. JS performed data analysis with support by MR and GJ. JS wrote the manuscript with the assistance of GJ, MR, JVC and ÅB. BA, NL, OJ, PM, RB and DG contributed samples and clinical information. All authors approved the final manuscript.
Electronic supplementary material
13058_2009_2532_MOESM1_ESM.XLS
Additional file 1: Patient and tumor characteristics for HER2 -amplified tumors. An Excel table containing clinical and experimental data on the 200 HER2-amplified tumors. (XLS 77 KB)
13058_2009_2532_MOESM2_ESM.DOC
Additional file 2: Supplementary methods. A Word document containing supplementary information about used methods and data processing. (DOC 33 KB)
13058_2009_2532_MOESM3_ESM.DOC
Additional file 3: Patient and tumor characteristics for HER2-negative tumors. A Word document containing a table of clinical data for HER2- tumors in the reference breast cancer data set. (DOC 50 KB)
13058_2009_2532_MOESM4_ESM.PDF
Additional file 4: HER2 copy number evaluation and TOP2A mRNA expression levels. A pdf file containing figures of the result of the comparison of HER2 copy number estimates between aCGH and FISH for 13 FFPE samples (S1A) and mRNA expression levels and S-phase fractions for HER2+ and HER2- tumors in the Jönsson et al. data set (S1B). (PDF 248 KB)
13058_2009_2532_MOESM5_ESM.XLS
Additional file 5: Significant GISTIC regions in HER2-amplified breast cancer. An Excel table presenting the 117 GISTIC regions, including genes significantly correlated between mRNA expression levels and copy numbers, and frequency of high-level amplifications on chromosome 17q. Additionally, the frequency of GISTIC regions in HER2+ tumors overall and stratified by ER status is presented and compared to corresponding frequencies in HER2- tumors stratified by ER status. (XLS 137 KB)
13058_2009_2532_MOESM6_ESM.DOC
Additional file 6: Recurrent high-level amplifications in HER2-amplified breast cancer. A Word file describing recurrent high-level amplifications, excluding chromosome 17, in HER2+ breast tumors including genes in amplicons significantly correlated between mRNA expression levels and copy numbers. (DOC 54 KB)
13058_2009_2532_MOESM7_ESM.XLS
Additional file 7: Frequency of GISTIC regions in HER2 -amplified and HER2 -negative breast cancer. An Excel table containing the results from the comparison of frequencies of the identified GISTIC regions in HER2-amplified breast cancer compared to HER2- tumors classified according to gene expression subtypes. (XLS 40 KB)
13058_2009_2532_MOESM8_ESM.PDF
Additional file 8: Expression of ER gene expression modules in HER2 -amplified and HER2 -negative breast cancer. A pdf file containing two subpanels illustrating: (1) differences in expression of two ER gene expression modules in the Jönsson et al. data set for HER2+ tumors stratified according to ER status, and (2) HER2- tumors classified according to gene expression subtypes. (PDF 271 KB)
13058_2009_2532_MOESM9_ESM.PDF
Additional file 9: Frequency of GISTIC regions in HER2-amplified and HER2-negative breast cancer according to gene expression subtypes. A pdf file containing two panels illustrating: (1) GISTIC regions significantly different between HER2+ tumors classified to the ERBB2 gene expression subtype, compared to HER2- tumors classified as basal-like, luminal A, luminal B and normal-like subtype, and (2) CNA frequency in HER2- basal-like classified tumors. (PDF 205 KB)
13058_2009_2532_MOESM10_ESM.PDF
Additional file 10: Copy number alterations in HER2-amplified breast cancer stratified by ER status. A pdf file containing two subpanels illustrating CNA frequencies in HER2+/ER- tumors and HER2+/ER+ tumors, respectively. (PDF 328 KB)
13058_2009_2532_MOESM11_ESM.XLS
Additional file 11: Global correlation of mRNA expression and gene dosage in HER2-amplified breast cancer. An Excel table listing genes that are found significantly correlated between gene expression data and aCGH in HER2+ tumors using a global matching. (XLS 361 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Staaf, J., Jönsson, G., Ringnér, M. et al. High-resolution genomic and expression analyses of copy number alterations in HER2-amplified breast cancer. Breast Cancer Res 12, R25 (2010). https://doi.org/10.1186/bcr2568
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/bcr2568