
Abstract
Estrogen and its receptor (ER) are critical for development and progression of breast cancer. This pathway is targeted by endocrine therapies that either block ER functions or deplete ER's estrogen ligand. While endocrine therapies are very effective, de novo and acquired resistance are still common. Laboratory and clinical data now indicate that bidirectional molecular crosstalk between nuclear or membrane ER and growth factor receptor pathways such as HER2/neu is involved in endocrine resistance. Preclinical data suggest that blockade of selected growth factor receptor signaling can overcome this type of resistance, and this strategy is already being tested in clinical trials
Similar content being viewed by others

Introduction
Estrogen receptors (ERs) play a key role in normal breast development and in the development and progression of breast cancer. Endocrine therapy for this disease, which had its start more than 100 years ago when patients were first treated with ovarian ablation [1], blocks the function of ERs and thereby represents the first molecularly targeted therapy for cancer. Long-term studies of patients treated with adjuvant endocrine therapy suggest that this therapy is curative in many patients. Now, many types of endocrine therapy are available that inhibit the function of ERs in different ways. In addition to ovarian ablation, selective ER modulators such as tamoxifen bind ERs and partially block their function [2]. Selective ER downregulators such as fulvestrant completely block the function of ERs and down-regulate the receptor proteins [3]. Finally, aromatase inhibitors in postmenopausal women reduce estrogen to very low levels – a strategy that may be the most effective endocrine therapy used today [4, 5].
However, endocrine therapies do not always work in patients, despite the presence of ERs in their tumors [6]. Some tumors are initially inhibited by endocrine therapy only to develop acquired resistance with prolonged treatment. Other tumors are de novo resistant to endocrine therapy. Clinical experience indicates that tumors resistant to one form of therapy may still be sensitive to other hormonal treatments [7], suggesting that resistance does not always indicate global estrogen independence; resistance can be treatment specific. Insights into the mechanisms of endocrine therapy resistance have come from provocative studies on the biology of ERs and the various signaling pathways in the cell with which they communicate. Potential strategies to overcome this resistance have been successful in preclinical models and are now being tested in patients.
Structure and function of estrogen receptors
ERs are members of a large family of nuclear transcriptional regulators that are activated by steroid hormones such as estrogen [8]. The ERs exist as two isoforms, α and β, that are encoded by two different genes [9]. These receptor proteins have a similar structure but evidence indicates that they have very different functions. ER proteins consist of a DNA binding region and at least two transcriptional activation functions (AFs) known as AF-1 and AF-2 [10]. A ligand binding domain that interacts with estradiol is also located in the AF-2 region [10, 11]. The DNA binding domain of ERs allows the receptors to bind to specific sequences in the promoters of estrogen-regulated genes. AF-1 and AF-2 are important domains for regulating transcription of these genes; AF-1 is constitutively active but is enhanced by growth factor signaling [12], whereas AF-2 is ligand-depend-active with estrogen but not with tamoxifen [13].
Although ER-α and ER-β are both expressed in normal mammary gland, it appears that ER-α, but not ER-β, is critical for normal gland development [14]. A dramatic increase in ER-α content is observed in premalignant hyperproliferative breast lesions and in many breast cancers compared with normal mammary glands [15]. These observations are consistent with the crucial role of the ER-α isoform in breast cancer etiology and progression, and with the role played by estrogens as tumor promoters. Growing evidence suggests that ER-β may antagonize the function of ER-α, and high levels of ER-β are associated with a more favorable response to tamoxifen treatment [16]. However, the overall role of ER-β in regulating breast cancer is yet to be defined, and therefore further discussion in this review focuses mostly on ER-α.
Classical ER-α function
Classically, ER-α regulates the expression of specific genes in the nucleus. This function is sometimes referred to as nuclear initiated steroid signaling [17]. Estrogen binding to ER-α results in phosphorylation of the receptor protein, dissociates chaperone proteins such as heat shock protein 90, and induces a change in protein conformation [11]. This 'activated' ER-α then dimerizes with another receptor protein and the dimer binds to specific DNA sequences sometimes called estrogen response elements in the promoter region of estrogen responsive genes (Fig. 1a) [10]. This complex then recruits a variety of coregulatory proteins such as amplified in breast cancer 1 (AIB1 or SRC3), which function as coactivators to enhance transcription of genes [18]. However, estrogen bound ER-α is also capable of downregulating expression of many genes [19]. Downregulation of gene expression by estrogen may be explained by the recruitment of corepressor proteins to the ER complex at certain gene promoter sequences.

Nuclear ER-α functions. (a) Enhanced transcription of genes with an estrogen response element (ERE) in the promoter. (b) Enhanced transcription of genes by modifying activity of transcription complexes at other promoter sites (activator protein [AP]-1). AIB, amplified in breast cancer; At, acetyltransferase; E, estradiol; ER, estrogen receptor-α.
The coregulatory proteins complexed with ER-α on the promoters of target genes are very important for the function of ER-α [20, 21]. Coactivators recruit other proteins including acetyltransferases, which alter chromatin structure to facilitate transcription [20–22]. AIB1 is a very important ER-α coactivator protein and could even function as an oncogene when overexpressed in preclinical models [23]. Reducing the level of AIB1 impedes ER-α function and can inhibit the growth of human breast tumor xenografts in mice [24]. AIB1 is also important in breast cancer in patients. It is overexpressed in 65% of breast cancers and is occasionally gene-amplified, suggesting that it plays an important role in the development and progression of breast cancer [25, 26]. Preclinical studies and clinical studies in patients suggest that high levels of AIB1 may contribute to tamoxifen resistance by enhancing the estrogen agonist activity of the drug [27–29]. This may be particularly true in tumors that also express high levels of HER2. Kinases activated by HER2 phosphorylate both ER and AIB1, resulting in an increase in ER-α activity [29]. In this situation, tamoxifen-bound ER-α may recruit coactivators such as AIB1 rather than corepressors, resulting in tamoxifen-stimulated growth as a mechanism for resistance to the drug.
ER-α has also been shown to modulate gene expression in other ways. Through protein–protein interactions ER can bind to transcriptional complexes on other regulatory DNA sequences such as activating protein (AP)-1, Sp1, and upstream stimulatory factor (USF) sites, as well as other poorly defined promoter regions that are not typical estrogen response elements (Fig. 1b) [30–33]. In this circumstance ER-α is not the primary transcription factor binding DNA but it is tethered to other transcription factor proteins such as c-jun or c-fos. It then functions as a coactivator protein by stabilizing DNA binding of the transcription complex or by recruiting other coactivator proteins to these complexes [34, 35]. Several genes important for tumor cell proliferation and survival may be regulated by estrogen in this way, and data suggest that modulation of gene expression at AP-1 sites may be the most critical element in estrogen-mediated tumor growth [30, 36].
Through these direct transcriptional mechanisms, estrogen regulates the expression of hundreds of genes, many of which are important for cell proliferation, antiapoptosis, invasion and metastasis, and angiogenesis. c-Myc, vascular endothelial growth factor, bcl-2, insulin-like growth factor (IGF)-R1, insulin receptor substrate-1, transforming growth factor-α, cyclin-D1, and IGF-2 are all regulated by estrogen [34, 37–41]. Downregulation of expression of these and other genes and/or upregulation of genes related to apoptosis or cell cycle inhibition may explain the curative potential of adjuvant endocrine therapy. Altered gene expression may also contribute to certain forms of endocrine therapy resistance. As mentioned above, the ER-α coactivator AIB1 can bind to tamoxifen-bound ER-α complexes under some circumstances, potentially reducing the estrogen antagonist activity of the drug [29]. Increased AP-1 transcriptional activity has been identified in preclinical models and in patients with tamoxifen resistance [42, 43]. More recent data suggest that ER-α crosstalk with growth factor receptor signaling pathways can modulate ER-α transcriptional activity and may also contribute to resistance to tamoxifen or other endocrine therapies [37, 38].
Modification of transcriptional function of estrogen receptor-α by growth factor receptor signaling
Numerous growth factors and signaling molecules including IGF-1, epidermal growth factor (EGF), heregulin, transforming growth factor-α, dopamine, cyclic AMP, and membrane permeable phosphatase inhibitors can all potentiate the nuclear transcriptional effects of ER-α [40, 44–46]. This functional activation of ER-α results from direct phosphorylation of the ER-α protein at multiple sites, mainly at the AF-1 domain of the receptor [47], or from phosphorylation of ER-α coactivators and corepressors [48]. This potentiation of ER-α transcriptional activity due to phosphorylation of the ligand independent AF-1 domain has been termed ligand independent activation of ER-α, because it can occur in the absence of estrogen. Multiple kinases involved in growth factor, stress, or cytokine induced signaling can phosphorylate ER-α or its coregulatory proteins. These include extracellular signal regulated kinase (ERK)1/2 and p38 mitogen-activated protein kinases (MAPKs), cyclin A/CDK-2, CDK-7, c-Src, protein kinase A, pp90rsk1, Akt, IκB kinase, and Jun amino terminal kinase [12, 49–57]. Phosphorylation of ER-α on these various sites affects protein dimerization, DNA binding, ligand binding, nuclear localization, interactions with coactivators and corepressors, and transcriptional activation of genes [53, 54, 58–60]. Phosphorylation of ER-α coregulatory proteins augments their activity on ER-α dependent transcription, alters nuclear localization, affects their binding to ER-α, and enhances their ability to recruit other transcriptional coregulators [56, 57, 61]. Phosphorylation of ER-α corepressors such as N-CoR also changes their affinity for ER-α binding and modifies nuclear localization [62–64]. Thus, estrogen regulated gene expression is a coordinated effort that depends on the receptor itself, its coregulatory molecules, specific gene promoter sequences, the presence or absence of other transcription factors, and input from a variety of intracellular signaling pathways.
Membrane initiated (nongenomic) steroid signaling
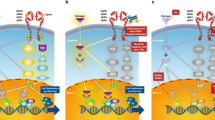
ER-α functions not only as a transcription factor in the nucleus; new data suggest that it can activate intracellular signaling pathways outside the nucleus in the cytoplasm or the plasma membrane (Fig. 2) [65]. It has been observed for decades that estrogens can have very rapid effects on cells, too rapid to invoke transcriptional mechanisms [66]. ERs have been localized outside the nucleus by biochemical analyses and by direct visualization using immunocytochemistry or more sophisticated microscopy [67, 68]. This nongenomic mode of action of ERs, as well as of other steroid receptors, is also known as membrane-initiated steroid signaling [17]. Many studies have now shown that non-nuclear ER-α can exist in complexes with several signaling molecules. Estrogen treatment of cells induces the interaction between ER-α and IGF-1 receptor, which activates signaling via ERK1/2 MAPK [69]. Estrogen also stimulates association between ER-α and the p85 subunit of phosphatidylinositol-3-OH kinase, resulting in its activation [70]. ER-α interacts with other IGF signaling intermediates including insulin receptor substrate 1 [71], which then translocates to the nucleus where it may exist in transcription complexes. ER-α can also interact with Shc, which links the receptor to other signaling intermediates in the membrane [72]. Thus, ER can complex with many elements in the IGF-1R signaling cascade, thereby activating this pathway in response to estrogen.

Membrane or nongenomic functions of ER-α. Estrogen (E) or tamoxifen binds membrane or cytoplasmic estrogen receptor (ER)-α to trigger complexes with growth factor signaling molecules (such as insulin-like growth factor [IGF]-R1, insulin receptor substrate [IRS]-1, or phosphatidylinositol-3-OH kinase [PI3K]), which activate mitogen-activated protein kinase (MAPK) or AKT. ER-α also acting as a G-protein-coupled receptor activates Src, which activates matrix metalloproteinase (MMP)-2, which cleaves heparin-binding epidermal growth factor (Hb-EGF) from the membrane to bind and activate EGF receptor (EGFR). EGFR also activates MAPK and AKT, which in turn can phosphorylate and activate nuclear ER-α and amplified in breast cancer (AIB)1, or directly contribute to tumor growth and survival (dotted line).
ER-α has also been shown to act as a G-protein-coupled receptor at the membrane, where it binds to caveolin-1 (Fig. 2) [73–75]. In this way, ER-α can activate c-Src, in turn leading to activation of metalloproteinases that then cleave heparin binding EGF from the cell. This growth factor, acting in an autocrine manner, binds EGF receptors on the cell membrane, thereby activating downstream kinases such as ERK1/2 MAPK and phosphatidylinositol-3-OH kinase. The observation that tamoxifen, like estrogen, can activate membrane ER suggests a plausible mechanism for tamoxifen resistance in tumors in which this pathway is active. Pure ER antagonists such as fulvestrant do not activate membrane ER [69, 75]. This type of ER-α activity may not be prominent in most breast cancers that lack strong receptor tyrosine kinase activity, but this activity can be potentiated in tumors that overexpress tyrosine kinase receptors [29, 76] or in those that expresshigh levels of other proteins that bind ER-α and sequester it in the cytoplasm or membrane. Such proteins as modulator of nongenomic activity of ER (MNAR) [77–79] and members of the metastasis associated gene family (MTA) [80, 81] may be important in membrane initiated ER-α signaling by trapping it outside the nucleus.
Thus, ER-α may stimulate breast cancer growth in at least two ways. It increases the expression of growth regulatory genes via its transcriptional effects in the nucleus and it activates the growth factor signaling cascade via its nongenomic or membrane activity outside the nucleus. In some tumors, particularly those with amplification of HER2 or overexpression of other growth factor receptor tyrosine kinases, a vicious cycle of cell proliferative and cell survival stimuli is initiated by the binding of estrogen to its receptor. ER-α by a variety of mechanisms activates the growth factor signaling cascade, while at the same time the growth factor receptor kinase cascade phosphorylates and activates ER-α and its coregulating proteins. Membrane or nonnuclear ER-α signaling has been described in experimental model systems and needs to be confirmed in clinical breast cancer. Tumors with active ER-α/growth factor receptor crosstalk, especially those that are largely dependent on the membrane/non-nuclear ER-α signaling, could be expected to be very dependent on estrogen for growth and might then be highly responsive to treatment with estrogen deprivation therapy, such as aromatase inhibition. These tumors should also be responsive to selective ER downregulators, such as fulvestrant, which function as complete antagonists on both nuclear and non-nuclear ER activities. Tamoxifen, on the other hand, because it activates membrane ER-α signaling might not be as effective, and in fact it might even stimulate growth of such tumors [29]. As described below, clinical data do support these paradigms. An understanding of these ER activities at the molecular level may yield new strategies to prevent or overcome resistance to tamoxifen and other forms of treatment.
Blocking estrogen receptor/growth factor receptor crosstalk to overcome treatment resistance
The molecular functions of ER-α and its crosstalk with growth factor receptors are exploitable for treatment purposes. Preclinical experimental models first suggested that tumors overexpressing HER2, in addition to ER-α, might be tamoxifen resistant [82]. Since then, the bulk of clinical evidence indicates that patients with HER2-overexpressing ER-α-positive breast cancers derive less benefit from tamoxifen than do those with HER2-negative breast cancers [29, 83–85]. A recent clinical report [86] also suggested that activation of ER-α function by growth factor signaling is important for tamoxifen resistance. Tamoxifen-treated patients whose tumors expressed high levels of both AIB1 and HER2 had extremely poor disease-free survival compared with patients whose tumors overexpressed neither or only one of the two proteins. This clinical observation makes biologic sense because kinases activated downstream from HER2 phosphorylate and functionally activate AIB1 and ER-α, thereby reducing the antagonist activity of the tamoxifen-bound receptor.
Prospective trials in patients with metastatic disease also suggest that tamoxifen resistance is associated with high levels of expression of receptor tyrosine kinases such as HER2 and EGF receptor [87]. Furthermore, because resistance to selective ER modulators such as tamoxifen is mediated, at least in part, via activation of the nongenomic actions of ER, other hormonal treatments that can block this ER activity should be effective second-line therapies for patients whose tumors acquired resistance to tamoxifen. Importantly, aromatase inhibitors and the ER downregulator fulvestrant, which can deactivate both the genomic and the nongenomic effects of ER, are indeed effective therapies in tamoxifen resistant patients [7]. Finally, three studies in which patients were given neoadjuvant endocrine therapy as initial treatment further support these hypotheses [88–90]. Two of these trials randomized patients to tamoxifen or an aromatase inhibitor. Both found that tumor response to tamoxifen was about half of that observed with the aromatase inhibitor in tumors overexpressing HER2. The response rates were more similar for tumors that were negative for HER2. In fact, the response rates to the aromatase inhibitor in these two trials, as well as in a third nonrandomized trial, were at least as high or higher in tumors overexpressing HER2 than in HER2 negative tumors. These neoadjuvant trials do not provide data on the duration of response to the aromatase inhibitors in HER2 positive disease. Preclinical studies suggest that growth factor receptor signaling can also contribute to acquired resistance to estrogen deprivation therapies [91]. Nevertheless, the substantial initial tumor response in the HER2 positive subset of patients suggests that these tumors are indeed very dependent on estrogen for growth. Analysis of completed large adjuvant trials with patients classified by ER-α, progesterone receptor, and HER2 status are needed to confirm the superior efficacy of aromatase inhibitors compared with tamoxifen in such patients.
If the growth factor receptor tyrosine kinase signaling pathway is involved in resistance to hormonal therapy, then an obvious question is whether blockade of these pathways can delay or overcome treatment resistance (Fig. 3). Compeling data from in vitro and in vivo experimental models suggest that this strategy is worth testing in patients. In a xenograft model of an ER-α positive, HER2 overexpressing tumor, tamoxifen-stimulated growth mediated by growth factor receptor activation by nongenomic ER-α signaling is the mechanism for de novo resistance [29]. Simultaneous treatment of mice with the EGF receptor tyrosine kinase inhibitor gefitinib blocked activation of HER2 signaling by tamoxifen, reduced phosphorylation of ER-α and AIB1, and completely restored tamoxifen's antagonist activity on tumor growth. Similar data have been reported in other experimental models [85]. In a recent study [92], lapatinib – a dual EGF receptor/HER2 tyrosine kinase inhibitor – effectively inhibited the growth of non-HER2 overexpressing tumors that had developed acquired resistance to tamoxifen. Gefitinib and lapatinib are now in clinical trials to test the concept that simultaneous treatment with growth factor inhibitors combined with ER-targeted therapy is more effective than either treatment by itself.

Blockage of growth factor receptor signaling pathways to overcome tamoxifen resistance. Tamoxifen (T) bound estrogen receptor (ER)-α in the membrane can activate epidermal growth factor receptor (EGFR), setting up a signaling cascade that leads to tamoxifen resistance. Blocking EGFR activation would block activation of mitogen-activated protein kinase (MAPK), AKT, ER-α and amplified in breast cancer (AIB)1, and would restore tamoxifen's antagonist activity on tumor growth.
Conclusion
Although ER-α was discovered nearly 40 years ago, scientists continue to unravel its complex role in cellular function. It is clear that ER-α has multiple activities and that it often does not act alone but rather in concert with other signaling pathways that can modify its output. Some of these pathways lead to specific changes in receptor activity that contribute to resistance to hormonal therapies. Because there are many signaling molecules that can modulate the function of ERs, an accurate molecular profile of each patient's tumor will be required in the future to design the optimal therapeutic approach by identifying the major pathways that are working together to drive the growth of a particular tumor. The fact that targeting ER-α by itself offers long-term disease control in many patients that is tantamount to cure indicates that the mechanisms regulating breast cancer growth may not be as complicated as some may have thought, and that accurate identification of the pathway or pathways that regulate tumor cell survival and proliferation and then blocking only those pathways may prove to be an extraordinarily successful strategy.
Note
This article is part of a review series on Endocrinology and hormone therapy in breast cancer, edited by James N Ingle and V Craig Jordan.
Other articles in the series can be found online at http://breast-cancer-research.com/articles/review-series.asp?series=bcr_endocrinology
Abbreviations
- AF:
-
activation function
- AIB:
-
amplified in breast cancer
- AP:
-
activating protein
- EGF:
-
epidermal growth factor
- ER:
-
estrogen receptor
- ERK:
-
extracellular signal-regulated kinase
- IGF:
-
insulin-like growth factor
- MAPK:
-
mitogen-activated protein kinase
- USF:
-
upstream stimulatory factor.
References
Beatson GT: On the treatment of inoperable cases of carcinogen of the mamma: suggestions for a new method of treatment with illustrative cases. Lancet. 1896, 2: 104-107. 162-167.
Osborne CK, Zhao H, Fuqua SA: Selective estrogen receptor modulators: structure, function, and clinical use. J Clin Oncol. 2000, 18: 3172-3186.
Howell A, Osborne CK, Morris C, Wakeling AE: ICI 182,780 (Faslodex): development of a novel, 'pure' antiestrogen. Cancer. 2000, 89: 817-825. 10.1002/1097-0142(20000815)89:4<817::AID-CNCR14>3.0.CO;2-6.
Winer EP, Hudis C, Burstein HJ, Wolff AC, Pritchard KI, Ingle JN, Chlebowski RT, Gelber R, Edge SB, Gralow J, et al: American Society of Clinical Oncology technology assessment on the use of aromatase inhibitors as adjuvant therapy for postmenopausal women with hormone receptor-positive breast cancer: status report 2004. J Clin Oncol. 2005, 23: 619-629. 10.1200/JCO.2005.09.121.
Howell A, Dowsett M: Endocrinology and hormone therapy in breast cancer: aromatase inhibitors versus antioestrogens. Breast Cancer Res. 2004, 6: 269-274. 10.1186/bcr945.
Osborne CK, Fuqua SA: Mechanisms of tamoxifen resistance. Breast Cancer Res Treat. 1994, 32: 49-55. 10.1007/BF00666205.
Carlson RW: Sequencing of endocrine therapies in breast cancer–integration of recent data. Breast Cancer Res Treat. 2002, 75: S27-S32. 10.1023/A:1020361700012. discussion S33-S25.
Parker MG: Steroid and related receptors. Curr Opin Cell Biol. 1993, 5: 499-504. 10.1016/0955-0674(93)90016-J.
Mosselman S, Polman J, Kijkema R: Identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996, 392: 49-53. 10.1016/0014-5793(96)00782-X.
Schiff R, Fuqua SAW: The importance of the estrogeb receptor in breast cancer. Breast Cancer: Prognosis, Treatment, and Prevention. Edited by: Pasqualini JR. 2002, New York, Basel: Marcel Dekker, Inc, 149-186.
Osborne CK, Schiff R, Fuqua SAW, Shou J: Estrogen receptor: current understanding of its activation and modulation. Clin Cancer Res. 2001, 7: 4338-4342.
Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, et al: Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995, 270: 1491-1494.
Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL: The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998, 95: 927-937. 10.1016/S0092-8674(00)81717-1.
Bocchinfuso WP, Korach KS: Mammary gland development and tumorigenesis in estrogen receptor knockout mice. J Mammary Gland Biol Neoplasia. 1997, 2: 323-334. 10.1023/A:1026339111278.
Allred DC, Mohsin SK, Fuqua SA: Histological and biological evolution of human premalignant breast disease. Endocr Relat Cancer. 2001, 8: 47-61. 10.1677/erc.0.0080047.
Hopp TA, Weiss H, Parra I, Cui Y, Osborne CK, Fuqua SA: Low levels of estrogen receptor beta protein predict resistance to tamoxifen therapy in breast cancer. Clin Cancer Res. 2004, 10: 7490-7499.
Nemere I, Pietras RJ, Blackmore PF: Membrane receptors for steroid hormones: signal transduction and physiological significance. J Cell Biochem. 2003, 88: 438-445. 10.1002/jcb.10409.
McKenna NJ, Nawaz Z, Tsai SY, Tsai M-J, O'Malley BW: Distinct steady-state nuclear receptor coregulator complexes exist in vivo. Proc Natl Acad Sci USA. 1998, 95: 11697-11702. 10.1073/pnas.95.20.11697.
Dobrzycka KM, Townson SM, Jiang S, Oesterreich S: Estrogen receptor corepressors: a role in human breast cancer?. Endocr Relat Cancer. 2003, 10: 517-536. 10.1677/erc.0.0100517.
McKenna NJ, Lanz RB, O'Malley BW: Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999, 20: 321-344. 10.1210/er.20.3.321.
Horwitz KB, Jackson TA, Bain DL, Richer JK, Takimoto GS, Tung L: Nuclear receptor coactivators and corepressors. Mol Endocrinol. 1996, 10: 1167-1177. 10.1210/me.10.10.1167.
Fuqua SAW, Schiff R: The biology of estrogen receptors. Diseases of the Breast. Edited by: Harris JR, Lippman ME, Morrow M, Osborne CK. 2004, Philadelphia, PA: Lippincott Williams & Wilkins, 585-602. 3
Torres-Arzayus MI, De Mora JF, Yuan J, Vazquez F, Bronson R, Rue M, Sellers WR, Brown M: High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell. 2004, 6: 263-274. 10.1016/j.ccr.2004.06.027.
List HJ, Lauritsen KJ, Reiter R, Powers C, Wellstein A, Riegel AT: Ribozyme targeting demonstrates that the nuclear receptor coactivator AIB1 is a rate-limiting factor for estrogen-dependent growth of human MCF-7 breast cancer cells. J Biol Chem. 2001, 276: 23763-23768. 10.1074/jbc.M102397200.
Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan XY, Sauter G, Kallioniemi OP, Trent JM, Meltzer PS: AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science. 1997, 277: 965-968. 10.1126/science.277.5328.965.
Bouras T, Southey MC, Venter DJ: Overexpression of the steroid receptor coactivator AIB1 in breast cancer correlates with the absence of estrogen and progesterone receptors and positivity for p53 and HER2/neu. Cancer Res. 2001, 61: 903-907.
Smith CL, Nawaz Z, O'Malley BW: Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol. 1997, 11: 657-666. 10.1210/me.11.6.657.
Takimoto GS, Graham JD, Jackson TA, Tung L, Powell RL, Horwitz LD, Horwitz KB: Tamoxifen resistant breast cancer: coregulators determine the direction of transcription by antagonist-occupied steroid receptors. J Steroid Biochem Mol Biol. 1999, 69: 45-50. 10.1016/S0960-0760(98)00148-4.
Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, Schiff R: Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004, 96: 926-935.
Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P: Estrogen receptor pathways to AP-1. Steroid Biochem Mol Biol. 2000, 74: 311-317. 10.1016/S0960-0760(00)00108-4.
Safe S: Transcriptional activation of genes by 17 beta-estradiol through estrogen receptor-Sp1 interactions. Vitam Horm. 2001, 62: 231-252.
Xing W, Archer TK: Upstream stimulatory factors mediate estrogen receptor activation of the cathepsin D promoter. Mol Endocrinol. 1998, 12: 1310-1321. 10.1210/me.12.9.1310.
Ray P, Ghosh SK, Zhang DH, Ray A: Repression of interleukin-6 gene expression by 17 beta-estradiol: inhibition of the DNA-binding activity of the transcription factors NF-IL6 and NF-kappa B by the estrogen receptor. FEBS Lett. 1997, 409: 79-85. 10.1016/S0014-5793(97)00487-0.
Sanchez R, Nguyen D, Rocha W, White JH, Mader S: Diversity in the mechanisms of gene regulation by estrogen receptors. Bioessays. 2002, 24: 244-254. 10.1002/bies.10066.
Wang F, Duan R, Chirgwin J, Safe SH: Transcriptional activation of cathepsin D gene expression by growth factors. J Mol Endocrinol. 2000, 24: 193-202. 10.1677/jme.0.0240193.
Liu Y, Ludes-Meyers J, Zhang Y, Munoz-Medellin D, Kim HT, Lu C, Ge G, Schiff R, Hilsenbeck SG, Osborne CK, Brown PH: Inhibition of AP-1 transcription factor causes blockade of multiple signal transduction pathways and inhibits breast cancer growth. Oncogene. 2002, 21: 7680-7689. 10.1038/sj.onc.1205883.
Schiff R, Massarweh S, Shou J, Osborne CK: Breast cancer endocrine resistance: how growth factor signaling and estrogen receptor coregulators modulate response. Clin Cancer Res. 2003, 9: 447S-454S.
Nicholson RI, McClelland RA, Robertson JF, Gee JM: Involvement of steroid hormone and growth factor cross-talk in endocrine response in breast cancer. Endocr Relat Cancer. 1999, 6: 373-387. 10.1677/erc.0.0060373.
Huynh H, Yang X, Pollak M: Estradiol and antiestrogens regulate a growth inhibitory insulin-like growth factor binding protein 3 autocrine loop in human breast cancer cells. J Biol Chem. 1996, 271: 1016-1021. 10.1074/jbc.271.2.1016.
Lee AV, Cui X, Oesterreich S: Cross-talk among estrogen receptor, epidermal growth factor, and insulin-like growth factor signaling in breast cancer. Clin Cancer Res. 2001, 7: 4429s-4435s. discussion 4411s-4412s.
Klinge CM: Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29: 2905-2919. 10.1093/nar/29.14.2905.
Johnston SR, Lu B, Scott GK, Kushner PJ, Smith IE, Dowsett M, Benz CC: Increased activator protein-1 DNA binding and c-Jun NH2-terminal kinase activity in human breast tumors with acquired tamoxifen resistance. Clin Cancer Res. 1999, 5: 251-256.
Schiff R, Reddy P, Ahotupa M, Coronado-Heinsohn E, Grim M, Hilsenbeck SG, Lawrence R, Deneke S, Herrera R, Chamness GC, et al: Oxidative stress and AP-1 activity in tamoxifen-resistant breast tumors in vivo. J Natl Cancer Inst. 2000, 92: 1926-1934. 10.1093/jnci/92.23.1926.
Smith CL, Conneely OM, O'Malley BW: Modulation of the ligand-independent activation of the human estrogen receptor by hormone and antihormone. Proc Natl Acad Sci USA. 1993, 90: 6120-6124.
Aronica SM, Katzenellenbogen BS: Stimulation of estrogen receptor-mediated transcription and alteration in the phosphorylation state of the rat uterine estrogen receptor by estrogen, cyclic adenosine monophosphate, and insulin-like growth factor-I. Mol Endocrinol. 1993, 7: 743-752. 10.1210/me.7.6.743.
Auricchio F, Di Domenico M, Migliaccio A, Castoria G, Bilancio A: The role of estradiol receptor in the proliferative activity of vanadate on MCF-7 cells. Cell Growth Differ. 1995, 6: 105-113.
Ali S, Coombes RC: Endocrine-responsive breast cancer and strategies for combating resistance. Nat Rev Cancer. 2002, 2: 101-112. 10.1038/nrc721.
Schiff R, Massarweh SA, Shou J, Bharwani L, Mohsin SK, Osborne CK: Cross-talk between estrogen receptor and growth factor pathways as a molecular target for overcoming endocrine resistance. Clin Cancer Res. 2004, 10: 331S-336S.
Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H: Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem. 2001, 276: 9817-9824. 10.1074/jbc.M010840200.
Lee H, Jiang F, Wang Q, Nicosia SV, Yang J, Su B, Bai W: MEKK1 activation of human estrogen receptor alpha and stimulation of the agonistic activity of 4-hydroxytamoxifen in endometrial and ovarian cancer cells. Mol Endocrinol. 2000, 14: 1882-1896. 10.1210/me.14.11.1882.
Rogatsky I, Trowbridge JM, Garabedian MJ: Potentiation of human estrogen receptor alpha transcriptional activation through phosphorylation of serines 104 and 106 by the cyclin A-CDK2 complex. J Biol Chem. 1999, 274: 22296-22302. 10.1074/jbc.274.32.22296.
Chen D, Washbrook E, Sarwar N, Bates GJ, Pace PE, Thirunuvakkarasu V, Taylor J, Epstein RJ, Fuller-Pace FV, Egly JM, et al: Phosphorylation of human estrogen receptor alpha at serine 118 by two distinct signal transduction pathways revealed by phosphorylation-specific antisera. Oncogene. 2002, 21: 4921-4931. 10.1038/sj.onc.1205420.
Arnold SF, Obourn JD, Jaffe H, Notides AC: Phosphorylation of the human estrogen receptor on tyrosine 537 in vivo and by src family tyrosine kinases in vitro. Mol Endocrinol. 1995, 9: 24-33. 10.1210/me.9.1.24.
Le Goff P, Montano MM, Schodin DJ, Katzenellenbogen BS: Phosphorylation of the human estrogen receptor. Identification of hormone-regulated sites and examination of their influence on transcriptional activity. J Biol Chem. 1994, 269: 4458-4466.
Joel PB, Smith J, Sturgill TW, Fisher TL, Blenis J, Lannigan DA: pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol Cell Biol. 1998, 18: 1978-1984.
Wu RC, Qin J, Hashimoto Y, Wong J, Xu J, Tsai SY, Tsai MJ, O'Malley BW: Regulation of SRC-3 (pCIP/ACTR/AIB-1/RAC-3/TRAM-1) Coactivator activity by I kappa B kinase. Mol Cell Biol. 2002, 22: 3549-3561. 10.1128/MCB.22.10.3549-3561.2002.
Wu RC, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ, O'Malley BW: Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic reponses to multiple cellular signaling pathways. Mol Cell. 2004, 15: 937-949. 10.1016/j.molcel.2004.08.019.
Ali S, Metzger D, Bornert JM, Chambon P: Modulation of transcriptional activation by ligand-dependent phosphorylation of the human oestrogen receptor A/B region. EMBO J. 1993, 12: 1153-1160.
Chen D, Pace PE, Coombes RC, Ali S: Phosphorylation of human estrogen receptor alpha by protein kinase A regulates dimerization. Mol Cell Biol. 1999, 19: 1002-1015.
Lee H, Bai W: Regulation of estrogen receptor nuclear export by ligand-induced and p38-mediated receptor phosphorylation. Mol Cell Biol. 2002, 22: 5835-5845. 10.1128/MCB.22.16.5835-5845.2002.
Font de Mora J, Brown M: AIB1 is a conduit for kinase-mediated growth factor signaling to the estrogen receptor. Mol Cell Biol. 2000, 20: 5041-5047. 10.1128/MCB.20.14.5041-5047.2000.
Lavinsky RM, Jepsen K, Heinzel T, Torchia J, Mullen TM, Schiff R, Del-Rio AL, Ricote M, Ngo S, Gemsch J, et al: Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proc Natl Acad Sci USA. 1998, 95: 2920-2925. 10.1073/pnas.95.6.2920.
Hong SH, Privalsky ML: The SMRT corepressor is regulated by a MEK-1 kinase pathway: inhibition of corepressor function is associated with SMRT phosphorylation and nuclear export. Mol Cell Biol. 2000, 20: 6612-6625. 10.1128/MCB.20.17.6612-6625.2000.
Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG: Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-amyloid precursor protein. Cell. 2002, 110: 55-67. 10.1016/S0092-8674(02)00809-7.
Levin ER: Cellular functions of the plasma membrane estrogen receptor. Trends Endocrinol Metab. 1999, 10: 374-377. 10.1016/S1043-2760(99)00192-7.
Selye H: Correlations between the chemical structure and the pharmacological actions of the steroids. Endocrinology. 1942, 30: 437-453.
Razandi M, Pedram A, Greene GL, Levin ER: Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERalpha and ERbeta expressed in Chinese hamster ovary cells. Mol Endocrinol. 1999, 13: 307-319. 10.1210/me.13.2.307.
Pappas TC, Gametchu B, Watson CS: Membrane estrogen receptors identified by multiple antibody labeling and impeded-ligand binding. FASEB J. 1995, 9: 404-410.
Kahlert S, Nuedling S, van Eickels M, Vetter H, Meyer R, Grohe C: Estrogen receptor alpha rapidly activates the IGF-1 receptor pathway. J Biol Chem. 2000, 275: 18447-18453. 10.1074/jbc.M910345199.
Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK: Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000, 407: 538-541. 10.1038/35035131.
Morelli C, Garofalo C, Bartucci M, Surmacz E: Estrogen receptor-alpha regulates the degradation of insulin receptor substrates 1 and 2 in breast cancer cells. Oncogene. 2003, 22: 4007-4016. 10.1038/sj.onc.1206436.
Song RX, McPherson RA, Adam L, Bao Y, Shupnik M, Kumar R, Santen RJ: Linkage of rapid estrogen action to MAPK activation by ERalpha-Shc association and Shc pathway activation. Mol Endocrinol. 2002, 16: 116-127. 10.1210/me.16.1.116.
Anderson RG: The caveolae membrane system. Annu Rev Biochem. 1998, 67: 199-225. 10.1146/annurev.biochem.67.1.199.
Razandi M, Alton G, Pedram A, Ghonshani S, Webb P, Levin ER: Identification of a structural determinant necessary for the localization and function of estrogen receptor alpha at the plasma membrane. Mol Cell Biol. 2003, 23: 1633-1646. 10.1128/MCB.23.5.1633-1646.2003.
Razandi M, Pedram A, Park ST, Levin ER: Proximal events in signaling by plasma membrane estrogen receptors. J Biol Chem. 2003, 278: 2701-2712. 10.1074/jbc.M205692200.
Chung YL, Sheu ML, Yang SC, Lin CH, Yen SH: Resistance to tamoxifen-induced apoptosis is associated with direct interaction between Her2/neu and cell membrane estrogen receptor in breast cancer. Int J Cancer. 2002, 97: 306-312. 10.1002/ijc.1614.
Vadlamudi RK, Wang RA, Mazumdar A, Kim Y, Shin J, Sahin A, Kumar R: Molecular cloning and characterization of PELP1, a novel human coregulator of estrogen receptor alpha. J Biol Chem. 2001, 276: 38272-38279.
Balasenthil S, Vadlamudi RK: Functional interactions between the estrogen receptor coactivator PELP1/MNAR and retinoblastoma protein. J Biol Chem. 2003, 278: 22119-22127. 10.1074/jbc.M212822200.
Wong CW, McNally C, Nickbarg E, Komm BS, Cheskis BJ: Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. Proc Natl Acad Sci USA. 2002, 99: 14783-14788. 10.1073/pnas.192569699.
Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W: NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998, 2: 851-861. 10.1016/S1097-2765(00)80299-3.
Kumar R: Another tie that binds the MTA family to breast cancer. Cell. 2003, 113: 142-143. 10.1016/S0092-8674(03)00274-5.
Benz CC, Scott GK, Sarup JC, Johnson RM, Tripathy D, Coronado E, Shepard HM, Osborne CK: Estrogen-dependent, tamoxifen-resistant tumorigenic growth of MCF-7 cells transfected with HER2/neu. Breast Cancer Res Treat. 1993, 24: 85-95.
Mass R: The role of HER-2 expression in predicting response to therapy in breast cancer. Semin Oncol. 2000, 27: 46-52. discussion 92-100.
Ciocca DR, Elledge R: Molecular markers for predicting response to tamoxifen in breast cancer patients. Endocrine. 2000, 13: 1-10. 10.1385/ENDO:13:1:1.
Nicholson RI, Hutcheson IR, Harper ME, Knowlden JM, Barrow D, McClelland RA, Jones HE, Wakeling AE, Gee JM: Modulation of epidermal growth factor receptor in endocrine-resistant, estrogen-receptor-positive breast cancer. Ann N Y Acad Sci. 2002, 963: 104-115.
Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM, Schiff R: Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/ neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2003, 95: 353-361.
Arpino G, Green SJ, Allred DC, Lew D, Martino S, Osborne CK, Elledge RM: HER-2 amplification, HER-1 expression, and tamoxifen response in estrogen receptor-positive metastatic breast cancer: a southwest oncology group study. Clin Cancer Res. 2004, 10: 5670-5676.
Ellis MJ, Coop A, Singh B, Mauriac L, Llombert-Cussac A, Janicke F, Miller WR, Evans DB, Dugan M, Brady C, et al: Letrozole is more effective neoadjuvant endocrine therapy than tamoxifen for ErbB-1- and/or ErbB-2-positive, estrogen receptor-positive primary breast cancer: evidence from a phase III randomized trial. J Clin Oncol. 2001, 19: 3808-3816.
Smith IE, Dowsett M: Comparison of anastrozole vs tamoxifen alone and in combination as neoadjuvant treatment of estrogen receptor-positive (ER+) operable breast cancer in postmenopausal women: the IMPACT trial. Breast Cancer Res. 2003, 82: S6-
Zhu L, Chow LW, Loo WT, Guan XY, Toi M: Her2/neu expression predicts the response to antiaromatase neoadjuvant therapy in primary breast cancer: subgroup analysis from celecoxib antiaromatase neoadjuvant trial. Clin Cancer Res. 2004, 10: 4639-4644.
Masamura S, Santner SJ, Heitjan DF, Santen RJ: Estrogen deprivation causes estradiol hypersensitivity in human breast cancer cells. J Clin Endocrinol Metab. 1995, 80: 2918-2925. 10.1210/jc.80.10.2918.
Chu I, Blackwell K, Chen S, Slingerland J: The dual ErbB1/ErbB2 inhibitor, lapatinib (GW572016), cooperates with tamoxifen to inhibit both cell proliferation- and estrogen-dependent gene expression in antiestrogen-resistant breast cancer. Cancer Res. 2005, 65: 18-25.
Acknowledgements
This work was supported in part by a breast cancer Specialized Program of Research Excellence grant (P50 CA58183) from the NCI, and by a research grant from AstraZeneca.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Schiff, R., Osborne, C.K. Endocrinology and hormone therapy in breast cancer: New insight into estrogen receptor-α function and its implication for endocrine therapy resistance in breast cancer. Breast Cancer Res 7, 205 (2005). https://doi.org/10.1186/bcr1287
Published:
DOI: https://doi.org/10.1186/bcr1287