
Statement of findings
The kinetics of apoptosis and the apoptosis-regulating gene p53 in adjuvant arthritis (AA) were investigated to assess the value of the AA rat model for testing apoptosis-inducing therapies. Very few terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate (dUTP) nick end-labeling (TUNEL)-positive cells were detected during the early phases of AA, but on day 23 (chronic arthritis) the percentage of TUNEL-positive cells was significantly increased. Expression of p53 in synovial tissue gradually increased from days 5-23, which was markedly higher than p53 levels in rheumatoid arthritis (RA) synovium. Significant apoptosis only occurs late in rat AA and is concordant with marked p53 overexpression, making it useful model for testing proapoptotic therapies, but rat AA is not the best model for p53 gene therapy because dramatic p53 overexpression occurs in the latter stages of the disease.
Similar content being viewed by others


Synopsis
Introduction
RA is a chronic inflammatory disorder that is characterized byinflammation and proliferation of synovial tissue. The amount of DNAfragmentation is significantly increased in rheumatoid synovium. Only lownumbers of apoptotic cells are present in rheumatoid synovial tissue, however.The proportion of cells with DNA strand breaks is so great that this disparitysuggests impaired apoptosis. Therefore, the development of novel therapeuticstrategies that are aimed at inducing apoptosis in rheumatoid synovial tissueis an attractive goal.
Although animal models for arthritis only approximate RA, theyprovide a useful test system for the evaluation of apoptosis-inducingtherapies. AA in rats is among the most commonly used animal models for RA. Forthe interpretation of such studies, it is essential to characterize the extentto which apoptosis occurs during the natural course of the disease. Therefore,we evaluated the number of apoptotic cells and the expression of p53 in variousphases of AA.
Materials and methods
In order to generate the AA rat model, Lewis rats were immunizedwith Mycobacterium tuberculosis in mineral oil on day 0. Paw swellingusually started around day 10. For the temporal analysis rats were sacrificedon days 0, 5 (prearthritis), 11 (onset of arthritis), 17 (acceleratingarthritis), or 23 (chronic arthritis).
For the detection of apoptotic cells, the hind paws were harvestedon days 0(n=6),5 (n=6), 11 (n=6), 17 (n=6),or 23 (n=4). The right ankle joints were fixed in formalin,decalcified in ethylenediaminetetra-acetic acid, embedded in paraffin, andsectioned. The TUNEL method was applied. The percentage of TUNEL-positive cellsof the total inflammatory cell infiltrate was noted.
For Western blot analysis, hind paws were harvested on days0 (n=2), 5 (n=3), 11 (n=4), 17 (n=4), or 23(n=4). In addition, hind paws of normal rats (n=2) werestudied. The right ankle joints were snap frozen and pulverized. Synovialtissue was also obtained by arthroscopy of three patients with longstanding(>5 years) RA. After protein extraction in lysis buffer, equal amounts ofprotein samples from lysates were pooled and examined by Western bolt analysisusing anti-p53 monoclonal antibody D07, which recognizes wild-type and mutantp53 from rodents and humans.
For immunohistochemical analysis, six rats were sacrificed on day23 after immunization and synovial tissue of the right ankle joints was snapfrozen and evaluated by immunohistochemistry using anti-p53-pan. The sectionswere evaluated semi-quantitatively using a 0-4 scale.
The kruskal-Wallis test for several group means was used tocompare the percentage of TUNEL-positive cells at different time points.
Results
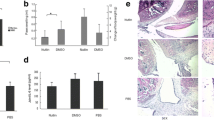
The percentages of TUNEL-positive cells were strongly dependent onthe stage of the disease. Very few TUNEL-positive cells were detected in normalrats or in the early phases of AA; the number of TUNEL-positive cells was 1% orless of the total cell infiltrate, including neutrophils, from days 0-17 (Table1). On day 23, however, the percentage of TUNEL-positivecells was significantly increased [15.8±5.1% (mean ± standard errorof the mean); P=0.01]. TUNEL-positive cells were observed in theintimal lining layer and synovial sublining of the invasive front, as well asin the articular cartilage (Fig. 1).

(Left) Swelling of the left hind paws (mean ± standard error of the mean) in different phases of adjuvant arthritis (AA). Swelling usually starts around day 10 after immunization, followed by the development of accelerating arthritis (day 17) and chronic arthritis (day 23). (Right) Distribution of TUNEL-positive cells (red, indicated by arrows) in joints of AA rats on day 11 (a and c) and on day 23 (b and d). Very few TUNEL-positive cells were detected in normal rats or during the onset of arthritis, whereas the number of TUNEL-positive cells were significantly increased in the synovium (and also in cartilage) of rats with chronic AA. TUNEL method counterstrained with Mayer's hemalum. Original magnification 250x.
Subsequently, we examined expression of the tumor suppressor genep53, because this is a key regulator of apoptosis. Expression of p53in pooled rat AA joint extracts gradually increased from day 0 (6 arbitraryunits) to day 23 (173 arbitrary units), which was markedly higher than p53levels in RA synovium (32 arbitrary units; Table 1).Overexpression of p53 protein on day 23 was confirmed by immunohistochemistryin a separate experiment in six rats with AA. Overexpression of p53 wasobserved in the intimal lining layer and synovial sublining in all rats on day23. In all cases a semiquantitative score of 4 was assigned, indicating that51% or more of the cells were positive, whereas control sections werenegative.
Discussion
The results presented here reveal that the number ofTUNEL-positive cells remained very low until chronic arthritis developed. Thisindicates that, although there was sufficient DNA damage to cause an incrementin p53 expression in the early phases, DNA strand breaks that can be detectedby TUNEL assays only occurred in chronic AA. The observation thatTUNEL-positive cells were nearly absent in early AA clearly indicates that onlyvery few cells were undergoing programmed cell death. This is an importantobservation, which makes it possible to study the effects of apoptosis-inducingtherapies in situ in early and accelerating AA. An effective therapywould obviously increase the number of TUNEL-positive cells.
There is already some overexpression of p53 in the preclinicalphase and during the onset of the arthritis, with an additional increment inp53 expression during accelerating and chronic arthritis. Presumably, this iswild-type p53, because the disease duration is likely too short to allow forthe development of p53 mutations. Transcription of p53 is probablyincreased in response to the toxic environment of the inflamed joint. Theincreased expression of p53 in the joints of rats with chronic AA was evengreater than that observed in synovial tissue of RA patients with long-standingdisease.
Overexpression of p53 and increased numbers of apoptotic cells didnot occur simultaneously in this model; rather p53 overexpression precededincreased apoptosis. Activation of p53 leads to induction of cellgrowth arrest, allowing time for DNA repair. It appears that DNA damage is onlyextensive enough to induce apoptosis in the latter stages of AA. Factors otherthan p53 may also play an important role in the actual induction ofapoptosis
Taken together, significant apoptosis only occurs late in AA andit follows marked p53 overexpression, making it a useful model fortesting proapoptotic therapies. AA is not the best model for p53 genetherapy, however, because dramatic p53 overexpression occurs in the latterstages of the disease.
Introduction
RA is a chronic inflammatory disorder that is characterized by inflammation and proliferation of synovial tissue. The disease is still associated with long-term morbidity and early mortality, despite treatment with antirheumatic drugs. Inadequate apoptosis appears to contribute toward prolonged survival and constitutive activation of specialized cells in rheumatoid synovium [1,2]. The amount of DNA fragmentation is significantly increased in rheumatoid synovium [3,4], which is presumably due to the toxic environment of the chronically inflamed joint [5]. Only low numbers of apoptotic cells are present in rheumatoid synovial tissue, however [4,6,7,8]. The proportion of cells with DNA strand breaks is so great that this disparity suggests impaired apoptosis. The observation that mice with the lymphoproliferative or generalized lymphoproliferative disorder, which have mutations that inactivate Fas and Fas ligand, respectively, develop pathology similar to that observed in immune-mediated diseases [9,10] illustrates that reduced apoptosis may play an important role in the pathogenesis of synovitis.
The p53 tumor suppressor is a key regulator of DNA repair and cell replication [11]. DNA damage activates p53, thereby inducing cell growth arrest to allow time for DNA repair. When DNA damage is extensive the cells may undergo apoptosis. Inactivation of the p53 gene renders cells less susceptible to undergo apoptosis [12]. The p53 system ensures that cells with damaged DNA either die or are repaired. We have previously proposed that impaired apoptosis in rheumatoid synovial tissue might be explained in part by the development of permanent genetic changes in the p53 tumor suppressor gene [5,13]. In addition, other factors may be involved, such as protection against apoptosis by nuclear factor-κB activation [14,15,16], a relative deficiency of functional Fas ligand in the RA joint [17], and expression of antiapoptotic molecules, such as bcl-2 [3] and sentrin [18]. Therefore, the development of novel therapeutic strategies aimed at inducing apoptosis in rheumatoid synovial tissue is an attractive goal.
Although animal models of arthritis only approximate RA, they provide a useful test system for the evaluation of apoptosis-inducing therapies. AA in rats is among the most commonly used animal models for RA [19,20]. This model has recently been used to investigate the effects of bisindolylmaleimide, a compound that facilitates Fas-mediated apoptosis [21]. Rat AA might also provide a useful screening model for the evaluation of gene therapies that are aimed at induction of apoptosis, because the size of the joints permits relatively easy intra-articular injection [22]. For the interpretation of such studies, however, it is essential to characterize the extent to which apoptosis occurs during the natural course of the disease. Therefore, we evaluated the number of apoptotic cells and the expression of p53 in various phases of AA.
Materials and methods
Adjuvant arthritis model
Male Lewis rats (150-200 g) were immunized at the base of the tail with 1 mg Mycobacterium tuberculosis H37RA (Difco, Detroit, MI, USA) in 0.1 ml mineral oil on day 0 [23]. Paw swelling usually started around day 10. For the temporal analyses, rats were killed on days 0, 5 (prearthritis), 11 (onset of arthritis), 17 (accelarating arthritis), or 23 (chronic arthritis) by carbon dioxide narcosis. All animals were handled in accordance with University of California San Diego Animal Subjects Committee and United States Department of Agriculture guidelines.
Detection of apoptotic cells
The hind paws were harvested on days 0 (n=6), 5 (n=6), 11 (n=6), 17 (n=6), or 23 (n=4). The right ankle joints were fixed in formalin, decalcified for 4 weeks in 15% ethylenediaminetetra-acetic acid in phosphate-buffered saline, embedded in paraffin, and sectioned. For detection of apoptotic cells the TUNEL method was applied, based on terminal deoxynucleotidyl transferase-mediated labeling of free 3'-hydroxy termini exposed in cells that exhibit DNA strand breaks. An in situ cell death detection alkaline phosphatase kit from Boehringer Mannheim (Indianapolis, IN, USA) was used according to the manufacturer's instructions. For detection of alkaline phosphatase activity we used the alkaline phosphatase substrate kit I (Fast Red) from Vector Laboratories (Burlingame, CA, USA). The percentage of TUNEL-positive cells of the total inflammatory cell infiltrate was noted.
Western blot analysis
Hind paws were harvested on days 0 (n=2), 5 (n=3), 11 (n=4), 17(n=4), or 23 (n=4). In addition, hind paws of normal rats (n=2) were studied. After removal of skin and muscle tissue, the right ankle joints were snap frozen in liquid nitrogen and pulverized. Synovial tissue was also obtained by anthroscopy of three patients with longstanding (>5 years) rheumatoid factor-positive, erosive RA; these patients have been described previously [24]. All patients had active arthritis in a knee joint and elevated serum levels of C-reactive protein. The patients were treated with nonsteroidal anti-inflammatory drugs. None were treated with corticosteroids or immunosuppressive drugs, such as azathioprine, methotrexate, or cyclophophamide, within 3 months before study entry [24]. After protein extraction in lysis buffer, equal amounts of protein samples (in total 20 μg/lane) from lysates were pooled and run on a gel in order to normalize for differences in synovial cellularity [24]. The pooled samples were then transferred onto a nitrocellulose membrane, and p53 protein detected with 0.25 μg/ml of the Immunoglobulin G2b mouse anti-p53 monoclonal anti-body DO7 (Novocastra Laboratories Ltd, Newcastle, UK), which recognizes wild-type and mutant p53 from rodents and humans. After incubation with horseradish peroxidase-conjugated goat-antimouse antibody, horseradish peroxidase activity was detected using hydrogen peroxide as the substrate and visualized by chemiluminescence. Densitometry was performed with Image software version 1.57 (National Institutes of Health, Bethesda, MD, USA). Results are expressed as arbitrary densitometry units.
Immunohistochemistry
Six rats were sacrificed on day 23 immunization, and synovial tissue of the right ankle joints was snap frozen in Tissue-Tek OCT (Miles Diagnostics, Elkhart, IN, USA) by immersion in methylbutane (-70°C). All slides were stained in one procedure. Endogenous peroxidase activity was inhibited using 0.1% sodium azide and 0.3% hydrogen peroxide in phosphate-buffered saline for 30 min. The biotinylated anti-p53-pan (Boehringer Mannheim) was diluted to a final concentration of 2μg/ml and incubated for 60 min. In negative control sections the primary anti-body was omitted or irrelevant antibody was applied at the same concentration as the primary antibody. This was followed by incubation with avidin-biotin-peroxidase complex (Vectastain ABC Kit; Vector Laboratories), biotinylated tyramine, and horseradish peroxidase-conjugated streptavidin, as previously described [24]. Horseradish peroxidase activity was detected using hydrogen peroxide as substrate and 3,3'-diaminobenzidine (DAB; Vector Laboratories) as dye. Sections were coded and randomly analyzed [24]. The sections were evaluated semiquantitatively using a 0-4 scale as follows: 0, no staining; 1, rare positive staining or trace staining (1-5%); 2, scattered clusters of positive cells (6-15%); 3, moderate staining in a specific region (16-50%) and 4, extensive staining throughout a region (51-100%) [24].
Statistical analysis
The Kruskal-Wallis test for several group means was used to compare the percentage of TUNEL-positive cells at different time points.
Results
Apoptosis in different phases of adjuvant arthritis
Detection of apoptotic cells was performed on the basis of in situ labeling of DNA strand breaks. Representative examples of the TUNEL stainings in relation to the paw volumes in various phases of the disease are shown in Figure 1. The percentages of TUNEL-positive cells were strongly dependent on the stage of the disease. Very few TUNEL-positive cells were detected in normal rats or during the early phases of AA; the number of TUNEL-positive cells was 1% or less of the total cell infiltrate, including neutrophils, from days 0-17 (Table 1). On day 23, however, the percentage of TUNEL-positive cells was significantly increased (15.8±5.1% [mean ± standard error of the mean]; P=0.01). TUNEL-positive cells were observed in the intimal lining layer and synovial sublining of the invasive front as well as in the articular cartilage (Fig. 1).
Expression of p53 in different phases of adjuvant arthritis
Subsequently, we examined expression of the tumor suppressor gene p 53, because this is a key regulator of apoptosis. Expression of p53 in pooled rat AA joint extracts gradually increased from day 0 (6 arbitrary units) to day 23 (173 arbitrary units), which was markedly higher than p53 levels in RA synovium (32 arbitrary units; Table 1 and Fig. 2). Overexpression of p53 protein on day 23 was confirmed by immunohistochemistry in a separate experiment in six rats with AA. Overexpression of p53 was observed in the intimal lining layer and synovial sublining in all rats on day 23 (Fig. 3). In all cases a semiquantitative score of 4 was assigned, indicating that 51% or more of the cells were positive, whereas control sections were negative.

Western blot analysis showing immunoreactive p53 in pooled joint extracts of rats with AA and pooled synovial tissue samples of patients with RA. Expression of p53 gradually increased from days 0-23 in rat AA (see also Table 1). Overexpression on day 23 (173 arbitrary units) was markedly higher than p53 levels in RA synovium (32 arbitrary units).

Representative synovial tissue from a rat with adjuvant arthritison day 23, showing marked p53 overexpression [(a), indicated by arrows].Both cytoplasmic and nuclear staining was noted in the intimal lining layer (L)and in the synovial sublining (S). Staining was absent in the negative controlsection (b). Monostaining peroxidase technique with tyramine enhancementcounterstained with Mayer's hemalum. Original magnification 400x.
Discussion
Rat AA is a T-cell dependent disease, which is characterized by paw swelling, joint erosions and ankylosis, as well as systemic manifestations. Infiltration of the synovium by leukocytes precedes the development of clinical signs and symptoms of arthritis [25,26]. There is also an increase in the numbers of CD8+ T cells and B cells in the regional lymph nodes in the preclinical phase [27]. As shown in the present study, clinical signs of arthritis usually appear by days 10-12. Subsequently, paw volume markedly increases as a result of cellular infiltration and edema of synovial tissue [28].
The results presented here reveal that the number of TUNEL-positive cells remained very low until chronic arthritis developed. Severe disease and marked paw swelling characterize this phase. The results indicate that, although there was sufficient DNA damage to cause an increment in p53 expression in the early phase, DNA strand breaks that can be detected by TUNEL assays only occurred in chronic AA. In general, the results may be false positive, because TUNEL-positive cells are not necessarily apoptotic [2]. The observation that TUNEL-positive cells were nearly absent in early AA, however, clearly indicates that only very few cells were undergoing programmed cell death. We have recently shown that there is increased oxidative stress in chronic arthritis [29], which may play a pivotal role in the induction of DNA strand breaks [5]. The results in the AA model suggest that the production of reactive oxygen and nitrogen species is not sufficiently increased in the earliest phases of arthritis to lead to induction of apoptosis. This is an important observation, which makes it possible to study the effects of apoptosis-inducing therapies in situ in early and accelerating AA. An effective therapy would obviously increase the number of TUNEL-positive cells.
There is already some overexpression of p53 in the preclinical phase and during the onset of the arthritis, with an additional increment in p53 expression during accelerating and chronic arthritis. Presumably, this is wild-type p53, because the disease duration is probably too short to allow for the development of p53 mutations. Transcription of p53 is probably increased in response to the toxic environment of the inflamed joint, with local production of oxygen radicals, nitric oxide [30], and cytokines. Similarly, exposure of human fibroblasts to nitric oxide generated from a nitric oxide donor or from overexpression of inducible nitric oxide synthase may result in accumulation of wild-type p53 [31]. There are also several examples of overexpression of wild-type p53 in human inflammatory diseases, which include the following: inflammation in atherosclerotic plaques [32], idiopathic pulmonary fibrosis [33], Helicobacter pylori-associated gastritis [34,35], ulcerative colitis [36], Crohn's disease [36], chronic pancreatitis [37], infectious colitis [38], lymphocytic thyroiditis [39], and RA [1,24,40,41]. The increased expression of p53 in the joints of rats with chronic AA was even greater than that observed in synovial tissue of RA patients with long-standing disease. Because there is already dramatic p53 overexpression in the latter stages of the disease, AA is probably not the best model to evaluate p53 gene therapy. Indeed, we could achieve only a marginal additional increase in p53 expression in vivo gene transfer (unpublished data).
Overexpression of p53 and increased numbers of apoptotic cells did not occur simultaneously in this model; rather p53 overexpression preceded increased apoptosis. Activation of p53 leads to induction of cell growth arrest, allowing time for DNA repair. It appears that DNA damage is only extensive enough to induce apoptosis in the latter stages of AA. Factors other than p53 may also play an important role in the actual induction of apoptosis, such as tumor necrosis factor-α, interactions between Fas and Fas ligand, and degranulation of granules that contain granzymes and perforin.
Taken together, significant apoptosis only occurs late in AA and it follows marked p53 overexpression, making it is useful model for testing proapoptotic therapies. AA is not the best model for p53 gene therapy, however, because dramatic p53 overexpression occurs in the latter stages of the disease.
References
Firestein GS, Nguyen K, Aupperle KR, Yeo M, Zvaifler NJ: Apoptosis in rheumatoid arthritis: p53 overexpression in rheumatoid arthritis synovium. Am J Pathol. 1996, 149: 2143-2151.
Tak PP, Firestein GS: Apoptosis in rheumatoid arthritis. Apoptosis and Inflammation. Edited by Winkler JD. Basel: Birkhauser PublishingLtd. 1999, : 149-162.
Firestein GS, Yeo M, Zvaifler NJ: Apoptosis in rheumatoid arthritis synovium. J Clin Invest. 1995, 96: 1631-1638.
Nakajima T, Aono H, Hasunuma T, et al: Apoptosis and functional Fas antigen in rheumatoid arthritis synoviocytes. Arthritis Rheum. 1995, 38: 485-491.
Tak PP, Zvaifler NJ, Green DR, Firestein GS: Rheumatoid arthritis and p53: how oxidative stress might alter the course of inflammatory diseases. Immunol Today. 2000, 21: 78-82. 10.1016/S0167-5699(99)01552-2.
Matsumoto S, Muller-Ladner U, Gay RE, Nishioka K, Gay S: Ultrastructural demonstration of apoptosis, Fas and Bcl-2 expression of rheumatoid synovial fibroblasts. J Rheumatol. 1996, 23: 1345-1352.
Sugiyama M, Tsukazaki T, Yonekura A, et al: Localisation of apoptosis and expression of apoptosis related proteins in the synovium of patients with rheumatoid arthritis. Ann Rheum Dis. 1996, 55: 442-449.
Ceponis A, Hietanen J, Tamulaitiene M, et al: A comparative quantitative morphometric study of cell apoptosis in synovial membranes in psoriatic, reactive and rheumatoid arthritis. Rheumatology (Oxford). 1999, 38: 431-440. 10.1093/rheumatology/38.5.431.
Nagata S, Suda T: Fas and Fas ligand: Ipr and gld mutations. Immunol Today. 1995, 16: 39-43. 10.1016/0167-5699(95)80069-7.
Ito MR, Terasaki S, Itoh J, et al: Rheumatic diseases in an MRL strain of mice with a deficit in the functional Fas ligand. Arthritis Rheum. 1997, 40: 1054-1063.
Sionov RV, Haupt Y: Apoptosis by p53: mechanisms, regulation, and clinical implications. Springer Semin Immunopathol. 1998, 19: 345-362.
Aupperle KR, Boyle DL, Hendrix M, et al: Regulation of synoviocyte proliferation, apoptosis and invasion by the p53 tumor suppressor gene. Regulation of synoviocyte proliferation, apoptosis, and invasion by the p53 tumor suppressor gene. Am J Pathol. 1998, 152: 1091-1098.
Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR: Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc Natl Acad Sci USA. 1997, 94: 10895-10900. 10.1073/pnas.94.20.10895.
Wang CY, Mayo MW, Baldwin AS: TNF-α and cancer therapy-induced apoptosis: potentiation by inhibition of NF-κB. Science. 1996, 274: 784-787. 10.1126/science.274.5288.784.
Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM: Suppression of TNF-alpha induced apoptosis by NF-κB. Science. 1996, 274: 787-789. 10.1126/science.274.5288.787.
Miagkov AV, Kovalenko DV, Brown CE, et al: NF-kappaB activation provides the potential link between inflammation and hyperplasia in the arthritic joint. Proc Natl Acad Sci USA. 1998, 95: 13859-13864. 10.1073/pnas.95.23.13859.
Cantwell MJ, Hua T, Zvaifler NJ, Kipps TJ: Deficient Fas ligand expression by synovial lymphocytes from patients with rheumatoid arthritis. Arthritis Rheum. 1997, 40: 1644-1652.
Franz JK, Hummel KM, Aicher WK, et al: Invasive synovial fibroblasts express the novel anti-apoptotic molecule sentrin in the SCID mouse model of rheumatoid arthritis [abstract]. Arthritis Rheum. 1999, 42 (suppl): S238-
Klareskog L, Ronnelid J, Holm G: Immunopathogenesis and immunotherapy in rheumatoid arthritis: an area in transition. J Intern Med. 1995, 238: 191-206.
Oliver SJ, Brahn E: Combination therapy in rheumatoid arthritis: the animal model perspective. J Rheumatol Suppl. 1996, 44: 56-60.
Zhou T, Song L, Yang P, et al: Bisindolylmaleimide VIII facilitates Fas-mediated apoptosis and inhibits T cell-mediated autoimmune diseases. Nature Med. 1999, 5: 42-48. 10.1038/4723.
Nguyen KHY, Boyle DL, McCormack JE, et al: Direct synovial gene transfer with retroviral vectors in rat adjuvant arthritis. J Rheumatol. 1998, 25: 1118-1125.
Firestein GS: Anti-inflammatory effects of adenosine kinase inhibitors in acute and chronic inflammation. Drug Dev Res. 1996, 39: 371-376. 10.1002/(SICI)1098-2299(199611/12)39:3/4<371::AID-DDR18>3.3.CO;2-#.
Tak PP, Smeets TJM, Boyle DL, et al: p53 overexpression in synovial tissue from patients with early and longstanding rheumatoid arthritis compared with patients with reactive arthritis and osteoarthirits. Arthritis Rheum. 1999, 42: 948-953. 10.1002/1529-0131(199905)42:5<948::AID-ANR13>3.0.CO;2-L.
Taurog JD, Argentieri DC, McReynolds RA: Adjuvant arthritis. Methods Enzymol. 1998, 162: 339-355.
Halloran MM, Szekanecz Z, Barquin N, Haines GK, Koch AE: Cellular adhesion molecules in rat adjuvant arthritis. Arthritis Rheum. 1996, 39: 810-819.
Rodriguez-Palmero M, Pelegri C, Ferri MJ, et al: Alterations of lymphocyte populations in lymph nodes but not in spleen during the latency period of adjuvant arthritis. Inflammation. 1999, 23: 153-165. 10.1023/A:1020293012793.
Borah B, Francis MD, Hovancik K, Boyce JT, Szeverenyi NM: A quantitative one-dimensional magnetic resonance imaging technique in adjuvant arthritis: the assessment of disease progression and indomethacin efficacy. J Rheumatol. 1995, 22: 855-862.
Maurice MM, Nakamura H, Gringhuis S, et al: Expression of the thioredoxin-thioredoxin reductase system in the inflamed joints of patients with rheumatoid arthritis. Arthritis Rheum. 1999, 42: 2430-2439. 10.1002/1529-0131(199911)42:11<2430::AID-ANR22>3.0.CO;2-6.
Evans CH: Nitric oxide: what role does it play in inflammation and tissue destruction?. Agents Actions Suppl. 1995, 47: 107-116.
Forrester K, Ambs S, Lupold SE, et al: Nitric oxide-induced p53 accumulation and regulation of inducible nitric oxide synthase expression by wild-type p53. Proc Natl Acad Sci USA. 1996, 93: 2442-2447. 10.1073/pnas.93.6.2442.
Ihling C, Menzel G, Wellens E, et al: Topographical association between the cyclin-dependent kinases inhibitor P21, p53 accumulation, and cellular proliferation in human atherosclerotic tissue. Arterioscler Thromb Vasc Biol. 1997, 17: 2218-2224.
Kuwano K, Kunitake R, Kawasaki M, et al: P21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1996, 154: 477-483.
Hahm KB, Lee KJ, Kim JH, Cho SW, Chung MH: Helicobacter pylori infection, oxidative DNA damage, gastric carcinogenesis, and reversibility by rebamipide. Dig Dis Sci. 1998, 43: 72S-77S. 10.1023/A:1018825532059.
Hibi K, Mitomi H, Koizumi W, et al: Enhanced cellular proliferation and p53 accumulation in gastric mucosa chronically infected with Helicobacter pylori. Am J Clin Pathol. 1997, 108: 26-34.
Krishna M, Woda B, Savas L, Baker S, Banner B: Expression of p53 antigen in inflamed and regenerated mucosa in ulcerative colitis and Crohn's disease. Mod Pathol. 1995, 8: 654-657.
Maacke H, Kessler A, Schmiegel W, et al: Overexpression of p53 protein during pancreatitis. Br J Cancer. 1997, 75: 1501-1504.
Islam D, Veress B, Bardhan PK, Lindberg AA, Christensson B: In situ characterization of inflammatory responses in the rectal mucosae of patients with shigellosis. Infect Immun. 1997, 65: 739-749.
Okayasu I, Osakabe T, Onozawa M, Mikami T, Fujiwara M: p53 and p21 (WAF1) expression in lymphocytic thyroiditis and thyroid tumors. Clin Immunol Immunopathol. 1998, 88: 183-191. 10.1006/clin.1998.4572.
Nickels A, Selter H, Pfreundschuh M, Montenarh M, Koch B: Detection of p53 in inflammatory tissue and lymphocytes using immunohistology and flow cytometry: a critical comment. J Clin Pathol. 1997, 50: 654-660.
Reme T, Travaglio A, Gueydon E, et al: Mutations of the p53 tumour suppressor gene in erosive rheumatoid synovial tissue. Clin Exp Immunol. 1998, 111: 353-358. 10.1046/j.1365-2249.1998.00508.x.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Tak, P.P., Klapwijk, M.S., Broersen, S.F. et al. Apoptosis and p53 expression in rat adjuvant arthritis. Arthritis Res Ther 2, 229 (2000). https://doi.org/10.1186/ar92
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar92