
Abstract
Tissue factor, the primary initiator of the coagulation cascade, maintains vascular integrity in response to injury. It is now recognised that, in addition to the role as a procoagulant activator, tissue factor participates in many tumour-related processes that contribute to malignant disease progression. The present review details the recent evidence supporting a role for tissue factor in tumour haemostasis, angiogenesis, metastasis and malignant cell survival. Furthermore, future research directions are discussed that may enhance our understanding of the role and regulation of this protein, which could ultimately lead to the innovative design and development of new anticancer therapies.
Similar content being viewed by others

Introduction
Angiogenesis, the development of new blood vessels from the existing vasculature, and haemostasis, the coagulation cascade leading to clot formation, are among the most consistent host responses associated with cancer. Tissue factor (TF) normally safeguards the vascular integrity of tissues by initiating the coagulation cascade following vessel injury. Hypercoagulability is exhibited by most cancer patients and contributes to the pathogenesis of tumour growth and metastasis by promoting angiogenesis. Haemostasis and angiogenesis are therefore interrelated processes with important implications for cancer therapy.
TF, similar to a number of haemostatic proteins, participates in many tumour-related processes, including tumour angiogenesis, metastasis, hypercoagulability and tumour cell survival; processes that all contribute to malignant disease progression. The molecular mechanisms responsible for the actions of TF are only just beginning to be elucidated, but it is thought that they occur by the action of intracellular signalling, resulting in gene transcription and subsequent protein synthesis.
Tissue factor
TF – also known as coagulation factor III, thromboplastin, or CD142 – is a 47 kDa transmembrane glycoprotein first cloned independently by four different groups in 1987 [1–4]. The human TF gene spans 12.4 kbp, has six exons and is located on chromosome 1, p21–p22. The TF protein consists of a 219-amino-acid extracellular domain, a 23-amino-acid transmembrane segment and a 21-amino-acid cytoplasmic tail that does not bear significant homology with other proteins [5]. In silico studies have resulted in TF being classified as a member of the class II cytokine/haematopoietic growth factor family [6]. The extracellular domain of TF contains factor VII/activated factor VII (FVIIa) binding sites, but the transmembrane domain plays a crucial role in anchoring the TF–FVIIa complex to the cell surface in addition to complete expression of the procoagulant activity [7].
TF gene expression is complex and is regulated by a number of transcription factors that may be sensitive to hypoxia or anoxia, including activator protein (AP-1), nuclear factor-κB (NF-κB), Sp-1 and early growth response gene-1 (Egr-1) [8, 9]. In addition, heparanase and platelet endothelial cell adhesion molecule 1 both participate in the regulation of TF gene expression (via activation of the p38 signalling pathway) [10, 11].
Although TF expression can be transiently upregulated in monocytes or macrophages and endothelial cells (ECs) by growth factors and cytokines, vascular ECs and intravascular cells do not express TF in normal physiological situations. Constitutive TF expression is restricted to subendothelial cells (such as pericytes, smooth muscle cells and fibroblasts) that only interact with blood when vascular integrity is compromised [12]. However, it is clear that during tumourigenesis, this strict regulation of TF expression is lost. Upregulation of TF protein by tumour cells and associated stromal cells has been well documented in breast cancer and other malignant tumours [13–17]. TF is now known to exist in several locations: in association with cells (intracellular or surface location) and within the circulation, either associated with microparticles [18] or in a free, soluble form [19, 20]. TF-expressing microparticles are membrane vesicles derived from haematopoietic cells (for example, monocytes and platelets) that play a putative role in haemostasis activation in cancer patients [21, 22].
Cryptic TF refers to the part of the cellular TF pool that is noncoagulant but retains functional cell signalling. Cryptic TF contains unpaired cysteine thiols and activation involves the formation of the disulphide bond Cys186–Cys209 [23]. Extracellular protein disulphide isomerase has been proposed to target this disulphide bond, inactivating the procoagulant activity of TF [24] while enhancing TF coagulant activity on microparticles shed from cells [25]. Protein disulphide isomerase has therefore been suggested to facilitate a dynamic and reversible switch (conformational change) between two distinct functional TF species: one that initiates coagulation, and an encrypted form that does not – this theory, however, is currently controversial [26].
In normal physiological conditions, initiation of the extrinsic coagulation pathway occurs when TF is exposed to the bloodstream, either following damage to the vascular system integrity or upon activation of monocytes or ECs. FVIIa then binds to TF on the cell surface. Sequential downstream activation of haemostatic protease complexes leads to the generation of thrombin, with subsequent platelet activation and the formation of a fibrin clot that restores vessel integrity (Figure 1) (reviewed in [27]). There is now increasing evidence that, in addition to initiating haemostasis, binding of FVIIa to TF directly cleaves protease-activated receptor (PAR)-2 and results in phosphorylation of the TF cytoplasmic domain. This subsequently inhibits the negative regulatory control of PAR-2-mediated signalling, thereby promoting angiogenesis (Figure 2) [28–30].

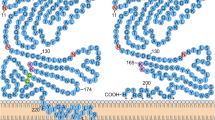
Tissue factor is the primary initiator of the coagulation cascade. In normal physiological conditions, following vessel damage or trauma, tissue factor (TF) forms a complex with activated factor VII (FVIIa) in the presence of calcium ions on an appropriate phospholipid membrane, and allosterically enhances the enzyme activity of this protease to catalyse the activation of factor X (FX) to FXa. Generation of FXa by the TF–FVIIa complex triggers the proteolytic conversion of prothrombin (factor II) to thrombin (activated factor II [FIIa]). This key step in the coagulation cascade is effected by the catalytic activity of the prothrombinase complex, which consists of FXa and the nonenzymatic cofactor activated factor V (FVa) in a 1:1 stoichiometric complex. The resulting burst in thrombin activity activates localised platelets and catalyses the conversion of circulating fibrinogen into an insoluble fibrin clot, thereby restoring vessel integrity.

Tissue factor-induced protease-activated receptor 2 signalling. (a) When the cytoplasmic domain is nonphosphorylated, tissue factor (TF) exerts a negative regulatory control on protease-activated receptor (PAR)-2 and inhibits signalling. (b) Binding of activated factor VII (FVIIa) to TF results in proteolytic cleavage of PAR-2 and phosphorylation of the cytoplasmic domain of TF via activation of phosphatidylcholine-specific phospholipase C (PC-PLC) and protein kinase C (PKC) alpha. Phosphorylation releases the negative regulatory control of PAR-2-mediated signalling, resulting in activation of several mitogen-activated protein kinase (MAPK) pathways and subsequent gene transcription. Phosphatidylinositol 3-kinase (PI3K) and Rac1 activation also constitute important signal transduction cascades, mainly related to antiapoptotic and migration processes. The constitutive association of α3β1 integrin with aggressive breast cancer cells (for example, MDA-MB-231 cells) implies that tumour cells have lost the ability to sense extracellular gradients of coagulation proteases and constitutively couple TF and integrin signalling. Although the precise mechanism for this coupling is unknown, it may play a role in tumour cell invasiveness. The overall activation of these signalling pathways is responsible for the induction of angiogenesis, malignant cell survival and metastasis, resulting in tumour growth and disease progression. BTCM, breast tumour cell membrane; Cyr61, cysteine-rich angiogenic inducer 61; CTGF, connective tissue growth factor; EC, endothelial cell; Egr-1, early growth response gene 1; ERK, extracellular signal-related kinase; JNK, c-jun N-terminal kinase; PKB, protein kinase B; TSP, thrombospondin; uPAR, urokinase-type plasminogen activator receptor; VEGF, vascular endothelial growth factor.
Tissue factor and tumour haemostasis
Many cancer patients display a hypercoagulable state leading to frequent serious complications, such as thromboembolism and haemorrhage. Moreover, the risk of tumour-associated hypercoagulability is compounded by the effects of chemotherapy and radiotherapy on the coagulation cascade. The annual incidence of venous thromboembolism in patients receiving chemotherapy is estimated to be 11% and is increased two-threefold with hormone therapies, such as tamoxifen, currently used to treat breast carcinomas [31]. In contrast to standard chemotherapeutic agents, antiangiogenic agents generally elicit few toxic side effects. An unexpected high incidence of both arterial and venous thrombosis has been reported, however, in a number of clinical trials in which patients received both antiangiogenic agents and standard chemotherapy [32–34].
The specific mechanisms that can cause exacerbation of prothrombotic events in cancer patients in response to these combination therapies are unknown, but are likely to be multifactorial and to involve the two major opposing influences that control haemostasis: TF and tissue factor pathway inhibitor (TFPI; an endogenous inhibitor of TF-induced coagulation [35]). The vascular endothelial growth factor (VEGF) receptor-2 inhibitor SU541 in combination with high doses of chemotherapeutic drugs (especially gemcitabine and cisplatin) recently triggered a procoagulant conversion of ECs in vitro (as measured by dramatic increases in their surface TFPI expression (ie, the coagulation index). In contrast, low (metronomic) concentrations of the agents significantly attenuated the coagulation index increase induced by these drugs, suggesting that use of low-dose chemotherapy regimens (rather than traditional maximum-tolerated dose regimens) might reduce the incidence of adverse clotting events associated with antiangiogenic/chemotherapy combination therapies [35].
The pathogenesis of a systemic hypercoagulable state is complex, but principally relates to the procoagulant properties of tumour cells, tumour-associated ECs and host inflammatory cells, in addition to the activation and adhesive interactions of these cells. In particular, upregulation of TF plays a key role [36]. TF expression is an independent predictive factor of venous thromboembolism development in ovarian cancer [37] and is associated with venous thromboembolism in pancreatic cancer [38]. Furthermore, the quantity of TF+ microparticles in platelets significantly correlates with plasma D-dimer levels (a marker that strongly predicts venous thromboembolism events in cancer patients) [21] and appears to predict a propensity to develop venous thromboembolism in malignant disease [22]. This evidence suggests that TF+ microparticles play a role in the hyper-coagulable state of many cancer patients.
In human breast cancer cell lines, clotting assays have demonstrated that the rate of clot formation is factor VII dependent and that it is completely inhibited by an anti-TF monoclonal antibody, indicating that TF is the only significant procoagulant present in these cancer cells [39]. Furthermore, tumour hypoxia and loss of the phosphatase and tensin homolog (mutated in multiple advanced cancers-1 (PTEN) tumour suppressor gene appears to cooperate in upregulating TF expression and increasing the procoagulant activity of malignant glioma cells [40]. Experiments by Yu and colleagues in 2005 suggested a genetic cause for both local and systemic hypercoagulability [41]. TF expression in colorectal cancer cells in vitro was controlled by the activation of K-ras oncogene and the inactivation of p53 tumour suppressor gene, in a manner dependent on mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K). Furthermore, upregulation of TF in tumour cells led to systemic TF release, demonstrating that K-ras and p53 influence not only cell surface TF expression, but also the total amount of TF procoagulant activity released from cancer cells into their local microenvironment and the circulation [41]. It has yet to be demonstrated whether K-ras and p53 are involved in TF upregulation in breast cancer.
Bogdanov and colleagues were the first to show that alternatively spliced TF mRNA, lacking exon 5, expresses a protein [42]. This alternatively spliced TF mRNA has been found in monocytes, ECs and cancer cells [20, 43, 44], although at much lower levels than the fully spliced TF mRNA. The procoagulant activity of alternatively spliced TF is currently controversial; some reports have shown a coagulant function [20], while other studies have not [19]. Clearly, further studies are needed to clarify the functional significance of alternatively spliced TF and the role it plays in tumour haemostasis.
Tissue factor and tumour angiogenesis
Since the work of Judah Folkman in the 1970s, it is now widely accepted that angiogenesis is essential for tumour growth since tumours cannot grow beyond 2 mm3 without developing a new vasculature [45]. TF can contribute to angiogenesis by different pathways: indirectly, via clotting-dependent mechanisms or by regulating the angiogenic properties of tumour cells by altering the production of growth regulatory molecules that act on vascular ECs; or directly, by clotting-independent mechanisms.
Indirect regulation of angiogenesis
Clotting-dependent induction of tumour angiogenesis is primarily mediated by TF-induced generation of thrombin and the subsequent deposition of cross-linked fibrin, which provides a provisional pro-angiogenic matrix that facilitates blood vessel infiltration [27]. Activation of coagulation may also indirectly support angiogenesis by releasing pro-angiogenic factors from the α-granules of activated platelets and generating additional angiogenic pathways that are dependent on activated factor X (FXa), thrombin, and the G protein-coupled PARs (PAR-1–PAR-4), such as thrombin signalling through PAR-1 on ECs [46].
Experiments conducted by Zhang and colleagues in 1994 provided the first in vivo evidence that TF could regulate angiogenesis by altering the expression of angiogenic molecules (by a mechanism distinct from activation of the coagulation system). Overexpression of TF in fibrosarcoma [47], in gastric cancer cells [48] and in melanoma cells [12] enhances tumour growth by diminishing the transcription of antiangiogenic thrombospondins and/or by increasing the transcription of pro-angiogenic VEGF. Although it is still controversial whether thrombospondins are directly regulated by TF overexpression in tumour cells [41, 47], the cytoplasmic domain of TF appears essential for the production of VEGF in human gastric cancer [48] and melanoma [12] cells. In a positive feedback mechanism, TF induces upregulation of VEGF on tumour cells, further increasing TF expression [47]. A significant relationship between TF expression and micro-vessel density (a surrogate marker of angiogenesis) exists in breast cancer (Bluff JE, Menakuru SR, Cross SS, Brown NJ, Reed MWR, Staton CA, manuscript in preparation) and other cancers [38]. Furthermore, the significant correlation between TF and VEGF in breast cancer [15], the colocalisation of TF and VEGF in breast cancer cells [15], and the correlation of increased TF, VEGF and microvessel density with disease progression in breast carcinoma [14] all provide further evidence that TF may modify the angiogenic properties of tumour cells by altering the production of growth regulatory molecules that act on the endothelium.
Direct regulation of angiogenesis
In addition to the indirect role that TF plays in tumour angiogenesis, an intracellular function of TF has now been hypothesised. Mice lacking TF die in utero (between embryonic days 8.5 and 10.5) due to haemorrhaging of embryonic blood from both extraembryonic and embryonic vessels [49]. Furthermore, specific inhibitors to the TF–FVIIa–FXa complex and to FXa alone demonstrate that the angiogenic process is dependent on FVIIa but not on FXa [50]. Angiogenesis therefore appears to be dependent on TF–FVIIa intracellular function.
The clotting-independent pathway of TF-induced tumour angiogenesis has been characterised primarily via PARs. Although thrombin cleaves PAR-1, PAR-3 and PAR-4, proteases such as FXa and the TF–FVIIa complex can activate PAR-2. PAR-1 can also be cleaved and activated by FVIIa, FXa, plasmin, activated protein C and matrix metallo-proteinase 1 (reviewed in [51]). Multiple proteases can clearly cleave and activate these receptors; thus, the particular PAR and protease that function in a specific cellular setting remain largely undefined.
TF–FVIIa signalling via PAR-2 was recently shown to play a crucial role in breast tumour growth in a murine orthotopic breast cancer model [28]. Additionally, angiogenesis mediated by TF–FVIIa signalling has been demonstrated to be regulated via PAR-2 signalling, tightly controlled by the TF cytoplasmic domain (Figure 2) [29, 30]. Genetic deletion of the TF cytoplasmic domain resulted in both accelerated physiological and pathological angiogenesis in mice. Loss of negative regulatory control by the TF cytoplasmic domain is therefore a pathway by which pro-angiogenic signalling of PAR-2 can be turned on. Interestingly, platelet-derived growth factor BB synergises with the enhanced PAR-2-dependent angiogenesis observed in mice that lack the TF cytoplasmic domain. Therefore it may be possible to target deregulated TF cytoplasmic domain signalling to suppress pathological PAR-2 and platelet-derived growth factor-dependent angiogenesis, without interfering with the physiological vascular functions of platelet-derived growth factor [30].
Furthermore, Ahamed and Ruf demonstrated that PAR-2 signalling specifically targets the TF cytoplasmic domain by inducing TF phosphorylation at multiple serine residues [52]. Protein kinase C-dependent phosphorylation of Ser253 enhances subsequent Ser258 phosphorylation by a proline-directed kinase. Thio-ester modification of the cytoplasmic cysteine, however, counteracts TF phosphorylation [53]. PAR-2 expression and changes in the palmitoylation status of TF therefore emerge as key regulators of cytoplasmic domain phosphorylation. Although the ternary TF–FVIIa–FXa complex similarly activates PAR-1 or PAR-2 (based on efficiency of PAR internalisation and extracellular-signal related kinase phosphorylation), it is PAR-2 signalling not PAR-1 signalling that leads to prolonged protein kinase C alpha activation and downstream TF cytoplasmic domain phosphorylation. Activation of phosphatidylcholine-specific phospholipase C upstream of protein kinase C alpha accounts for this unique signalling response following PAR-2 activation [52].
The proteolytic activity of the TF–FVIIa complex induces activation of various MAPK family members – such as p42/p44 MAPK, p38 MAPK and c-jun N-terminal kinase – which, at least in some cell types, form parts of the signalling cassettes together with Src-like kinases and the lipid protein/phosphatidylinositol 3-kinase [54]. Interestingly, inhibition of the MAPK pathway was shown to significantly prevent VEGF-driven upregulation of TF on ECs, indicating the critical role of MAPK activation on TF production [55]. In contrast, negative regulation of TF production has been shown to occur via activation of the phosphatidylinositol 3-kinase/Akt pathway [41, 55]. In combination, these experiments suggest that upregulated TF expression on ECs may be the result of diminished phosphatidylinositol 3-kinase activity or the result of an increase in MAPK activity.
Although the physiological relevance of TF–FVIIa-induced activation of MAPK is not fully understood, the MAPK family has been increasingly associated with cell proliferation, suggesting that TF–FVIIa might contribute to the angiogenic process simply by stimulating cell division. The mitogenic capacity of TF–FVIIa remains disputable, however, and a more probable mechanism involves the production of angiogenesis-stimulating proteins. MAPK are well-known mediators of gene transcription via the phosphorylation of transcription factors.
Exposure of TF-expressing cells (invasive breast cancer cells, fibroblasts, pancreatic tumour cells and keratinocytes) to FVIIa led to increased gene expression of VEGF [56, 57], urokinase-type plasminogen activator receptor [58], early growth response gene 1 [59], IL-8 [56, 60], cysteine-rich angiogenic inducer 61 and connective tissue growth factor [56]. These genes encode proteins that function either as growth factors, as inducers of angiogenesis or as extra-cellular matrix signalling proteins that facilitate the process of new vessel development or regulate a myriad of cellular functions, such as cell adhesion, proliferation and migration, which are essential for tumour angiogenesis and metastasis. PAR-2 also activates Rac and recruits a β-arrestin scaffolding complex for extracellular signal-related kinase localisation, thus promoting cell motility [61]. Additionally, the initiation of TF–FVIIa signalling is further complicated by activation of epidermal growth factor receptor with subsequent phosphorylation of the epidermal growth factor receptor cytoplasmic domain [62].
Tissue factor and tumour metastasis
Metastasis is the result of multiple pathways acting in concert to provide tumour cells with the capacity to migrate, to gain access to the bloodstream or lymphatic vessels, and to ultimately home to distant sites. It is now clear that these processes are highly dependent on components of the blood coagulation cascade, such as thrombin [63, 64]. Moreover, it has been shown that TF supports metastasis through a fibrin(ogen)-dependent and platelet-dependent restriction in natural killer cell-mediated clearance of micrometastases [63].
TF is highly expressed in metastatic breast carcinoma cells, in contrast to nonmetastatic breast carcinoma cells [65]. However, it is unclear how TF on tumour cells contributes to tumour metastasis; the TF–FVIIa complex may play a direct role and/or the downstream generation of active coagulation factors may be implicated in this process. There is, however, good evidence that TF–FVIIa signalling stimulates cell motility. Experimental work has shown that extracellular ligation of the TF molecule results in the specific interaction of the cytoplasmic domain with actin-binding protein 280, which supports cell adhesion and migration in in vitro assays [66]. In fibroblasts, TF–FVIIa signalling enhanced platelet-derived growth factor BB-stimulated migration, suggesting signalling cross-talk between PARs and tyrosine kinase receptors in this process [67].
PAR-1 expression is minimal or absent in benign and normal breast tissue and noninvasive carcinoma, but thrombin activation of PAR-1 has been implicated in tumour cell growth, in invasion and metastasis of breast cancer and in other human malignant diseases [64]. Little is known regarding the role of tumour cell PAR-2 in this process. Signalling of the TF–FVIIa–FXa complex, however, has been shown to enhance the migration of breast cancer cells in vitro, via a PAR-2-dependent mechanism [68]; and anti-PAR-2 antibodies inhibit the indirect and promigratory effect of IL-8 in breast carcinoma cells [60]. Furthermore, small interfering RNA targeting PAR-1 and PAR-2 demonstrated impaired migration of invasive breast cancer cells deficient in PAR-2, but not deficient in PAR-1, once again demonstrating the critical role for PAR-2 in breast cancer cell migration and invasion [69].
The role of the TF cytoplasmic domain as a negative regulator of chemotaxis has been supported by the recent observation that TF expression in keratinocytes and melanoma cells suppresses α3β1-dependent migration on laminin 5. This effect was reversed by PAR-2-dependent phosphorylation of the TF cytoplasmic domain, demonstrating that phosphorylation regulates the function of TF in cell motility (Figure 2) [70]. Other reports have also indicated the mandatory function of the cytoplasmic TF domain for cell migration of porcine aortic ECs [71] and human bladder carcinoma cells, through the activation of p38 and Rac1 [72]. Interestingly, TF has recently been shown to be constitutively associated with α3β1 on highly aggressive breast cancer cells (MDA-MB-231) [28], although the precise mechanism for this coupling remains to be elucidated.
TF expression correlates strongly with metastatic potential in many human tumours, and the expression of functional TF in in vivo metastasis models has confirmed that TF plays an important role in this process [73–76]. The metastatic potential of two subsets of melanoma cells, expressing high levels and low levels of human TF, was compared by intravenous injection of the tumour cells into severe combined immunodeficiency mice. Metastatic lung tumours were detected in 86% of the mice injected with the high-TF-expressing lines, but in only 5% of the mice injected with the low-TF-expressing lines, indicating that a high TF level promotes lung metastasis of human melanoma in vivo [75]. A blocking anti-TF monoclonal antibody and Fab fragments thereof significantly inhibited the growth of pulmonary metastases in this model [76], dependent on both signalling of the TF cytoplasmic domain [75, 77] and on the extracellular proteolytic activity of the active TF–FVIIa complex [75]. Moreover, PAR-1 enhanced the metastatic potential of TF in this model [78]. More recently, in a similar model of metastasis, small interfering RNAs targeting TF on melanoma cells inhibited melanoma cell colonisation of the lung [74]; it has since been proposed that silencing of tissue factor pathway inhibitor 2 may be involved in tumour cell metastasis [79]. A humanised anti-TF monoclonal antibody (CNTO 859) also inhibited experimental lung metastasis of invasive breast cancer cells (MDA-MB-231), by over 99% [73], suggesting a role for TF in breast cancer as well as melanoma cell metastasis.
Tissue factor and tumour cell survival
It is possible that defects in apoptosis or activation of antiapoptotic pathways promote tumour growth and survival, which may be linked to tumour metastasis. Indeed, resistance to apoptosis is a key factor in the survival of malignant cells [51]. FVIIa is known to induce the activation of both P42/44 MAPK and protein kinase B signalling – pathways known to inhibit apoptosis [80]. The effect of TF–FVIIa-mediated signalling on apoptosis has recently been investigated in a human breast cancer cell line (MCF-7) that had been trans-fected to express TF [81]. The formation of the TF–FVIIa–FXa complex prevented apoptosis in these cells by a thrombin-independent pathway. Moreover, the antiapoptotic effect of this signalling pathway involved phosphorylation of both p44/42 MAPK and protein kinase B/Akt, and may be mediated, in part, by an increase in the antiapoptotic protein survivin. Additional studies have demonstrated that FVIIa appears to be capable of inducing cell survival in serum-deprived baby hamster kidney and Chinese hamster ovary cells that overexpressed TF [82]. Furthermore, FVIIa inhibits apoptosis induced by the loss of adhesion [83], and TF–FVIIa signalling can induce cell survival through STAT5-dependent BclxL production and Jak2-dependent activation of protein kinase B [84].
It has been hypothesised that the expression of TF by disseminated tumour cells confers protection against immune recognition and cytotoxicity. When challenged with peripheral blood mononuclear cells, TF-expressing colon carcinoma cells (HT29) partially evaded cellular cytotoxicity by approximately 40% and this protective function was dependent on the cytoplasmic domain of TF [85]. TF may therefore be capable of promoting immune evasion in tumour cells, which could lead to increased cell survival and metastasis of cancer cells. The evidence for antiapoptotic effects of FVIIa, however, is limited to cells transfected to overexpress TF. The anti-apoptotic effect of TF–FVIIa signalling in tumour and/or associated stromal cells has therefore not yet been confirmed.
Future directions for tissue factor research
The direct targeting of TF with, for example, monoclonal antibodies or inactivating peptides has already been demonstrated efficacious in several models in vivo [69]. Moreover, inhibition of various procoagulant and TF signalling effects using antagonists (for example, tissue factor pathway inhibitor) [86], inducing selective intratumoral thrombosis (tumour infarction) [87] or inducing immune attack [88] are currently being explored. One of the major anticipated adverse effects of these approaches in humans are problems associated with the haemostatic system. Although a recent phase I clinical trial with a monoclonal antibody to TF reported no major bleeding events, the frequency of minor haemorrhage increased in a dose-dependent manner, characterised by oral mucosal bleeding, spontaneous tongue haematomas and cutaneous bleeding [89]. Interestingly, most animal models have reported no apparent side effects using different anti-TF approaches, perhaps reflecting the focus on agents that act locally – for example, strategies specifically targeted to TF expressed within tumours – thereby causing minimal disturbance of systemic haemostasis. Phase I direct TF-targeting clinical studies are now in progress, but many important questions remain unanswered. What causes the upregulation of TF in cancer cells, with the subsequent increase in their malignancy? Do oncogene-directed therapies impact on expression of TF in vivo and do TF-targeting agents synergise with oncogene-targeting agents, antiangiogenics or anticancer chemotherapy?
Broad generalisations regarding the role of TF in cancer progression and metastasis may be premature. This is likely to depend on the biological context such as cell, tissue and disease type, as well as on the expression of other genes related to the malignant process; this clearly warrants more extensive and thorough investigation.
There is still no direct evidence that TF, as a true receptor, induces cell signalling via the cytoplasmic tail, and an increasing number of studies suggest a direct role for TF and FVIIa in TF-mediated oncogenic processes through the activation of signal transduction pathways via the proteolytic cleavage of PAR-2. However, it is largely unclear which other cell membrane components, such as integrins, proteoglycans and growth factor receptors, may also be involved in transmitting or regulating TF–FVIIa signalling. Analysis of other potential cell surface receptors mediating TF–FVIIa signalling and investigations into how the spatial organisation of participating signalling components regulates TF–FVIIa signalling are important areas requiring further study. Characterisation of ligand binding to the phosphorylated and nonphosphorylated TF cytoplasmic domain will be instrumental in understanding the signalling complexes that associate with TF. It has been proposed that epitopes unique to the TF–FVIIa complex may allow specific targeting of TF–FVIIa signalling associated with cancer progression, without compromising haemostasis [23–25, 28].
Versteeg and colleagues recently employed a unique pair of isotype-matched anti-TF antibodies that specifically block either direct TF–FVIIa signalling (mAb 10H10) or coagulation (mAb 5G9). These authors showed that mAb 10H10 (and also inhibition of PAR-2 signalling) was effective in suppressing aggressive human breast cancer cell growth in both murine subcutaneous and orthotopic models, with minimal effects on TF coagulant function [28]. What is unclear, however, is whether the possibility exists in vivo that TF–FVIIa is active without concomitant FXa generation and FXa/thrombin-mediated PAR signalling; this approach therefore needs validation.
Detailed investigations into the mechanisms of TF-induced signal transduction and the regulation and function of TF will therefore be instrumental for devising rational therapy aimed at inhibiting TF-dependent tumour vascularisation, hyper-coagulability and metastasis. Furthermore, TF is quantifiable in plasma and serum, and tumour cells are known to shed TF into the circulation. Furthermore, TF is quantifiable in plasma and serum and tumour cells are known to shed TF into the circulation; clearly, more studies are required to evaluate circulating TF (microparticles) as a diagnostic and/or prognostic marker of tumour progression or pathological angiogenesis in cancer.
Conclusion
Substantial evidence exists supporting a role for TF acting as both a regulatory target and as an important mediator of oncogenic processes, specifically tumour metastasis and tumour-associated angiogenesis and hypercoagulability. Furthermore, TF expression by cancer cells and tumour-associated stromal cells, but not by benign tumours, makes TF an attractive therapeutic target. Like most therapies, anti-TF strategies may have their caveats – but preclinical studies with TF-targeted cancer therapeutics have demonstrated efficacy that may translate to the clinical setting, offering renewed hope for the control of cancer growth and metastasis. The development of specific inhibitors that suppress TF–FVIIa signalling in pathological angiogenesis, but not TF–FVIIa coagulant function, could provide opportunities to design specific drugs with therapeutic value for the treatment of cancer. Furthermore, as TF–FVIIa is a multitasking complex, numerous relevant potential targets for therapeutic intervention have yet to be explored.
Abbreviations
- EC:
-
endothelial cell
- FVIIa:
-
activated factor VII
- FXa:
-
activated factor X
- IL:
-
interleukin
- mAb:
-
monoclonal antibody
- MAPK:
-
mitogen-activated protein kinase
- NF:
-
nuclear factor
- PAR:
-
protease-activated receptor
- TF:
-
tissue factor
- VEGF:
-
vascular endothelial growth factor.
References
Fisher KL, Gorman CM, Vehar GA, O'Brien DP, Lawn RM: Cloning and expression of human tissue factor cDNA. Thromb Res. 1987, 48: 89-99. 10.1016/0049-3848(87)90349-5.
Morrissey JH, Fakhrai H, Edgington TS: Molecular cloning of the cDNA for tissue factor, the cellular receptor for the initiation of the coagulation protease cascade. Cell. 1987, 50: 129-135. 10.1016/0092-8674(87)90669-6.
Scarpati EM, Wen D, Broze GJ, Miletich JP, Flandermeyer RR, Siegel NR, Sadler JE: Human tissue factor: cDNA sequence and chromosome localization of the gene. Biochemistry. 1987, 26: 5234-5238. 10.1021/bi00391a004.
Spicer EK, Horton R, Bloem L, Bach R, Williams KR, Guha A, Kraus J, Lin TC, Nemerson Y, Konigsberg WH: Isolation of cDNA clones coding for human tissue factor: primary structure of the protein and cDNA. Proc Natl Acad Sci USA. 1987, 84: 5148-5152. 10.1073/pnas.84.15.5148.
Mackman N: Alternatively spliced tissue factor – one cut too many?. Thromb Haemost. 2007, 97: 5-8.
Camerer E, Kolsto AB, Prydz H: Cell biology of tissue factor, the principal initiator of blood coagulation. Thromb Res. 1996, 81: 1-41. 10.1016/0049-3848(95)00209-X.
Morrissey JH, Neuenschwander PF, Huang Q, McCallum CD, Su B, Johnson AE: Factor VIIa–tissue factor: functional importance of protein–membrane interactions. Thromb Haemost. 1997, 78: 112-116.
Rong Y, Hu F, Huang R, Mackman N, Horowitz JM, Jensen RL, Durden DL, Van Meir EG, Brat DJ: Early growth response gene-1 regulates hypoxia-induced expression of tissue factor in glioblastoma multiforme through hypoxia-inducible factor-1-independent mechanisms. Cancer Res. 2006, 66: 7067-7074. 10.1158/0008-5472.CAN-06-0346.
Moll T, Czyz M, Holzmuller H, Hofer-Warbinek R, Wagner E, Winkler H, Bach FH, Hofer E: Regulation of the tissue factor promoter in endothelial cells. Binding of NF kappa B-, AP-1-, and Sp1-like transcription factors. J Biol Chem. 1995, 270: 3849-3857. 10.1074/jbc.270.29.17064.
Zhang JJ, Kelm RJ, Biswas P, Kashgarian M, Madri JA: PECAM-1 modulates thrombin-induced tissue factor expression on endothelial cells. J Cell Physiol. 2007, 210: 527-537. 10.1002/jcp.20908.
Nadir Y, Brenner B, Zetser A, Ilan N, Shafat I, Zcharia E, Goldshmidt O, Vlodavsky I: Heparanase induces tissue factor expression in vascular endothelial and cancer cells. J Thromb Haemost. 2006, 4: 2443-2451. 10.1111/j.1538-7836.2006.02212.x.
Abe K, Shoji M, Chen J, Bierhaus A, Danave I, Micko C, Casper K, Dillehay DL, Nawroth PP, Rickles FR: Regulation of vascular endothelial growth factor production and angiogenesis by the cytoplasmic tail of tissue factor. Proc Natl Acad Sci USA. 1999, 96: 8663-8668. 10.1073/pnas.96.15.8663.
Bluff JE, Brown NJ, Reed MWR, Staton CA: Assessment of angiogenesis, vascular endothelial growth factor and tissue factor in human breast Cancer [abstract]. Microcirculation. 2007, 14: P638-
Ueno T, Toi M, Koike M, Nakamura S, Tominaga T: Tissue factor expression in breast cancer tissues: its correlation with prognosis and plasma concentration. Br J Cancer. 2000, 83: 164-170. 10.1054/bjoc.2000.1272.
Shoji M, Hancock WW, Abe K, Micko C, Casper KA, Baine RM, Wilcox JN, Danave I, Dillehay DL, Matthews E, Contrino J, Morrissey JH, Gordon S, Edgington TS, Kudryk B, Kreutzer DL, Rickles FR: Activation of coagulation and angiogenesis in cancer: immunohistochemical localization in situ of clotting proteins and vascular endothelial growth factor in human cancer. Am J Pathol. 1998, 152: 399-411.
Contrino J, Hair G, Kreutzer DL, Rickles FR: In situ detection of tissue factor in vascular endothelial cells: correlation with the malignant phenotype of human breast disease. Nat Med. 1996, 2: 209-215. 10.1038/nm0296-209.
Staton CA, Chetwood AS, Cameron IC, Cross SS, Brown NJ, Reed MW: The angiogenic switch occurs at the adenoma stage of the adenoma carcinoma sequence in colorectal cancer. Gut. 2007, 56: 1426-1432. 10.1136/gut.2007.125286.
Abid Hussein MN, Boing AN, Biro E, Hoek FJ, Vogel GM, Meuleman DG, Sturk A, Nieuwland R: Phospholipid composition of in vitro endothelial microparticles and their in vivo thrombogenic properties. Thromb Res. 2007.
Censarek P, Bobbe A, Grandoch M, Schror K, Weber AA: Alternatively spliced human tissue factor (asHTF) is not pro-coagulant. Thromb Haemost. 2007, 97: 11-14.
Szotowski B, Antoniak S, Poller W, Schultheiss HP, Rauch U: Procoagulant soluble tissue factor is released from endothelial cells in response to inflammatory cytokines. Circ Res. 2005, 96: 1233-1239. 10.1161/01.RES.0000171805.24799.fa.
Hron G, Kollars M, Weber H, Sagaster V, Quehenberger P, Eichinger S, Kyrle PA, Weltermann A: Tissue factor-positive microparticles: cellular origin and association with coagulation activation in patients with colorectal cancer. Thromb Haemost. 2007, 97: 119-123.
Tesselaar ME, Romijn FP, Van Der Linden IK, Prins FA, Bertina RM, Osanto S: Microparticle-associated tissue factor activity: a link between cancer and thrombosis?. J Thromb Haemost. 2007, 5: 520-527. 10.1111/j.1538-7836.2007.02369.x.
Chen VM, Hogg PJ: Allosteric disulfide bonds in thrombosis and thrombolysis. J Thromb Haemost. 2006, 4: 2533-2541. 10.1111/j.1538-7836.2006.02236.x.
Ahamed J, Versteeg HH, Kerver M, Chen VM, Mueller BM, Hogg PJ, Ruf W: Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc Natl Acad Sci USA. 2006, 103: 13932-13937. 10.1073/pnas.0606411103.
Versteeg HH, Ruf W: Tissue factor coagulant function is enhanced by protein-disulfide isomerase independent of oxidoreductase activity. J Biol Chem. 2007, 282: 25416-25424. 10.1074/jbc.M702410200.
Pendurthi UR, Ghosh S, Mandal SK, Rao LV: Tissue factor activation: is disulfide bond switching a regulatory mechanism?. Blood. 2007, 110: 3900-3908. 10.1182/blood-2007-07-101469.
Rak J, Milsom C, May L, Klement P, Yu J: Tissue factor in cancer and angiogenesis: the molecular link between genetic tumor progression, tumor neovascularization, and cancer coagulopathy. Semin Thromb Hemost. 2006, 32: 54-70. 10.1055/s-2006-933341.
Versteeg HH, Schaffner F, Kerver M, Petersen HH, Ahamed J, Felding-Habermann B, Takada Y, Mueller BM, Ruf W: Inhibition of tissue factor signaling suppresses tumor growth. Blood. 2008, 111: 190-199. 10.1182/blood-2007-07-101048.
Uusitalo-Jarvinen H, Kurokawa T, Mueller BM, Andrade-Gordon P, Friedlander M, Ruf W: Role of protease activated receptor 1 and 2 signaling in hypoxia-induced angiogenesis. Arterioscler Thromb Vasc Biol. 2007, 27: 1456-1462. 10.1161/ATVBAHA.107.142539.
Belting M, Dorrell MI, Sandgren S, Aguilar E, Ahamed J, Dorfleutner A, Carmeliet P, Mueller BM, Friedlander M, Ruf W: Regulation of angiogenesis by tissue factor cytoplasmic domain signaling. Nat Med. 2004, 10: 502-509. 10.1038/nm1037.
Goldhaber SZ: Tamoxifen: preventing breast cancer and placing the risk of deep vein thrombosis in perspective. Circulation. 2005, 111: 539-541. 10.1161/01.CIR.0000156099.83394.A7.
Zangari M, Barlogie B, Anaissie E, Saghafifar F, Eddlemon P, Jacobson J, Lee CK, Thertulien R, Talamo G, Thomas T, Van Rhee F, Fassas A, Fink L, Tricot G: Deep vein thrombosis in patients with multiple myeloma treated with thalidomide and chemotherapy: effects of prophylactic and therapeutic anticoagulation. Br J Haematol. 2004, 126: 715-721. 10.1111/j.1365-2141.2004.05078.x.
Kuenen BC, Levi M, Meijers JC, van Hinsbergh VW, Berkhof J, Kakkar AK, Hoekman K, Pinedo HM: Potential role of platelets in endothelial damage observed during treatment with cisplatin, gemcitabine, and the angiogenesis inhibitor SU5416. J Clin Oncol. 2003, 21: 2192-2198. 10.1200/JCO.2003.08.046.
Marx GM, Steer CB, Harper P, Pavlakis N, Rixe O, Khayat D: Unexpected serious toxicity with chemotherapy and antiangiogenic combinations: time to take stock!. J Clin Oncol. 2002, 20: 1446-1448.
Ma L, Francia G, Viloria-Petit A, Hicklin DJ, du Manoir J, Rak J, Kerbel RS: In vitro procoagulant activity induced in endothelial cells by chemotherapy and antiangiogenic drug combinations: modulation by lower-dose chemotherapy. Cancer Res. 2005, 65: 5365-5373. 10.1158/0008-5472.CAN-04-3156.
Molnar S, Guglielmone H, Lavarda M, Rizzi ML, Jarchum G: Procoagulant factors in patients with cancer. Hematology. 2007.
Uno K, Homma S, Satoh T, Nakanishi K, Abe D, Matsumoto K, Oki A, Tsunoda H, Yamaguchi I, Nagasawa T, Yoshikawa H, Aonuma K: Tissue factor expression as a possible determinant of thromboembolism in ovarian cancer. Br J Cancer. 2007, 96: 290-295. 10.1038/sj.bjc.6603552.
Khorana AA, Ahrendt SA, Ryan CK, Francis CW, Hruban RH, Hu YC, Hostetter G, Harvey J, Taubman MB: Tissue factor expression, angiogenesis, and thrombosis in pancreatic cancer. Clin Cancer Res. 2007, 13: 2870-2875. 10.1158/1078-0432.CCR-06-2351.
Hu T, Bach RR, Horton R, Konigsberg WH, Todd MB: Procoagulant activity in cancer cells is dependent on tissue factor expression. Oncol Res. 1994, 6: 321-327.
Rong Y, Post DE, Pieper RO, Durden DL, Van Meir EG, Brat DJ: PTEN and hypoxia regulate tissue factor expression and plasma coagulation by glioblastoma. Cancer Res. 2005, 65: 1406-1413. 10.1158/0008-5472.CAN-04-3376.
Yu JL, May L, Lhotak V, Shahrzad S, Shirasawa S, Weitz JI, Coomber BL, Mackman N, Rak JW: Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumor progression and angiogenesis. Blood. 2005, 105: 1734-1741. 10.1182/blood-2004-05-2042.
Bogdanov VY, Balasubramanian V, Hathcock J, Vele O, Lieb M, Nemerson Y: Alternatively spliced human tissue factor: a circulating, soluble, thrombogenic protein. Nat Med. 2003, 9: 458-462. 10.1038/nm841.
Bajaj MS, Ghosh M, Bajaj SP: Fibronectin-adherent monocytes express tissue factor and tissue factor pathway inhibitor whereas endotoxin-stimulated monocytes primarily express tissue factor: physiologic and pathologic implications. J Thromb Haemost. 2007, 5: 1493-1499. 10.1111/j.1538-7836.2007.02604.x.
Chand HS, Ness SA, Kisiel W: Identification of a novel human tissue factor splice variant that is upregulated in tumor cells. Int J Cancer. 2006, 118: 1713-1720. 10.1002/ijc.21550.
Folkman J: What is the evidence that tumors are angiogenesis dependent?. J Natl Cancer Inst. 1990, 82: 4-6. 10.1093/jnci/82.1.4.
Griffin CT, Srinivasan Y, Zheng YW, Huang W, Coughlin SR: A role for thrombin receptor signaling in endothelial cells during embryonic development. Science. 2001, 293: 1666-1670. 10.1126/science.1061259.
Zhang Y, Deng Y, Luther T, Muller M, Ziegler R, Waldherr R, Stern DM, Nawroth PP: Tissue factor controls the balance of angiogenic and antiangiogenic properties of tumor cells in mice. J Clin Invest. 1994, 94: 1320-1327. 10.1172/JCI117451.
Zhang J, Ding J, Zhang X, Shao X, Hao Z: Regulation of vascular endothelial growth factor (VEGF) production and angiogenesis by tissue factor (TF) in SGC-7901 gastric cancer cells. Cancer Biol Ther. 2005, 4: 769-772.
Carmeliet P, Mackman N, Moons L, Luther T, Gressens P, Van Vlaenderen I, Demunck H, Kasper M, Breier G, Evrard P, Müller M, Risau W, Edgington T, Collen D: Role of tissue factor in embryonic blood vessel development. Nature. 1996, 383: 73-75. 10.1038/383073a0.
Hembrough TA, Swartz GM, Papathanassiu A, Vlasuk GP, Rote WE, Green SJ, Pribluda VS: Tissue factor/factor VIIa inhibitors block angiogenesis and tumor growth through a nonhemostatic mechanism. Cancer Res. 2003, 63: 2997-3000.
Rao LV, Pendurthi UR: Tissue factor–factor VIIa signaling. Arterioscler Thromb Vasc Biol. 2005, 25: 47-56.
Ahamed J, Ruf W: Protease-activated receptor 2-dependent phosphorylation of the tissue factor cytoplasmic domain. J Biol Chem. 2004, 279: 23038-23044. 10.1074/jbc.M401376200.
Dorfleutner A, Ruf W: Regulation of tissue factor cytoplasmic domain phosphorylation by palmitoylation. Blood. 2003, 102: 3998-4005. 10.1182/blood-2003-04-1149.
Versteeg HH, Hoedemaeker I, Diks SH, Stam JC, Spaargaren M, van Bergen En Henegouwen PM, van Deventer SJ, Peppelenbosch MP: Factor VIIa/tissue factor-induced signaling via activation of Src-like kinases, phosphatidylinositol 3-kinase, and Rac. J Biol Chem. 2000, 275: 28750-28756. 10.1074/jbc.M907635199.
Guba M, Yezhelyev M, Eichhorn ME, Schmid G, Ischenko I, Papyan A, Graeb C, Seeliger H, Geissler EK, Jauch KW, Bruns CJ: Rapamycin induces tumor-specific thrombosis via tissue factor in the presence of VEGF. Blood. 2005, 105: 4463-4469. 10.1182/blood-2004-09-3540.
Albrektsen T, Sorensen BB, Hjorto GM, Fleckner J, Rao LV, Petersen LC: Transcriptional program induced by factor VIIa–tissue factor, PAR1 and PAR2 in MDA-MB-231 cells. J Thromb Haemost. 2007, 5: 1588-1597. 10.1111/j.1538-7836.2007.02603.x.
Ollivier V, Bentolila S, Chabbat J, Hakim J, de Prost D: Tissue factor-dependent vascular endothelial growth factor production by human fibroblasts in response to activated factor VII. Blood. 1998, 91: 2698-26703.
Taniguchi T, Kakkar AK, Tuddenham EG, Williamson RC, Lemoine NR: Enhanced expression of urokinase receptor induced through the tissue factor–factor VIIa pathway in human pancreatic cancer. Cancer Res. 1998, 58: 4461-4467.
Camerer E, Rottingen JA, Gjernes E, Larsen K, Skartlien AH, Iversen JG, Prydz H: Coagulation factors VIIa and Xa induce cell signaling leading to up-regulation of the egr-1 gene. J Biol Chem. 1999, 274: 32225-32233. 10.1074/jbc.274.45.32225.
Hjortoe GM, Petersen LC, Albrektsen T, Sorensen BB, Norby PL, Mandal SK, Pendurthi UR, Rao LV: Tissue factor–factor VIIa-specific up-regulation of IL-8 expression in MDA-MB-231 cells is mediated by PAR-2 and results in increased cell migration. Blood. 2004, 103: 3029-3037. 10.1182/blood-2003-10-3417.
Ge L, Ly Y, Hollenberg M, DeFea K: A beta-arrestin-dependent scaffold is associated with prolonged MAPK activation in pseudopodia during protease-activated receptor-2-induced chemotaxis. J Biol Chem. 2003, 278: 34418-34426. 10.1074/jbc.M300573200.
Wiiger MT, Prydz H: The epidermal growth factor receptor (EGFR) and proline rich tyrosine kinase 2 (PYK2) are involved in tissue factor dependent factor VIIa signalling in HaCaT cells. Thromb Haemost. 2004, 92: 13-22.
Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, Hu Z, Barney KA, Degen JL: Tumor cell-associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell-dependent and-independent mechanisms. Blood. 2007, 110: 133-141. 10.1182/blood-2007-01-065995.
Booden MA, Eckert LB, Der CJ, Trejo J: Persistent signaling by dysregulated thrombin receptor trafficking promotes breast carcinoma cell invasion. Mol Cell Biol. 2004, 24: 1990-1999. 10.1128/MCB.24.5.1990-1999.2004.
Bluff JE, Mangera A, Brown NJ, Reed MWR, Staton CA: Tissue factor and vascular endothelial growth factor in breast cancer [abstract]. Angiogenesis. 2006, 9: P31-
Ott I, Fischer EG, Miyagi Y, Mueller BM, Ruf W: A role for tissue factor in cell adhesion and migration mediated by interaction with actin-binding protein 280. J Cell Biol. 1998, 140: 1241-1253. 10.1083/jcb.140.5.1241.
Siegbahn A, Johnell M, Rorsman C, Ezban M, Heldin CH, Ronnstrand L: Binding of factor VIIa to tissue factor on human fibroblasts leads to activation of phospholipase C and enhanced PDGF-BB-stimulated chemotaxis. Blood. 2000, 96: 3452-3458.
Jiang X, Bailly MA, Panetti TS, Cappello M, Konigsberg WH, Bromberg ME: Formation of tissue factor–factor VIIa–factor Xa complex promotes cellular signaling and migration of human breast cancer cells. J Thromb Haemost. 2004, 2: 93-101. 10.1111/j.1538-7836.2004.00545.x.
Morris DR, Ding Y, Ricks TK, Gullapalli A, Wolfe BL, Trejo J: Pro-tease-activated receptor-2 is essential for factor VIIa and Xa-induced signaling, migration, and invasion of breast cancer cells. Cancer Res. 2006, 66: 307-314. 10.1158/0008-5472.CAN-05-1735.
Dorfleutner A, Hintermann E, Tarui T, Takada Y, Ruf W: Crosstalk of integrin α3β1 and tissue factor in cell migration. Mol Biol Cell. 2004, 15: 4416-4425. 10.1091/mbc.E03-09-0640.
Siegbahn A, Johnell M, Sorensen BB, Petersen LC, Heldin CH: Regulation of chemotaxis by the cytoplasmic domain of tissue factor. Thromb Haemost. 2005, 93: 27-34.
Ott I, Weigand B, Michl R, Seitz I, Sabbari-Erfani N, Neumann FJ, Schomig A: Tissue factor cytoplasmic domain stimulates migration by activation of the GTPase Rac1 and the mitogen-activated protein kinase p38. Circulation. 2005, 111: 349-355. 10.1161/01.CIR.0000153333.52294.42.
Ngo CV, Picha K, McCabe F, Millar H, Tawadros R, Tam SH, Nakada MT, Anderson GM: CNTO 859, a humanized anti-tissue factor monoclonal antibody, is a potent inhibitor of breast cancer metastasis and tumor growth in xenograft models. Int J Cancer. 2007, 120: 1261-1267. 10.1002/ijc.22426.
Amarzguioui M, Peng Q, Wiiger MT, Vasovic V, Babaie E, Holen T, Nesland JM, Prydz H: Ex vivo and in vivo delivery of anti-tissue factor short interfering RNA inhibits mouse pulmonary metastasis of B16 melanoma cells. Clin Cancer Res. 2006, 12: 4055-4061. 10.1158/1078-0432.CCR-05-2482.
Bromberg ME, Konigsberg WH, Madison JF, Pawashe A, Garen A: Tissue factor promotes melanoma metastasis by a pathway independent of blood coagulation. Proc Natl Acad Sci USA. 1995, 92: 8205-8209. 10.1073/pnas.92.18.8205.
Mueller BM, Reisfeld RA, Edgington TS, Ruf W: Expression of tissue factor by melanoma cells promotes efficient hematogenous metastasis. Proc Natl Acad Sci USA. 1992, 89: 11832-11836. 10.1073/pnas.89.24.11832.
Mueller BM, Ruf W: Requirement for binding of catalytically active factor VIIa in tissue factor-dependent experimental metastasis. J Clin Invest. 1998, 101: 1372-1378. 10.1172/JCI930.
Bromberg ME, Bailly MA, Konigsberg WH: Role of protease-activated receptor 1 in tumor metastasis promoted by tissue factor. Thromb Haemost. 2001, 86: 1210-1214.
Nobeyama Y, Okochi-Takada E, Furuta J, Miyagi Y, Kikuchi K, Yamamoto A, Nakanishi Y, Nakagawa H, Ushijima T: Silencing of tissue factor pathway inhibitor-2 gene in malignant melanomas. Int J Cancer. 2007, 121: 301-307. 10.1002/ijc.22637.
Dimmeler S, Zeiher AM: Endothelial cell apoptosis in angiogenesis and vessel regression. Circ Res. 2000, 87: 434-439.
Jiang X, Guo YL, Bromberg ME: Formation of tissue factor–factor VIIa–factor Xa complex prevents apoptosis in human breast cancer cells. Thromb Haemost. 2006, 96: 196-201.
Sorensen BB, Rao LV, Tornehave D, Gammeltoft S, Petersen LC: Antiapoptotic effect of coagulation factor VIIa. Blood. 2003, 102: 1708-1715. 10.1182/blood-2003-01-0157.
Versteeg HH, Spek CA, Richel DJ, Peppelenbosch MP: Coagulation factors VIIa and Xa inhibit apoptosis and anoikis. Oncogene. 2004, 23: 410-417. 10.1038/sj.onc.1207066.
Versteeg HH, Spek CA, Slofstra SH, Diks SH, Richel DJ, Peppelenbosch MP: FVIIa:TF induces cell survival via G12/G13-dependent Jak/STAT activation and BclXL production. Circ Res. 2004, 94: 1032-1040. 10.1161/01.RES.0000125625.18597.AD.
Li C, Colman LM, Collier ME, Dyer CE, Greenman J, Ettelaie C: Tumour-expressed tissue factor inhibits cellular cytotoxicity. Cancer Immunol Immunother. 2006, 55: 1301-1308. 10.1007/s00262-006-0130-1.
Amirkhosravi A, Meyer T, Chang JY, Amaya M, Siddiqui F, Desai H, Francis JL: Tissue factor pathway inhibitor reduces experimental lung metastasis of B16 melanoma. Thromb Haemost. 2002, 87: 930-936.
Hu P, Yan J, Sharifi J, Bai T, Khawli LA, Epstein AL: Comparison of three different targeted tissue factor fusion proteins for inducing tumor vessel thrombosis. Cancer Res. 2003, 63: 5046-5053.
Tang Y, Borgstrom P, Maynard J, Koziol J, Hu Z, Garen A, Deisseroth A: Mapping of angiogenic markers for targeting of vectors to tumor vascular endothelial cells. Cancer Gene Ther. 2007, 14: 346-353. 10.1038/sj.cgt.7701030.
Morrow DA, Murphy SA, McCabe CH, Mackman N, Wong HC, Antman EM: Potent inhibition of thrombin with a monoclonal antibody against tissue factor (Sunol-cH36): results of the PROXIMATE-TIMI 27 trial. Eur Heart J. 2005, 26: 682-688. 10.1093/eurheartj/ehi094.
Acknowledgements
The authors would like to thank Yorkshire Cancer Research for funding.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Bluff, J.E., Brown, N.J., Reed, M.W. et al. Tissue factor, angiogenesis and tumour progression. Breast Cancer Res 10, 204 (2008). https://doi.org/10.1186/bcr1871
Published:
DOI: https://doi.org/10.1186/bcr1871