
Abstract
MicroRNAs (miRNAs) are a major class of small endogenous RNA molecules that post-transcriptionally inhibit gene expression. Many miRNAs have been implicated in several human cancers, including breast cancer. Here we describe the association between altered miRNA signatures and breast cancer tumorigenesis and metastasis. The loss of several tumor suppressor miRNAs (miR-206, miR-17-5p, miR-125a, miR-125b, miR-200, let-7, miR-34 and miR-31) and the overexpression of certain oncogenic miRNAs (miR-21, miR-155, miR-10b, miR-373 and miR-520c) have been observed in many breast cancers. The gene networks orchestrated by these miRNAs are still largely unknown, although key targets have been identified that may contribute to the disease phenotype. Here we report how the observed perturbations in miRNA expression profiles may lead to disruption of key pathways involved in breast cancer.
Similar content being viewed by others


Introduction
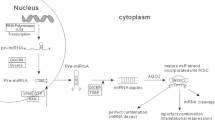
MicroRNAs (miRNAs) are an evolutionarily conserved class of small, approximately 22-nucleotide non-coding RNAs that decrease gene expression post-transcriptionally in a sequence-specific manner. Most miRNAs are transcribed in the nucleus by RNA polymerase II (although some miRNAs are also transcribed by RNA polymerase III [1]) as long primary transcripts (primiRNAs) that undergo processing by Drosha and DGCR8, resulting in an approximately 70-nucleotide stem-loop RNA (pre-miRNA). Pre-miRNAs are subsequently exported to the cytoplasm via Exportin 5 and cleaved by Dicer, giving rise to approximately 22-nucleotide RNA duplexes. The strand with decreased base-pairing at its 5' end is then selected to function as a mature miRNA, while the other strand (also referred to as the passenger strand) is typically degraded [2, 3]. The mature miRNA associates with Argonautes and other proteins to form the RNA-induced silencing complex (miRISC), which then binds to target mRNAs via partial complementarity. Many transcription factors that regulate mRNA transcription also control miRNA biogenesis. Although transcription plays a major role in miRNA biogenesis, additional mechanisms, such as DNA methylation, can also regulate miRNA expression [4]. Mammalian miRNAs predominantly act by binding to the 3' untranslated region (UTR) of cognate mRNAs. However, there is growing evidence that they can also downregulate the expression of some genes by base-pairing to the coding region [5–8] or the 5' UTR of some mRNAs [9].
Initially, miRNAs were thought to function mainly by suppressing mRNA translation [10]. However, two recent studies combined proteomics and microarrays to reveal that changes in protein expression mediated by a miRNA are usually associated with altered mRNA expression, suggesting that mRNA degradation may be the major component of mammalian miRNA repression [11, 12]. Although most studies suggest that miRNAs negatively regulate gene expression by base-pairing to the 3' UTR, a few recent examples have shown that miRNAs can also upregulate the translation of their target mRNAs [13].
According to miRBase release 14 (September 2009), more than 700 human miRNAs have been identified [14]. Regardless of the mechanism by which they regulate gene expression, each miRNA can potentially regulate the expression of hundreds of genes and a single transcript can be targeted by multiple miRNAs to concurrently downregulate multiple proteins in the same pathway. In fact, almost one-third of the protein-coding genes are susceptible to miRNA regulation [15] and, as a consequence, many miRNAs seem to play pivotal roles in important biological processes, including cellular proliferation, differentiation and apoptosis [16]. Not surprisingly, aberrant miRNA expression is a hallmark of several diseases, cancer in particular [17, 18]. In this review we will focus on recent advances on the functions of miRNAs in breast tumor development and metastasis. Finally, based on reported targets of specific breast cancer-associated miRNAs, we have built a direct gene interaction network to illustrate how the targets of these miRNAs interact with each other via protein-protein or protein-DNA interactions. This approach reveals that many genes involved in breast cancer are embedded in a miRNA network that controls their expression leading to breast cancer pathogenesis.
MicroRNAs as critical regulators of tumorigenesis
In normal cells, miRNAs control normal rates of cellular growth, proliferation, differentiation and apoptosis. Since miRNAs inhibit cell cycle progression and drive terminal differentiation, downregulation of some miRNAs may play an important role in the development or progression of cancer [19]. In this context, at least three observations early in the history of miRNAs suggested their potential role in cancer. First, the earliest miRNAs discovered in Caenorhabditis elegans and Drosophila were shown to regulate cellular proliferation and apoptosis [20, 21], suggesting that deregulation of these miRNAs may lead to proliferative diseases. Second, many miRNA loci frequently map to genomic regions that are commonly amplified or deleted in various human cancers [19, 22–25]. Third, tumor cell lines and malignant tumors were found to have widespread deregulation of miRNA expression [23, 26, 27]. Furthermore, expression of some miRNAs correlated with diverse clinicopathological parameters [23, 28, 29] and prognosis [30, 31]. Taken together, these findings highlighted a potential role of miRNAs as new diagnostic or prognostic biomarkers. Moreover, the evidence that specific miRNAs have tumor suppressor and oncogenic functions makes them novel targets for cancer therapy.
A global decrease in miRNA expression has been observed in human cancers, suggesting that most miRNAs may act as tumor suppressors [27]. Furthermore, poorly differentiated tumors have lower miRNA levels compared to more differentiated tumors, indicating that global changes in miRNA expression may indicate the degree of cellular differentiation. In support of this hypothesis, a recent study showed that most miRNAs were expressed at lower levels in human tumor-derived cell lines compared to the corresponding tissue [26]. However, it remained unclear if the reduced miRNA expression was the cause or the consequence of malignant transformation. Remarkably, another study demonstrated that the global loss in miRNA expression in cancer promoted tumorigenesis [32]. Global loss of miRNA expression achieved by knockdown of Drosha and Dicer, two proteins essential for miRNA biogenesis, enhanced cellular growth in vitro. When these cells were injected into nude mice, they formed faster-growing and more invasive tumors. Taken together, these results suggest that genome-wide loss of miRNAs enhances tumorigenesis. However, it remains to be seen if the loss of all miRNAs or only a subgroup of key tumor suppressor miRNAs is the key event that promotes tumorigenesis.
Roles of microRNAs in breast cancer
Over the past few years, miRNA profiling studies have led to the identification of miRNAs that are aberrantly expressed in human breast cancer. The function of only a handful of these miRNAs in breast cancer has been investigated. As in other cancers, some miRNAs can function as tumor suppressors and other miRNAs as oncogenes. Thus, tumor formation may arise from a reduction or deletion of a tumor suppressor miRNA and/or amplification or overexpression of an oncogenic miRNA. In addition, tumor metastasis may be promoted by enhanced expression of prometastatic and/or downregulation of antimetastatic miRNAs. The functions of these miRNAs in breast tumor progression and metastasis are discussed below.
Tumor suppressor miRNAs
In this section we discuss recent findings on seven well-known miRNAs that inhibit breast tumor formation and/or metastasis (Table 1).
miR-206
A potential role of miR-206 in suppressing breast cancer was first identified by employing miRNA microarrays to compare miRNA expression between normal and breast cancer tissues [28]. miR-206 was upregulated in estrogen receptor (ER)α-negative breast cancers, suggesting a role of miR-206 in regulation of the estrogen receptor gene ERα (ESR1). Indeed, miR-206 was recently shown to inhibit the expression of ESR1 mRNA through two binding sites in the ESR1 3' UTR [33]. The latter study also showed that miR-206 expression was strongly repressed by ERα agonists, but not by an ERβ agonist or progesterone, suggesting the existence of a feedback loop. Another study showed that miR-206 expression decreased in ERα-positive human breast cancer tissues and that miR-206 suppresses ESR1 expression and inhibits growth of MCF7 breast cancer cells. In addition to miR-206, the authors found that ESR1 mRNA was a direct target of miR-18a, miR-18b, miR-193b and miR-302c in breast cancer cells [34]. Moreover, miR-18a, miR-18b, miR-193b, miR-206 and miR-302c were also shown to induce cell cycle arrest and to inhibit estrogen-induced proliferation, with inhibition levels comparable to those achieved by ERα small interfering RNA (siRNA). Since dysregulation of ERα expression is a hallmark of most human breast cancers, these studies provide important insights into the molecular mechanisms that lead to breast cancer pathogenesis. The tumor suppressive role for miR-206 in breast cancer was further substantiated by the recent demonstration that miR-206, miR-335 and miR-126 are potently downregulated in metastatic breast cancer cells compared to parental cells [35]. Importantly, restoring the expression of these three miRNAs reduced their invasive capacity. The authors identified four direct targets of miR-335, PTPRN2, MERTK, TNC and SOX4; among them, TNC (encoding an extracellular matrix component) and SOX4 (encoding a transcription factor that is involved in tumorigenesis) were shown to be functional targets implicated in metastasis. Restoring miR-206 expression in metastatic cells did not influence their proliferation or sensitivity to apoptosis, but altered cellular morphology, possibly contributing to a decrease in cell motility that could limit the migration of metastatic cells [35]. These findings suggest that miR-206 could be a novel candidate for breast cancer therapy.
miR-17-5p
miR-17-5p, also known as miR-91, is located on chromosome 13q31, a genomic region that undergoes loss of heterozygosity in multiple cancers, including breast cancer [36]. The oncogene AIB1 (amplified in breast cancer) is a direct target of miR-17-5p [37]. The protein encoded by the AIB1 gene is a steroid receptor co-activator that enhances the transcriptional activity of ERα, E2F1 (which is also directly regulated by miR-17-5p) and other transcription factor genes. miR-17-5p represses the translation of AIB1 mRNA, thereby inhibiting the function of E2F1 and ERα. Downregulation of AIB1 by miR-17-5p results in the suppression of estrogen-stimulated proliferation and estrogen/ER-independent breast cancer cell proliferation [37]. In breast cancer cells, the gene cyclin D1 (CCND1), which is overexpressed in approximately 50% of human cancers, was recently identified as a direct target of miR-17-5p [38]. miR-17-5p inhibits the proliferation of breast cancer cells by suppressing cyclin D1 protein synthesis and this effect was abrogated by a CCND1 siRNA in cyclin D1-deficient breast cancer cells. This study also identified a regulatory mechanism in which cyclin D1 induces miR-17-5p expression, thus providing a negative feedback loop that limits cyclin D1 expression.
miR-125a and miR-125b
The HER2 (ERBB2) gene encodes a member of the epidermal growth factor receptor family of tyrosine kinases. This protein has no ligand-binding domain of its own and cannot, therefore, bind growth factors. However, it binds tightly to other ligand-bound epidermal growth factor receptor family members to form a heterodimer, stabilizing ligand-binding activity and enhancing kinase-mediated activation of downstream signaling pathways, such as those involving mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3 kinase [39]. Amplification and/or over expression of this gene has been reported in numerous cancers, including breast tumors. miR-125a and miR-125b are downregulated in HER2-amplified and HER2-overexpressing breast cancers [40]. These two miRNAs are potential tumor suppressors and their overexpression in SKBR3 cells (a HER2-dependent human breast cancer cell line) suppresses HER2 and HER3 mRNA and protein levels, leading to a reduction in anchorage-dependent growth, cell motility, and invasiveness [41]. However, this influence was subtle in non-transformed HER2-independent breast cancer cells (MCF10A).
miR-200 family
Invasion and metastasis are the hallmarks of malignant tumor progression. Increasing evidence indicates that activation of the embryonic program 'epithelial-mesenchymal transition' (EMT) promotes these processes by allowing detachment of cells from each other, thereby increasing tumor cell mobility and dissemination [42]. In recent years, it has become evident that aberrant activation of EMT is responsible for the malignant transformation of many human cancers. EMT is activated by EMT-inducing transcriptional repressors, including members of the Snail family and the ZFH family of transcription factors [43]. These proteins inhibit the transcription of epithelial genes, such as that encoding E-cadherin. Recently, ZEB1, a ZFH-family member, was shown to be a crucial EMT activator in human cancers, including breast cancer [44, 45], and to promote metastasis of tumor cells in a mouse xenograft model [46]. The miR-200 family of miRNAs has been shown to be crucial inducers of an epithelial phenotype by suppressing the expression of the EMT inducers ZEB1 and ZEB2. Gregory et al. [47] induced EMT in MDCK cells (human Madin Darby canine kidney epithelial cells) either by transforming growth factor (TGF)-β or by stable expression of the tyrosine phosphatase Pez. The authors found that all five members of the miR-200 family, which are clustered in two genomic locations, were downregulated upon TGF-β-induced EMT. Functional studies showed that inhibition of miR-200a, b, and c in MDCK cells triggered TGF-β-independent EMT that was associated with increased expression of vimentin, fibronectin and N-cadherin, and downregulation of E-cadherin. Conversely, overexpression of miR-200 family members in mesenchymal cells initiated mesenchymal-to-epithelial transition. Luciferase reporter assays under the control of ZEB1 and ZEB2 3' UTR verified that these genes are direct targets of the miR-200 family. The clinical relevance of these findings was further suggested by additional analyses of human breast cancer tissues showing that a decrease in miRNA-200 family miRNAs was associated with highly aggressive, metaplastic breast tumors. Their finding that ZEB1 and ZEB2 expression is controlled by the miR-200 family suggests that downregulation of these miRNAs is an essential early step in tumor metastasis.
Similar overlapping results were obtained in another study, which employed the NCI-60 panel of cell lines, representing 60 cell lines of different human tumor types [48]. By using two markers, the authors divided the panel into two major groups, characterized either by an epithelial phenotype (marked by E-cadherin) or a mesenchymal phenotype (marked by vimentin) and found that the miR-200 family was strongly associated with the epithelial phenotype. Overexpression of miR-200 induced epithelial differentiation in undifferentiated breast cancer cells (MDA-MB231) whereas antagonizing it induced an EMT phenotype in the colorectal cancer cell line HCT116. ZEB1 and ZEB2 were again detected as targets of the miR-200 family members. These and other recent reports on the miR-200 family [49, 50] add important miRNAs to the growing list of tumor-associated miRNAs. Furthermore, a recent study showed that the expression of the miR-200 family was decreased by Akt2, suggesting that, in many cases, breast cancer metastasis may be under the control of the Akt-miR-200-E-cadherin pathway [51]. Interestingly, a recent study showed that expression of miR-200 unexpectedly enhanced macroscopic metastases in mouse breast cancer cell lines [52]. Their results suggest that, for some tumors, tumor colonization at metastatic sites might be enhanced by mesenchymal-to-epithelial transition and that the epithelial nature of a tumor may not predict metastatic outcome. Although this paper is the first to show the direct enhancement of metastasis by the miR-200 family, changes in miR-200 family levels have been associated with enhanced tumorigenesis. For instance, the miR-200 family is upregulated in human ovarian cancers compared to normal ovarian tissue [29]. Moreover, overexpression of the miR-200 family significantly correlates with decreased survival. Patients with ovarian tumors with high miR-200a expression have been shown to have approximately 50% decrease in median survival time compared to those lacking significant miR-200a expression. Furthermore, the region on chromosome 1 that encodes the miR-200 cluster was also found to be amplified in several epithelial cancers - ovarian cancer, breast cancer and melanoma [53]. A new study [54] revealed the roles of three miRNA clusters (miR-200c-141, miR-200b-200a-429 and miR-183-96-182) in the regulation of self-renewal in cancer stem cells and normal stem cells. These miRNAs were downregulated in human breast cancer stem cells, normal human and mouse mammary stem/progenitor cells as well as embryonal carcinoma cells. The authors provided evidence that BMI1 (a gene that promotes stem cell self-renewal) is specifically inhibited by these miRNAs and showed that ectopic overexpression of miR-200c in embryonal carcinoma cells resulted in growth retardation and neural differentiation and suppressed tumorigenicity of breast cancer stem cells in vivo [55]. Taken together, these studies suggest that miRNAs of the miR-200 family play important roles in regulating tumor progression and metastasis.
let-7 family
let-7, one of the founding members of the miRNA family, was originally discovered to play a role in the developmental timing of C. elegans [56] and is conserved throughout the animal phyla. let-7 is poorly expressed or deleted in many human cancers. Recent data from both hematologic malignancies and solid tumors suggest that each includes minor populations of cells that are capable of tumor initiation [57]. These tumor-initiating cells (T-ICs) have properties of tumor stem cells and can undergo asymmetric cell division and self renewal. T-ICs comprise only a minor fraction among the bulk of the more differentiated cells in the tumor. Based on the cancer stem cell hypothesis, T-ICs are responsible for the initiation, progression, metastasis and resistance to therapy. Breast T-ICs (BT-ICs) can be enriched by purifying CD44+CD24-/low cells by sorting or by purifying spherical clusters of self-replicating cells (mammospheres) from cell suspensions.
A recent study compared miRNA expression in self-renewinzand differentiated cells from breast cancer lines and found that the expression of let-7 was strongly reduced in BT-ICs and increased with differentiation [58]. Introducing let-7 in BT-ICs reduced their proliferative capacity, their ability to form mammospheres and tumor formation and metastasis in vivo. Conversely, knocking down let-7 enhanced self-renewal of non-T-ICs in vitro. Known oncogenic targets of let-7, such as H-RAS and HMGA2, were downregulated by let-7 overexpression. Silencing H-RAS in a BT-IC-enriched cell line reduced self-renewal but had no effect on differentiation, while knocking down HMGA2 enhanced differentiation but did not affect self-renewal. Their results suggest that let-7 regulates multiple BT-IC stem cell-like properties and that let-7 may offer a unique opportunity to attack tumor stem cells using therapeutic RNA. Delivery of the let-7 miRNA to tumors could potentially deplete stem cells by inducing cellular differentiation.
A recent report linked the protein RKIP (Raf kinase inhibitory protein) to let-7 and breast cancer metastasis. RKIP (also called PEBP1) inhibits MAPK, G protein-coupled receptor kinase-2, and NF-κB signaling cascades [59]. The authors found that RKIP inhibited invasion by metastatic breast cancer cells and repressed breast tumor cell intravasation and bone metastasis in a mouse model. Inhibition of MAPK decreased transcription of LIN28 by MYC; in turn, downregulation of LIN28, an inhibitor of let-7 biogenesis, enhanced let-7 expression in breast cancer cells leading to decreased expression of HMGA2, a chromatin-remodeling protein that activates proinvasive and prometastatic genes, including Snail. Collectively, their findings indicate that RKIP suppresses invasion and metastasis via a signaling cascade that involves MAPK, MYC, LIN28, let-7, and downstream let-7 targets such as HMGA2.
miR-34a
miR-34a is one of several miRNAs that are downregulated in multiple cancers [26] and has been shown to be transcriptionally regulated by p53. In the context of breast cancer, only one study [60] has shown that miR-34a levels were lower in triple negative and mesenchymal breast cancer cell lines compared with normal epithelial lines and HER-2+ lines. The authors propose that p53 mutations in these subtypes of breast cancer might contribute to low miR-34a expression. To elucidate the relationship between miR-34a and radiosensitivity in these cells, the authors compared the sensitivity of a normal breast epithelial line (HMEC), a HER-2+ line (UACC812) and a mesenchymal line (MDA-MB-231) and found that the MDA-MB-231 cell line (with low miR-34a levels) was significantly more sensitive to radiation than the HMECs or UACC-812 (with high miR-34a levels). Increasing the levels of miR-34a protected the MDA-MB-231 cells from radiation-induced cell death, and downregulating it had the converse effect. These results show that miR-34a is necessary for the survival of MDA-MB-231 cells from non-apoptotic cell death and suggest that miR-34a may have therapeutic potential in breast cancers, since antagonizing miR-34a increases the sensitivity of breast cancer cells towards radiation.
miR-31
miR-31 was recently shown to prevent metastasis at multiple steps by inhibiting the expression of prometastatic genes [61]. miR-31 is expressed in normal breast cells and its abundance was shown to be dependent on the metastatic state of the tumor. It is moderately decreased in non-metastatic breast cancer cell lines and is almost undetectable in metastatic mouse and human breast cancer cell lines. Importantly, the authors demonstrated that introducing miR-31 in metastatic breast cancer cells suppressed metastasis-related functions (motility, invasion and resistance to anoikis) in vitro and metastasis in vivo. Notably, they found that although miR-31 over-expressing breast cancer cells formed larger and more proliferative tumors, these tumors were well encapsulated and thereby less invasive, suggesting that miR-31 inhibits metastasis in the early stages of the metastasis cascade. Injecting miR-31 overexpressing cells directly into the circulation impeded the ability of the cells to survive and form secondary tumors in the lung, suggesting that miR-31 inhibits metastasis at multiple steps of the metastatic cascade. Conversely, inhibition of miR-31 function increased invasiveness and promoted metastasis in vivo. To identify functionally relevant mRNA targets of miR-31 through which it might exert its antimetastatic effects, the authors employed Gene Ontology analysis on the putative targets of miR-31 predicted by the miRNA-target prediction algorithms TargetScan and PicTar. Using this approach, they validated six targets of miR-31 in human breast cancer cells: frizzled3 (Fzd3), integrin α-5 (ITGA5), myosin phosphatase-Rho-interacting protein (M-RIP), matrix metallopeptidase 16 (MMP16), radixin (RDX) and the ras homolog gene family member A (RhoA). Interestingly, they showed that re-expression of three of these genes, ITGA5, RDX and RhoA, in metastatic breast cancer cells abrogated the motility defects and reversed the impaired invasion and anoikis resistance defects conferred by miR-31, suggesting that these three genes are functionally important targets of miR-31. Taken together, their findings demonstrate that miR-31 may also be an attractive therapeutic target for breast cancer as it exerts its antimetastatic effect by targeting multiple prometastatic genes of the metastasis cascade.
Oncogenic miRNAs
The function of four well-established oncogenic and/or metastasis promoting miRNAs (Table 1) are discussed below.
miR-21
miR-21 has been found to be consistently overexpressed in many tumors, including breast cancer [28, 62]. Consistent with these findings, miR-21 was also shown to be highly upregulated in breast tumors compared to the matched normal breast tissues among 157 human miRNAs analyzed by real-time RT-PCR arrays [63], suggesting that miR-21 may function as an oncogene. The latter study also examined the oncogenic role of miR-21 in cell culture as well as in mouse xenografts. Although the influence of knocking down miR-21 on cell survival was marginal (25% decrease), the authors found that this growth inhibition increased when the transfected MCF-7 cells were treated with the anticancer drug topotecan, suggesting that suppression of miR-21 can sensitize tumor cells to anticancer agents. To further investigate whether suppression of miR-21 alone influences tumorigenesis, they antagonized miR-21 in MCF-7 cells and then injected them into mammary pads of female nude mice. Tumors derived from MCF-7 cells transfected with anti-miR-21 were one-half the size of those derived from the cells transfected with the negative control, while anti-Ki-67 indicated that the reduced tumor growth was likely due to a lower proliferation caused by anti-miR-21. These results show that miR-21 is an oncogenic miRNA that plays an important role in tumorigenesis. The authors also found that antagonizing miR-21 caused apoptosis in MCF-7 cells, which was associated with lower expression of Bcl-2 protein in the anti-miR-21-transfected MCF-7 cells and also in tumors derived from these cells, suggesting that Bcl-2 may be an indirect target of miR-21. To further elucidate the molecular mechanisms that regulate the function of miR-21, proteomic analysis of the above-mentioned xenograft tumors revealed that the tumor suppressor protein tropomyosin 1 (TPM1), which is known to be downregulated in breast cancer epithelial cell lines, was a target of miR-21 [64]. In the context of breast cancer, miR-21 has also been shown to inhibit the expression of the tumor suppressor PDCD4 (programmed cell death-4). Although depletion of PDCD4 protein in MCF-7 cells by PDCD4 siRNA had no effect on cellular proliferation, it significantly alleviated the anti-proliferative effect of miR-21 inhibition, underscoring an essential role for PDCD4 as a mediator of the biological effects of miR-21 in breast cancer cells [65]. Another important target of miR-21 is the tumor suppressor gene phosphatase and tensin homolog (PTEN) [66]. In addition to TPM1 and PDCD4, one study also identified Maspin as a direct target of miR-21 [67]. These three genes were shown to reduce invasiveness of a metastatic breast cancer cell line [64]. Their findings further establish miR-21 as an oncogenic miRNA and suggest that miR-21 has a role not only in tumor growth but also in invasion and tumor metastasis by targeting multiple anti-metastatic genes.
miR-155
miR-155 is over-expressed in a number of human malignancies, including breast cancer [28, 62]. A recent study has shown that miR-155 is upregulated in normal mouse mammary gland epithelial cells (NMuMG cells) by the TGF-β/Smad4 pathway and mediates TGF-β-induced EMT and cell invasion [68]. To investigate the role of miR-155 in TGF-β-induced EMT, the authors found that ectopic expression of miR-155 in NMuMG cells disrupted proper tight junction formation and promoted cell migration and invasion. Conversely, antagonizing miR-155 in NMuMG cells reduced the occurrence of TGF-β-induced EMT and cell migration and invasion. miR-155 directly inhibited the expression of RhoA, a gene that regulates many cellular processes, including cell adhesion, motility, and polarity, and is an important modulator of cell junction formation and stability. Importantly, miR-155-induced phenotypes were restored by expressing a miR-155 insensitive version of RhoA in miR-155 overexpressing cells. These findings suggest that miR-155 is regulated by the TGF-β/Smad4 pathway and downregulates RhoA protein expression to drive EMT progression. They further demonstrated that miR-155 is associated with cancer invasiveness in human primary breast carcinoma by showing that miR-155 is highly expressed in invasive tumors but not in noninvasive cancer tissues. These results implicate miR-155 in EMT and invasion as observed in NMuMG cells and suggest that miR-155 may play a critical role in breast cancer metastasis.
miR-10b
miR-10b was the first miRNA found to influence the metastatic potential of human cancer cells [69]. Unlike miR-155, which is overexpressed in many breast tumors, miR-10b was highly expressed only in metastatic cancer cells and was found to promote cell migration and invasion in vitro and initiate tumor invasion and metastasis in vivo. Expression of miR-10b was found to be induced by the transcription factor Twist; in turn, miR-10b inhibits the translation of the transcription factor homeobox D10 (HOXD10), resulting in a cascade of cellular alterations that include expression of the prometastatic gene RHOC (ras homologue gene family member C), a gene that promotes cancer cell migration and invasion.
miR-373/520c family
miRNAs of the miR-373/520c family were identified [60] as prometastatic using a forward genetic screen involving overexpression of almost 450 miRNAs in a nonmetastatic human breast cancer cell line (MCF-7 cells). MCF-7 cells were transduced with these miRNAs and subjected to a trans-well cell migration assay to identify miRNAs that stimulate cell migration. The authors found that miR-373 and miR-520c promoted cancer cell migration and invasion in vitro and in vivo. They also showed that some cancer cell lines required miR-373 for migration and that these miRNAs elicited a migratory phenotype by inhibiting expression of the metastasis repressor CD44. Importantly, ectopic overexpression of CD44 without its 3' UTR significantly reduced the migration of MCF-7 cells that express miR-373 or miR-520c, suggesting that downregulation of CD44 is required for the migration phenotype of MCF-7 cells expressing miR-373 and miR-520c. Finally, the authors showed that miR-373 was upregulated whereas CD44 was decreased in clinical breast cancer metastasis samples, further implicating its role in breast cancer metastasis.
Gene interaction network for breast cancer miRNA targets
The above-mentioned investigations on miRNAs implicated in breast cancers substantially advance our knowledge about the roles of specific miRNAs in breast cancer development and metastasis. It is clear that each of these miRNAs inhibit the expression of many genes, suggesting that comprehensive regulation can be achieved by antagonizing or over-expressing a single miRNA. Moreover, concomitant deregulation of these miRNAs would consequently alter the expression of many genes, thereby inducing tumorigenesis.
To better understand how the functions of these experimentally validated breast cancer miRNAs and their gene targets might be integrated within the pathogenesis of breast cancer, we have performed a gene interaction network analysis (Figure 1) similar to our previous study [70]. To do this, we generated a list of 34 genes known to be altered by the 11 miRNAs discussed above. Among these genes, 19 formed a well-connected gene interaction network in which MYC, a target of miR-34a, was the central node (10 connections). Other highly interacting genes were BCL-2, E2F1, CCND1 and ESR1, with seven, seven, seven and six interactions, respectively.

Gene interaction network analysis of miRNA targets in breast cancer. Determined using Ingenuity software, the direct interaction network of 34 published targets of 11 miRNAs implicated in breast cancer pathogenesis is shown: miR-206 (grey), miR-17-5p (blue), miR-125a/b (cyan), miR-200 (dark green), let-7 (green), miR-34 (yellow), miR-31 (orange), miR-21 (pink), miR-155 (red), and miR-373/520c (maroon). The targets form a highly connected network centered on MYC. Arrows indicate protein-protein or protein-DNA interactions, suggesting these targets coordinate the expression and/or function of one another.
These results suggest that breast cancer is associated with changes in the expression of multiple miRNAs that, in turn, disrupt a network of genes that either activate or inhibit each other's transcription or interact directly via protein-protein interactions. For instance, downregulation of miR-206 would enhance ESR1 expression, consequently increasing MYC transcription. MYC protein will be further elevated since loss of miR-34a would enhance its translation. Since CCND1, E2F1 and E2F3 are known to be activated by MYC, increased MYC would in turn enhance their expression. Moreover, down-modulation of miR-17-5p (regulates CCND1 and E2F1) and miR-34a (regulates E2F3, CCND1 and CDK6) would further elevate levels of these proteins. Our findings suggest that decreased expression of multiple tumor suppressor miRNAs leads to enhanced transcription and translation of these oncogenes in breast cancer via direct or indirect mechanisms.
The role of oncogenic or metastasis-promoting miRNAs is also important. For example, increased levels of miR-21, miR-31 and miR-373/520c would ensure that the protein synthesis of TPM1 (regulated by miR-21), CD44 (regulated by miR-373/520c), and MMP16 (regulated by miR-31) is repressed in breast cancer cells. In most cases, the protein encoded by a gene is up- or downregulated due to increased/decreased transcription and translation. Thus, tumor suppressor or oncogenic miRNAs regulate the transcription of some of these genes by targeting transcription factors and also provide an additional layer of regulation by regulating their translation. Although many of these gene regulatory events will likely occur at different stages of tumorigenesis and metastasis, it is clear that loss of miRNA regulation leads to a cascade of events that alter gene interaction networks important for breast cancer progression.
In addition to the above mentioned miRNAs, several other miRNAs, including miR-7, miR-128a, miR-210, miR-27b, miR-335, miR-126, miR-145 and miR-27a, have also been implicated in breast cancer. Uncovering the genes under the control of these miRNAs and how they integrate into the breast cancer gene interaction network will further aid our understanding of the disease. Furthermore, miRNA profiling is emerging as a powerful diagnostic tool to characterize features of different tumor types. This has been particularly useful in breast cancer, as miRNA signatures can unequivocally distinguish normal and malignant breast tissue and discriminate between breast cancer subtypes [71].
Conclusion
The usefulness of miRNA-based breast cancer therapy has been explored by these emerging studies that high-light their importance in breast cancer. A better understanding of the network of genes and cellular pathways regulated by these miRNAs will undoubtedly enable us to understand breast cancer pathogenesis and therapy. To accomplish this, identifying the genome-wide targets of these miRNAs is essential. Moreover, it is yet to be seen how these breast cancer-associated miRNAs will move from the laboratory bench into a clinical setting for the treatment of breast cancer. In this regard, the delivery of miRNA inhibitors or miRNA mimics specifically to tumor cells is one of the greatest challenges. Viral vector systems seem to be a reasonable option for efficient and organ-specific delivery [72], and it is likely that miRNA-based therapeutics will become a reality in the near future.
Abbreviations
- BT-IC:
-
breast tumor-initiating cell
- EMT:
-
epithelial-mesenchymal transition
- ER:
-
estrogen receptor
- MAPK:
-
mitogen-activated protein kinase
- MDCK:
-
Madin Darby canine kidney
- miRNA:
-
microRNA
- siRNA:
-
small interfering RNA
- TGF:
-
transforming growth factor
- T-IC:
-
tumor-initiating cell
- UTR:
-
untranslated region.
References
Borchert GM, Lanier W, Davidson BL: RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol. 2006, 13: 1097-1101. 10.1038/nsmb1167.
Tomari Y, Zamore PD: Perspective: machines for RNAi. Genes Dev. 2005, 19: 517-529. 10.1101/gad.1284105.
Filipowicz W, Bhattacharyya SN, Sonenberg N: Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight?. Nat Rev Genet. 2008, 9: 102-114. 10.1038/nrg2290.
Lehmann U, Hasemeier B, Christgen M, Müller M, Römermann D, Länger F, Kreipe H: Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J Pathol. 2008, 214: 17-24. 10.1002/path.2251.
Lal A, Kim HH, Abdelmohsen K, Kuwano Y, Pullmann R, Srikantan S, Subrahmanyam R, Martindale JL, Yang X, Ahmed F, Navarro F, Dykxhoorn D, Lieberman J, Gorospe M: p16(INK4a) translation suppressed by miR-24. PLoS ONE. 2008, 3: e1864-10.1371/journal.pone.0001864.
Tay Y, Zhang J, Thomson AM, Lim B, Rigoutsos I: MicroRNAs to Nanog, Oct4 and Sox2 coding regions modulate embryonic stem cell differentiation. Nature. 2008, 455: 1124-1128. 10.1038/nature07299.
Abdelmohsen K, Srikantan S, Kuwano Y, Gorospe M: miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels. Proc Natl Acad Sci USA. 2008, 105: 20297-20302. 10.1073/pnas.0809376106.
Forman JJ, Legesse-Miller A, Coller HA: A search for conserved sequences in coding regions reveals that the let-7 microRNA targets Dicer within its coding sequence. Proc Natl Acad Sci USA. 2008, 105: 14879-14884. 10.1073/pnas.0803230105.
Lytle JR, Yario TA, Steitz JA: Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5' UTR as in the 3' UTR. Proc Natl Acad Sci USA. 2007, 104: 9667-9672. 10.1073/pnas.0703820104.
Olsen PH, Ambros V: The lin-4 regulatory RNA controls developmental timing in Caenorhabditis elegans by blocking LIN-14 protein synthesis after the initiation of translation. Dev Biol. 1999, 216: 671-680. 10.1006/dbio.1999.9523.
Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP: The impact of microRNAs on protein output. Nature. 2008, 455: 64-71. 10.1038/nature07242.
Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N: Widespread changes in protein synthesis induced by microRNAs. Nature. 2008, 455: 58-63. 10.1038/nature07228.
Vasudevan S, Tong Y, Steitz JA: Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007, 318: 1931-1934. 10.1126/science.1149460.
Bartel DP: MicroRNAs: target recognition and regulatory functions. Cell. 2009, 136: 215-233. 10.1016/j.cell.2009.01.002.
Lewis BP, Burge CB, Bartel DP: Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005, 120: 15-20. 10.1016/j.cell.2004.12.035.
Zhao Y, Srivastava D: A developmental view of microRNA function. Trends Biochem Sci. 2007, 32: 189-197. 10.1016/j.tibs.2007.02.006.
Ventura A, Jacks T: MicroRNAs and cancer: short RNAs go a long way. Cell. 2009, 136: 586-591. 10.1016/j.cell.2009.02.005.
Esquela-Kerscher A, Slack FJ: Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006, 6: 259-269. 10.1038/nrc1840.
Croce CM: Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet. 2009, 10: 704-714. 10.1038/nrg2634.
Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM: bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003, 113: 25-36. 10.1016/S0092-8674(03)00231-9.
Lee RC, Feinbaum RL, Ambros V: The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993, 75: 843-854. 10.1016/0092-8674(93)90529-Y.
Calin GA, Liu CG, Sevignani C, Ferracin M, Felli N, Dumitru CD, Shimizu M, Cimmino A, Zupo S, Dono M, Dell'Aquila ML, Alder H, Rassenti L, Kipps TJ, Bullrich F, Negrini M, Croce CM: MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci USA. 2004, 101: 11755-11760. 10.1073/pnas.0404432101.
Calin GA, Croce CM: MicroRNA signatures in human cancers. Nat Rev Cancer. 2006, 6: 857-866. 10.1038/nrc1997.
Jiang J, Lee EJ, Gusev Y, Schmittgen TD: Real-time expression profiling of microRNA precursors in human cancer cell lines. Nucleic Acids Res. 2005, 33: 5394-5403. 10.1093/nar/gki863.
Murakami Y, Yasuda T, Saigo K, Urashima T, Toyoda H, Okanoue T, Shimotohno K: Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non-tumorous tissues. Oncogene. 2006, 25: 2537-2545. 10.1038/sj.onc.1209283.
Gaur A, Jewell DA, Liang Y, Ridzon D, Moore JH, Chen C, Ambros VR, Israel MA: Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res. 2007, 67: 2456-2468. 10.1158/0008-5472.CAN-06-2698.
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR: MicroRNA expression profiles classify human cancers. Nature. 2005, 435: 834-838. 10.1038/nature03702.
Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Ménard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM: MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65: 7065-7070. 10.1158/0008-5472.CAN-05-1783.
Iorio MV, Visone R, Di Leva G, Donati V, Petrocca F, Casalini P, Taccioli C, Volinia S, Liu CG, Alder H, Calin GA, Ménard S, Croce CM: MicroRNA signatures in human ovarian cancer. Cancer Res. 2007, 67: 8699-8707. 10.1158/0008-5472.CAN-07-1936.
Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, Iorio MV, Visone R, Sever NI, Fabbri M, Iuliano R, Palumbo T, Pichiorri F, Roldo C, Garzon R, Sevignani C, Rassenti L, Alder H, Volinia S, Liu CG, Kipps TJ, Negrini M, Croce CM: A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005, 353: 1793-1801. 10.1056/NEJMoa050995.
Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, Bowman ED, Yanaihara N, Yuen ST, Chan TL, Kwong DL, Au GK, Liu CG, Calin GA, Croce CM, Harris CC: MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008, 299: 425-436. 10.1001/jama.299.4.425.
Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T: Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007, 39: 673-677. 10.1038/ng2003.
Adams BD, Furneaux H, White BA: The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-alpha (ERalpha) and represses ERalpha messenger RNA and protein expression in breast cancer cell lines. Mol Endocrinol. 2007, 21: 1132-1147. 10.1210/me.2007-0022.
Leivonen SK, Mäkelä R, Ostling P, Kohonen P, Haapa-Paananen S, Kleivi K, Enerly E, Aakula A, Hellström K, Sahlberg N, Kristensen VN, Børresen-Dale AL, Saviranta P, Perälä M, Kallioniemi O: Protein lysate microarray analysis to identify microRNAs regulating estrogen receptor signaling in breast cancer cell lines. Oncogene. 2009, 28: 3926-3936. 10.1038/onc.2009.241.
Tavazoie SF, Alarcón C, Oskarsson T, Padua D, Wang Q, Bos PD, Gerald WL, Massagué J: Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008, 451: 147-152. 10.1038/nature06487.
Eiriksdottir G, Johannesdottir G, Ingvarsson S, Björnsdottir IB, Jonasson JG, Agnarsson BA, Hallgrimsson J, Gudmundsson J, Egilsson V, Sigurdsson H, Barkardottir RB: Mapping loss of heterozygosity at chromosome 13q: loss at 13q12-q13 is associated with breast tumour progression and poor prognosis. Eur J Cancer. 1998, 34: 2076-2081. 10.1016/S0959-8049(98)00241-X.
Hossain A, Kuo MT, Saunders GF: Mir-17-5p regulates breast cancer cell proliferation by inhibiting translation of AIB1 mRNA. Mol Cell Biol. 2006, 26: 8191-8201. 10.1128/MCB.00242-06.
Yu Z, Wang C, Wang M, Li Z, Casimiro MC, Liu M, Wu K, Whittle J, Ju X, Hyslop T, McCue P, Pestell RG: A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J Cell Biol. 2008, 182: 509-517. 10.1083/jcb.200801079.
Baselga J, Swain SM: Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009, 9: 463-475. 10.1038/nrc2656.
Mattie MD, Benz CC, Bowers J, Sensinger K, Wong L, Scott GK, Fedele V, Ginzinger D, Getts R, Haqq C: Optimized high-throughput microRNA expression profiling provides novel biomarker assessment of clinical prostate and breast cancer biopsies. Mol Cancer. 2006, 5: 24-10.1186/1476-4598-5-24.
Scott GK, Goga A, Bhaumik D, Berger CE, Sullivan CS, Benz CC: Coordinate suppression of ERBB2 and ERBB3 by enforced expression of micro-RNA miR-125a or miR-125b. J Biol Chem. 2007, 282: 1479-1486. 10.1074/jbc.M609383200.
Thiery JP, Acloque H, Huang RYJ, Nieto MA: Epithelial-mesenchymal transitions in development and disease. Cell. 2009, 139: 871-890. 10.1016/j.cell.2009.11.007.
Peinado H, Olmeda D, Cano A: Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype?. Nat Rev Cancer. 2007, 7: 415-428. 10.1038/nrc2131.
Aigner K, Dampier B, Descovich L, Mikula M, Sultan A, Schreiber M, Mikulits W, Brabletz T, Strand D, Obrist P, Sommergruber W, Schweifer N, Wernitznig A, Beug H, Foisner R, Eger A: The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene. 2007, 26: 6979-6988. 10.1038/sj.onc.1210508.
Dohadwala M, Yang SC, Luo J, Sharma S, Batra RK, Huang M, Lin Y, Goodglick L, Krysan K, Fishbein MC, Hong L, Lai C, Cameron RB, Gemmill RM, Drabkin HA, Dubinett SM: Cyclooxygenase-2-dependent regulation of E-cadherin: prostaglandin E(2) induces transcriptional repressors ZEB1 and snail in non-small cell lung cancer. Cancer Res. 2006, 66: 5338-5345. 10.1158/0008-5472.CAN-05-3635.
Spaderna S, Schmalhofer O, Wahlbuhl M, Dimmler A, Bauer K, Sultan A, Hlubek F, Jung A, Strand D, Eger A, Kirchner T, Behrens J, Brabletz T: The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res. 2008, 68: 537-544. 10.1158/0008-5472.CAN-07-5682.
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ: The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008, 10: 593-601. 10.1038/ncb1722.
Park S-M, Gaur AB, Lengyel E, Peter ME: The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22: 894-907. 10.1101/gad.1640608.
Hurteau GJ, Carlson JA, Roos E, Brock GJ: Stable expression of miR-200c alone is sufficient to regulate TCF8 (ZEB1) and restore E-cadherin expression. Cell Cycle. 2009, 8: 2064-2069.
Korpal M, Lee ES, Hu G, Kang Y: The miR-200 family inhibits epithelialmesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008, 283: 14910-14914. 10.1074/jbc.C800074200.
Iliopoulos D, Polytarchou C, Hatziapostolou M, Kottakis F, Maroulakou IG, Struhl K, Tsichlis PN: MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Sci Signal. 2009, 2: ra62-10.1126/scisignal.2000356.
Dykxhoorn DM, Wu Y, Xie H, Yu F, Lal A, Petrocca F, Martinvalet D, Song E, Lim B, Lieberman J: miR-200 enhances mouse breast cancer cell colonization to form distant metastases. PLoS ONE. 2009, 4: e7181-10.1371/journal.pone.0007181.
Zhang L, Huang J, Yang N, Greshock J, Megraw MS, Giannakakis A, Liang S, Naylor TL, Barchetti A, Ward MR, Yao G, Medina A, O'brien-Jenkins A, Katsaros D, Hatzigeorgiou A, Gimotty PA, Weber BL, Coukos G: microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci USA. 2006, 103: 9136-9141. 10.1073/pnas.0508889103.
Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, Dirbas FM, Somlo G, Pera RA, Lao K, Clarke MF: Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009, 138: 592-603. 10.1016/j.cell.2009.07.011.
Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, zur Hausen A, Brunton VG, Morton J, Sansom O, Schüler J, Stemmler MP, Herzberger C, Hopt U, Keck T, Brabletz S, Brabletz T: The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009, 11: 1487-1495. 10.1038/ncb1998.
Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G: The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000, 403: 901-906. 10.1038/35002607.
Clarke MF, Fuller M: Stem cells and cancer: two faces of eve. Cell. 2006, 124: 1111-1115. 10.1016/j.cell.2006.03.011.
Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J, Song E: let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007, 131: 1109-1123. 10.1016/j.cell.2007.10.054.
Dangi-Garimella S, Yun J, Eves EM, Newman M, Erkeland SJ, Hammond SM, Minn AJ, Rosner MR: Raf kinase inhibitory protein suppresses a metastasis signalling cascade involving LIN28 and let-7. EMBO J. 2009, 28: 347-358. 10.1038/emboj.2008.294.
Kato M, Paranjape T, Müller RU, Ullrich R, Nallur S, Gillespie E, Keane K, Esquela-Kerscher A, Weidhaas JB, Slack FJ: The mir-34 microRNA is required for the DNA damage response in vivo in C. elegans and in vitro in human breast cancer cells. Oncogene. 2009, 28: 2419-2424. 10.1038/onc.2009.106.
Valastyan S, Reinhardt F, Benaich N, Calogrias D, Szász AM, Wang ZC, Brock JE, Richardson AL, Weinberg RA: A pleiotropically acting microRNA, miR-31, inhibits breast cancer metastasis. Cell. 2009, 137: 1032-1046. 10.1016/j.cell.2009.03.047.
Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, Scarpa A, Vecchione A, Negrini M, Harris CC, Croce CM: A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006, 103: 2257-2261. 10.1073/pnas.0510565103.
Si M-L, Zhu S, Wu H, Lu Z, Wu F, Mo Y-Y: miR-21-mediated tumor growth. Oncogene. 2007, 26: 2799-2803. 10.1038/sj.onc.1210083.
Zhu S, Si M-L, Wu H, Mo Y-Y: MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol Chem. 2007, 282: 14328-14336. 10.1074/jbc.M611393200.
Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH: Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem. 2008, 283: 1026-1033. 10.1074/jbc.M707224200.
Qi L, Bart J, Tan LP, Platteel I, Sluis Tvd, Huitema S, Harms G, Fu L, Hollema H, Berg Avd: Expression of miR-21 and its targets (PTEN, PDCD4, TM1) in flat epithelial atypia of the breast in relation to ductal carcinoma in situ and invasive carcinoma. BMC Cancer. 2009, 9: 163-10.1186/1471-2407-9-163.
Zhu S, Wu H, Wu F, Nie D, Sheng S, Mo Y-Y: MicroRNA-21 targets tumor suppressor genes in invasion and metastasis. Cell Res. 2008, 18: 350-359. 10.1038/cr.2008.24.
Kong W, Yang H, He L, Zhao J-j, Coppola D, Dalton WS, Cheng JQ: MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol. 2008, 28: 6773-6784. 10.1128/MCB.00941-08.
Ma L, Teruya-Feldstein J, Weinberg RA: Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007, 449: 682-688. 10.1038/nature06174.
Lal A, Navarro F, Maher CA, Maliszewski LE, Yan N, O'Day E, Chowdhury D, Dykxhoorn DM, Tsai P, Hofmann O, Becker KG, Gorospe M, Hide W, Lieberman J: miR-24 Inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to "seedless" 3'UTR microRNA recognition elements. Mol Cell. 2009, 35: 610-625. 10.1016/j.molcel.2009.08.020.
Blenkiron C, Goldstein LD, Thorne NP, Spiteri I, Chin SF, Dunning MJ, Barbosa-Morais NL, Teschendorff AE, Green AR, Ellis IO, Tavaré S, Caldas C, Miska EA: MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007, 8: R214-10.1186/gb-2007-8-10-r214.
Kota J, Chivukula RR, O'Donnell KA, Wentzel EA, Montgomery CL, Hwang HW, Chang TC, Vivekanandan P, Torbenson M, Clark KR, Mendell JR, Mendell JT: Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009, 137: 1005-1017. 10.1016/j.cell.2009.04.021.
Mayr C, Hemann MT, Bartel DP: Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007, 315: 1576-1579. 10.1126/science.1137999.
Wu L, Fan J, Belasco JG: MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci USA. 2006, 103: 4034-4039. 10.1073/pnas.0510928103.
Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ: RAS is regulated by the let-7 microRNA family. Cell. 2005, 120: 635-647. 10.1016/j.cell.2005.01.014.
Christoffersen NR, Shalgi R, Frankel LB, Leucci E, Lees M, Klausen M, Pilpel Y, Nielsen FC, Oren M, Lund AH: p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ. 2010, 17: 236-245. 10.1038/cdd.2009.109.
Welch C, Chen Y, Stallings RL: MicroRNA-34a functions as a potential tumor suppressor by inducing apoptosis in neuroblastoma cells. Oncogene. 2007, 26: 5017-5022. 10.1038/sj.onc.1210293.
Sun F, Fu H, Liu Q, Tie Y, Zhu J, Xing R, Sun Z, Zheng X: Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett. 2008, 582: 1564-1568. 10.1016/j.febslet.2008.03.057.
Huang Q, Gumireddy K, Schrier M, le Sage C, Nagel R, Nair S, Egan DA, Li A, Huang G, Klein-Szanto AJ, Gimotty PA, Katsaros D, Coukos G, Zhang L, Puré E, Agami R: The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat Cell Biol. 2008, 10: 202-210. 10.1038/ncb1681.
Acknowledgements
We thank Myriam Gorospe (NIH) for careful reading of the article. Due to space constraints this is not an exhaustive review of the literature on the subject and we apologize for not citing all publications.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
O'Day, E., Lal, A. MicroRNAs and their target gene networks in breast cancer. Breast Cancer Res 12, 201 (2010). https://doi.org/10.1186/bcr2484
Published:
DOI: https://doi.org/10.1186/bcr2484