
Abstract
Primary Sjögren's syndrome (pSS) is an autoimmune disorder characterized by specific pathologic features and the production of typical autoantibodies. In addition, characteristic changes in the distribution of peripheral B cell subsets and differences in use of immunoglobulin variable-region genes are also features of pSS. Comparison of B cells from the blood and parotid gland of patients with pSS with those of normal donors suggests that there is a depletion of memory B cells from the peripheral blood and an accumulation or retention of these antigen-experienced B cells in the parotids. Because disordered selection leads to considerable differences in the B cell repertoire in these patients, the delineation of its nature should provide important further clues to the pathogenesis of this autoimmune inflammatory disorder.
Similar content being viewed by others


Introduction
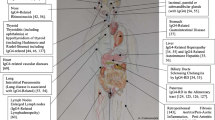
Primary Sjögren's syndrome (pSS) represents an idiopathic inflammatory exocrinopathy characterized by both organ-specific autoimmunity, preferentially affecting the salivary and/or lacrimal glands, and systemic manifestations. The characteristic hallmarks of pSS are focal lymphocytic infiltrates and subsequent destruction of the lacrimal and salivary glands, resulting in keratoconjunctivitis sicca and xerostomia. In addition, there is a broad variety of accompanying clinical and laboratory manifestations, emphasizing that pSS is a systemic disorder [1]. In most patients, these laboratory parameters include hypergammaglobulinemia, circulating immune complexes and autoantibodies, such as those against the Ro-SSA and/or La-SSB autoantigens and rheumatoid factors. The typical production of autoantibodies and polyclonal hypergammaglobulinemia indicates that abnormalities of humoral immunity are significant in pSS and have been included in the classification criteria [2]. However, the factors driving autoimmunity and leading to the differentiation of autoreactive lymphocytes into autoantibody-producing plasma cells remain largely unknown, although several epitope mapping studies have suggested that autoimmunity in pSS is driven by autoantigens.
The glandular infiltration in pSS is composed mainly of CD4+ T lymphocytes [3] but usually also contains a substantial number of B cells and plasma cells [4, 5]. The degree of glandular destruction and symptoms of dryness do not seem to be directly related to the number of infiltrating lymphocytes. Indeed, the mechanism of glandular damage remains incompletely delineated, although a role for CD4+ T cells has been proposed, either directly or through the action of secreted cytokines.
Although local autoantibody production in the glands has been suspected and autoantibodies have been found in the saliva [6], the pathogenetic role of the glandular B lymphocytic infiltrates remains largely unknown. In this regard, it is notable that autoantibodies to the M3 type of the muscarinergic acetylcholine receptor might function to inhibit salivary flow (reviewed in [7]) in a manner comparable to the antibody-mediated blockage of nicotinergic receptors in patients with myasthenia gravis. Cell clusters resembling germinal center (GC)-like structures have been reported in the focal lymphocytic sialadenitis of the minor (labial) salivary glands in patients with pSS [4], although it is not known whether they function completely as lymphoid GCs. Because similar ectopic GC-like reactions have also been observed in the synovium in rheumatoid arthritis (RA) [8, 9] as well as in a variety of other diseases (such as spondylarthopathies, myasthenia gravis and thyroiditis), it has been proposed that such potentially functional B cell aggregates can be induced in several extrafollicular tissues in autoimmune disorders [4, 8–10]. The formation of these aggregates seems to be clearly dependent on the interaction of chemokines and their receptors [10–12].
Cytokines and chemokines in pSS
In patients with pSS the salivary and lacrimal glands are the main target organs for the immune system and, at least in part, subsequent autoimmune-mediated tissue damage. Local production of cytokines by mononuclear cells and also epithelial cells might contribute to the immune-mediated destruction of exocrine glands in pSS [13]. pSS has therefore also been called 'autoimmune epithelitis' [14], emphasizing that epithelial cells are thought to be important in the immunopathogenesis.
Cytokines
Several cytokines have been demonstrated in inflamed tissue by using reverse-transcriptase-mediated polymerase chain reaction technologies as well as in animal models. Cytokines, such as tumor necrosis factor (TNF), lymphotoxin α (LTα) and interleukin (IL)-1β have been found to influence the destruction of the acinar structure in human salivary gland cell clones [15]. The most prominent cytokines detected in affected salivary glands of patients with pSS are IL-1, IL-6, IL-10, transforming growth factor (TGF)-β, interferon (IFN)-γ and TNF. Fox et al.[16] found that salivary gland CD4+ T cells produced over 40-fold more IL-2, IFN-γ and IL-10 than peripheral-blood CD4+ T cells from patients with SS or from controls. Moreover, salivary gland epithelial cells produced 40-fold more IL-1α, IL-6 and TNF mRNA than epithelial cells from individuals with histologically normal salivary glands.
It has therefore been suggested that T helper type 1 (Th1) cytokines, such as IFN-γ and IL-2, as well as IL-10, IL-6 and TGF-β, might be important in the induction and/or maintenance of pSS [10], whereas Th2 cytokines, detected in some cases in association with a striking B cell accumulation in the labial salivary glands, might be involved in the progression of the disease. Furthermore, it has been suggested that TGF-β, an important immunoregulatory cytokine whose absence can lead to systemic autoimmune disease [17], might be deficient in SS. In this regard, reduced levels of TGF-β have been found in SS glands with intense lymphocytic infiltrates [18]. Furthermore, mice that fail to express TGF-β develop an exocrinopathy resembling SS [17]. Although several studies have analyzed cytokines that seem to be involved in the pathogenesis of SS, reports on cytokine polymorphisms are very limited and do not allow any firm conclusion. It should be noted that the interaction between epithelial cells and infiltrating T cells has been characterized in detail, but the cytokines involved in local B cell activation remain largely unknown.
Chemokines
The infiltration of lymphocytes into glandular aggregates apparently has a crucial role in the tissue pathology of SS. This process seems to be tightly regulated at least in part by chemokines and the local expression of their receptors. Chemokines and the expression of chemokine receptors by the inflamed tissue as well as by lymphocytes are therefore likely to be of importance in the evolution of tissue pathology in pSS. Mice deficient in lymphoid-homing chemokine receptors CXCR4 and CXCR5 lack normal lymphoid organs [19–21], indicating that these receptors as well as the chemokines CXCL12 (stromal cell-derived factor 1; SDF-1) and CXCL13 (B-cell-attracting chemokine 1; BCA-1) are important for lymphoid organogenesis. In addition, studies in CXCL13 transgenic mice found that this chemokine, together with TNF and LTβ, is crucial in lymphoid organogenesis, whereas the LTβ knockout mouse lacks the formation of GC structures. A recent review reported that CXCL13 (BCA-1), CCL19 (ELC), CCL21 (SLC) and CXCL12 (SDF-1) all contribute to lymphoid homing and to the persistence of chronic inflammation in pSS [11]. Moreover, studies on RA demonstrated that CXCL12 [22] and CXCL13 [12] are involved in the formation of GC in the rheumatoid synovium. Thus, recent studies have shed light on factors involved in directing lymphocytes into inflamed tissue and maintaining inflammation in SS, whereas the etiologic factors of SS remain to be delineated.
Of potential importance is the fact that enhanced levels of B lymphocyte stimulator (BLyS; also known as B cell activating factor belonging to the TNF family [BAFF] or transmembrane activator and CAML interactor [TACI]) have been demonstrated in patients with SS [23], levels that are higher than previously identified in systemic lupus erythematosus (SLE) [24]. In addition, expression of BLyS has been found to be markedly enhanced in the inflamed salivary glands [23], indicating that activation of B cells might take place in the parotids. In this regard, it is notable that BLyS transgenic mice develop SS with age after the development of manifestations of lupus.
General aspects of Ig variable region (IgV) gene usage
Autoantibodies are features of most systemic autoimmune diseases, including pSS. The production and persistence of autoantibodies in autoimmune conditions is considered to occur because of immune dysregulation with a resultant break in tolerance, regardless of whether these autoantibodies are pathogenic [25]. Despite intensive work on the characterization of autoantigens, the cellular basis of their production and the strong association of certain autoantibodies with particular MHC class II alleles, and despite increasing knowledge about tissue B cell immunology and the role of cytokines and chemokines, little is known about the usage of IgV genes in autoimmune conditions and about the respective autoantibodies.
The observation that certain autoantibodies are frequently encoded by a limited number of IgV gene segments suggested that biases in the development of the B cell receptor repertoire might have a role in the tendency of specific individuals to develop these autoantibodies. Whether the usage of these specific gene segments is different in normals and patients with systemic autoimmune disorders remains controversial. Recent approaches have made it possible to address this issue and to estimate the differential impact of molecular and selective influences in shaping the IgV gene repertoire in normals and patients with autoimmune diseases, including those with pSS.
Previous analyses of autoimmune B cells focused almost exclusively on productive V gene rearrangements and therefore could not discern the impact of molecular and selective influences. Analysis of the nonproductive repertoire is especially important to assess the immediate impact of molecular processes, such as recombination and somatic hypermutation [26–32], because the nonproductive rearrangements do not encode an expressed Ig molecule and are therefore not influenced by selection. In contrast, the distribution of B cells and their productive IgV gene rearrangements can be influenced by a variety of selective events during development and subsequent antigenic stimulation because of the nature of the expressed heavy and/or light chain and the cognate (auto)antigen.
Recent analysis of the nonproductive V gene repertoire in patients with pSS and SLE has documented minimal abnormalities in the nonproductive VH, Vκ and Vλ gene repertoires [33–41]. Although only a few patients with pSS and with SLE have been analyzed with this approach to assess the overall repertoire, the data indicate that IgV gene usage in the nonproductive repertoire is not significantly different from normal, suggesting that the basic process of IgV gene recombination is largely normal in patients with these autoimmune diseases. This does not rule out the possibility that abnormalities in the usage of specific VH or VL genes might contribute to autoimmunity. However, it is apparent that there is unlikely to be a generalized abnormality in V(D)J recombination in these autoimmune diseases. A study by de Wildt et al.[42] confirmed on the mRNA level that the IgV gene usage in patients with SLE or mixed connective-tissue disease (MCTD) was comparable to normal. Moreover, the large number of VH genes that encode specific autoantibodies make it unlikely that an abnormality of V gene usage underlies autoimmunity in most patients.
Although there is no conclusive evidence for recombination biases predisposing to systemic autoimmunity in SLE and SS, there are some exceptions. In some circumstances, specific autoantibodies express marked biases in IgV gene usage. One example is the almost exclusive usage of VH4–34 in cold agglutinin disease [43]. In addition, the 16/6 idiotype expressed by some anti-DNA antibodies is encoded by VH3–23 and the 9G4 epitope encoded by VH4–34 is over-represented in anti-DNA antibodies in SLE. In these instances, autoantibodies preferentially employ specific VH genes for the variable regions. Despite the apparent increase in the use of these specific genes, the generalized higher frequency of somatic hypermutation, abnormal patterns of targeted mutations toward specific DNA motifs, indications of increased receptor editing and differences in the entire IgV gene repertoire of peripheral B cells from patients with SLE [34, 35, 37, 44] indicate that broad abnormalities in B cell selection are characteristic of SLE. B cell repertoire abnormalities are therefore not restricted to clones of autoreactive B cells in this autoimmune disease.
A recent study [45] demonstrated that VH4–34 is clearly negatively selected among post-GC B cells in normals, as already shown at the single-cell level for peripheral B cells [27] and post-switch tonsilar plasma cells [46]. By contrast, patients with SLE do not negatively select these cells appropriately in their GC, allowing their expansion after encountering antigen and receiving help from T cells. An enhanced frequency of VH4–34 has previously been observed in peripheral plasma cells of a patient with active SLE [33]. This contrasts markedly with the normal post-switch plasma cell repertoire, in which VH4–34 is strictly excluded [46]. This finding is consistent with the conclusion that disturbances in selection have a key role in SLE. Notably, different autoimmune diseases, such as pSS and SLE, seem to have characteristic abnormalities of particular censoring mechanisms.
IgV gene usage in patients with pSS is preferentially shaped by disordered selection
Distinct abnormalities in the B cell repertoire have been identified in pSS. Recent studies in three patients with pSS found that major abnormalities were related mainly to influences of selection, because the usage of VH and VL gene segments in the nonproductive repertoire was largely normal. However, significant abnormalities were found in the productive repertoires, especially by affecting VL distribution [36, 37]. Notably, four Vλ genes (2A2, 2B2, 2C and 7A) represented 56% of all functional Vλ joints [37]. In the productive Vκ repertoire, three Vκ genes (L12, O12/O2 and B3) comprised 43% of all amplified VκJκ joints [36]. It is of interest that VκA27, a gene frequently employed by autoantibodies, rheumatoid factor and lymphomas in SS, was found less frequently in the peripheral nonproductive repertoire of the patients with pSS than in normal controls (8% versus 14%; P < 0.05). By contrast, this gene was found at an increased frequency in the parotid gland (29%) compared with the blood (8%; P < 0.05) in the one patient examined [39]. Moreover, 2 of 15 VκA27–Jκ5 rearrangements found in the parotids were clonally related [38]. Furthermore, 11 clonally unrelated VκA27–Jκ2 rearrangements representing 34% of all productive Jκ2 using rearrangements were found in the parotid. Accumulation or local expansion of B cells expressing VκA27–Jκ2 rearrangements in the parotid glands seems to be a characteristic of pSS.
Receptor editing is a mechanism by which B cells are able to escape deletion by revising their autoreactive receptors. To the best of our knowledge, there are no reports addressing the role of editing in SS, whereas several studies did so for SLE. Formerly it was thought that expression of the IgB cell receptor (BCR) extinguished subsequent Ig rearrangements by downregulating the expression of recombination activating gene (RAG) 1 and RAG2 enzymes in the bone marrow. However, recent studies provide evidence that immature B cells outside the bone marrow [9, 47, 48] retain RAG activity and can therefore replace their receptors by secondary Ig gene recombination (receptor editing/revision). This is noted with increased frequency in secondary lymphoid organs [9, 46, 47, 49] and in the fetus [50]. The extent to which the presence of recombination enzymes is correlated with actual editing is uncertain.
There is a controversy over whether defects in receptor editing or secondary rearrangements are involved in shaping the B cell repertoire in autoimmunity. The possibility that deficiencies in central or peripheral receptor editing could have a role in generating autoimmunity has been suggested [51]. In addition, analysis of autoreactive hybridomas [52] generated from patients with SLE demonstrated an overusage of J-proximal Vκ1 genes and a preferential use of J elements proximal to Vκ, suggesting that receptor editing in SLE might be defective, because skewing towards the usage of Jκ, distal Vκ genes and Jκ5-expressing V gene products [9, 34, 53] has been taken as an indication of active receptor editing. Because receptor editing at the VL loci is thought to have a major role in rescuing autoreactive B cells from deletion [52], defects in receptor editing could have a role in the etiology of SLE [25, 34, 35, 49–52].
Recent studies in patients with RA [9, 54, 55] provided evidence that receptor editing/revision might also be more active in the synovium of these patients than in normals. In contrast with these patients with RA, patients with pSS seem to have decreased receptor editing/revision, as identified by an enhanced usage of V-proximal JL segments. It is possible that this reflects a defect or infrequent usage of receptor editing in pSS [36, 37, 39]. In this regard, a recent analysis of six monoclonal antibodies with rheumatoid factor activity obtained from the peripheral blood of patients with pSS showed that all used Vλ-proximal Jλ2/3 gene segments [56], which is consistent with the conclusion that receptor editing/revision might be defective in pSS. The role of abnormalities in this mechanism in permitting the emergence of autoimmunity remains to be fully delineated.
Influences of selection by direct comparison of the B cell receptor repertoire in the parotids versus blood in pSS
As already mentioned, recent studies [36–41] addressed the question of whether there are differences in the IgV chain gene repertoire of CD19+ B cells by comparing two immune compartments, the peripheral blood and the inflamed parotid gland, a target tissue in pSS. Although only one patient was analyzed, the data obtained provide new insights into this disease.
The underlying assumption of this study was that the peripheral circulating B cell repertoire reflects a complex group of cells expressing IgV genes that might have been influenced by a variety of immune compartments, whereas the IgV genes of B cells infiltrating the parotid might provide a more skewed population owing to the local selection and/or (antigen-dependent) proliferation. Alternatively, inflammation might have induced the migration of polyclonal B cells into the affected tissue [40, 56–58]. Whereas clonal B cell expansions in the target tissues of SS patients are well established [4, 5] and early studies examining anti-idiotypes have suggested that B cell infiltrations in pSS represent a highly selected population [59], a molecular analysis of 37 Ig heavy chain rearrangements from labial salivary gland biopsies in pSS [60] has shown a rather polyclonal pattern of IgV gene usage, comparable to that of circulating B cells from normals. Thus, the lack of direct comparison between the B cell repertoire in the blood and that in the parotids has prevented the drawing of firm conclusions about the extent to which selective pressures influence the repertoire in autoimmune diseases with characteristic extrafollicular germinal centers, such as pSS.
IgV gene usage
With the exception of the VH7 family, a single gene family that is known to be related to an insertion/deletion polymorphism of the human VH locus [61], members of all VH families were found in the patient's peripheral blood as well as in the parotid gland. Notably, however, there were specific differences in the VL gene repertoire when blood and parotid were compared [38]. Strong selective influences were detected in the parotid gland of the patient in that B cells with rearranged VκA27, VκA19 and Vλ2E as well as Vλ1C were markedly enriched. Furthermore, there was evidence of clonal expansion of VκA27–Jκ5 and VκA19–Jκ2 rearrangements in the patient's parotid gland as well as of Vλ1C–Jλ3 rearrangements in both blood and parotid gland. An increase in VκA27 preferentially rearranged to Jκ2 but not clonally related in the gland is noteworthy because there was a significantly lower frequency of VκA27 in the periphery of patients with pSS compared with normals. These data are consistent with the conclusion that there is clonal expansion within the salivary gland B cells as well as the selection of cells expressing particular light chains. In addition, a polyclonal population of B cells was present.
Positive selection of particular VL chain genes by foreign antigens or autoantigens present in the gland seems to shape the productive VL chain repertoire in the inflamed tissue. This is in contrast to the VH repertoire of the patient analyzed, which was similar in the peripheral blood and in the parotid gland. These results suggest an important role for VL chain gene usage in the immune activation of B cells within the parotid gland of the pSS patient studied. A restriction of the VL chain repertoire has been described after vaccination. As an example, antibodies against Haemophilus influenzae (Hib) B that develop as part of a TH2 response have been identified as being frequently encoded by VκA2, O8/O18, L11, A17 and A27 [62]. Moreover, Vλ genes of the Vλ2 and Vλ7 family were found in the Hib-antibody VL gene repertoire [62]. In addition, VκA27 and Vλ2C, 2E, 2A2 or 10A were also shown to encode antibodies against Streptococcus pneumoniae[63]. Interestingly, VκA27 and Vλ2E, which were frequently found in the parotid gland of this patient, with VκA27 expanded clonally, have also been shown to encode antibodies against rabies virus [64]. Thus, microbial antigens, including bacterial and viral epitopes that could be involved in the pathogenesis of pSS, might also be involved in the selective processes shaping the VL gene repertoires of B cells accumulated in the parotid gland of this patient with SS.
In contrast, it is possible that autoantigens might be involved in the accumulation of parotid gland B cells in this patient. In this regard, VκA27 was frequently used by rheumatoid factors in patients with RA [65]. Rheumatoid factor is typically present in the sera of patients with pSS and was also detected in the saliva or in salivary gland biopsies [66] of these patients. In this regard, Martin et al.[66] described two salivary-gland lymphomas that developed in patients with pSS from rheumatoid-factor-specific B cells. Moreover, VκA27 has been reported to be frequently employed by lymphomas developing in the salivary gland of patients with pSS [67]. Despite the presence of clonally expanded B cells expressing VκA27, the patient studied did not develop lymphoma during a follow-up period of 3 years after the examination, indicating that additional factors or further persistence of the chronic B cell proliferation are essential for the development of lymphoma.
Analysis of mutations
In the peripheral-blood B cells of patients with pSS, less than a third (28.3%) of the CD19+ B cells expressed somatically mutated productive VH rearrangements [39]. The frequency of mutations was lower than that previously reported for circulating CD19+ B cells of normals (1.4 versus 2.6%; P < 0.001) [26, 28]. Although a direct comparison of B cell subsets was not performed at the beginning of these analyses, decreased levels of memory CD27+ B cells in patients with pSS probably account for the difference in mutations [39, 40]. By contrast, the vast majority (about 80%) of the parotid B cells used mutated VH rearrangements, and both the nonproductive and productive glandular rearrangements exhibited significantly increased mutational frequencies compared with the blood counterpart. Because mutated IgV genes are characteristic of memory B cells [68], this finding indicates an accumulation of memory-type B cells in the inflamed parotid gland.
The mutational frequency and the percentage of mutated light-chain genes were also greater in the productive VL chain rearrangements of B cells from the parotid gland than in cells from the peripheral blood, but the VL rearrangements accumulated a large number of silent mutations. Interestingly, productively rearranged Vλ genes from the parotid gland (3.32%) exhibited a significantly greater mutational frequency than the Vκ gene rearrangements (2.35%; P < 0.001) [38].
Because GC-like structures have previously been described in the parotid gland [3, 4], this site might be able to act as a secondary lymphoid organ facilitating somatic hypermutation and selection of antigen-specific B cells. Antigen-driven germinal-center reactions might proceed within ectopic lymphoid follicles in the parotid gland, giving rise to highly mutated antigen-specific B cells. However, the migration of highly mutated antigen-specific B cells from the patient's blood to the parotid gland could also contribute to the observed differences in the mutational frequencies.
The analysis of the replacement/silent (R/S) ratio and the mutational 'hot spots' of productive VL chain rearrangements of peripheral and parotid gland B cells revealed no major abnormalities when compared with normal donors, indicating intact selective mechanisms with selection against R mutations in the frame work regions that might cause structural constraints of the Ig molecule. In the productive VL chain repertoire of B cells from the parotid gland, we found the frequency of S mutations to be increased, which was consistent with a reduced R/S ratio in the complementarity-determining regions (CDRs) [38, 39]. This is in accordance with the observations of other studies (Gellrich et al.[60], Stott et al.[4] and Miklos et al.[69]). Stott et al.[4] described a decreased R/S ratio in the CDRs of VH and VL gene rearrangements of B cells obtained by minor salivary-gland biopsies from two patients with SS. Detailed analyses of the frequency of the distribution of mutations revealed that mutations in nonproductive VL rearrangements of B cells from the parotid gland were less targeted towards the highly mutable RGYW [R(purine)/G/Y(pyrimidine)/W(A or T)] motifs. However, these targeted mutations of RGYW in VL gene rearrangements were highly selected in B cells from the parotid gland. Although no firm conclusion can be drawn, it is possible that these targeted mutations are generated in the parotids of the patient, with retention of particular mutated VL rearrangements.
CDR3 analysis of IgV gene rearrangements
Remarkably, the productive glandular VH rearrangements, but not VL rearrangements, were found to exhibit a significantly shorter CDR3 region than their peripheral productive counterparts [38, 39]. To a considerable extent, this was accounted for by a less frequent usage of the JH6 segment in the glandular rearrangements when compared with the patient's peripheral repertoire as well as with that reported previously for normals [27, 28]. It is noteworthy that the JH6 segment encodes the longest CDR3 component (29 nucleotides) of all JH genes and can thereby contribute to rearrangements with longer CDR3 regions. This confirms conclusions that JH6 is positively selected in the expressed preimmune repertoire [70] but negatively selected in the mutated repertoire. Moreover, selective influences on productive rearrangements seem to favor shorter CDR3 regions [27, 28]. The finding of VH sequences with shorter CDR3 regions is in accordance with the conclusion that memory B cells that accumulate in the parotids are recruited by antigen. The CDR3 region of VH rearrangements might be more important in reacting to parotid antigens than other regions of the VH molecule.
Evidence of clonal expansions
The major salivary glands are known to be the site of preferential B cell expansions and in some cases of lymphoproliferation in pSS. B cells from the parotid gland have been identified as a distinct population showing preferential expansion and somatic mutation of particular VL chain rearrangements, such as VκA27–Jκ5, VκA19–Jκ2 and Vλ1C–Jλ2/3, in comparison with peripheral B cells [38]. Clonal expansion was not associated with any evidence of intraclonal diversification. Thus, this glandular expansion might be derived from proliferation of the very small subset of proliferating mantle-zone (founder) B cells or from an early state of dark-zone germinal center cells [71]. However, it is uncertain whether these cells are able to leave the germinal center.
Accumulation of memory-type B cells in the inflamed parotid gland supports the conclusion that there is an enhanced influx/homing of particular memory-type B cells into the inflamed gland, rather than a proliferation of a few founder B cells entering the parotid GC structures in patients with pSS, despite evidence of clonally expanded B cells in the tissue.
Analysis of B cell subsets in Sjögren's syndrome allows differentiation from SLE
Several groups, including our own, have performed studies on the distribution of peripheral B cell subsets in systemic autoimmune diseases, such as SLE and pSS [33, 40, 57, 72]. In this regard, the identification of CD27 as a marker of memory B cells [33, 68, 73, 74] made it possible to characterize peripheral naive (IgM+/CD27-) and memory (CD27+) B cells. Interaction of CD27 with its ligand on T cells, CD70, serves as a pathway of differentiation of B cells into plasma cells [73, 75, 76]. Recently, homotypic interaction of CD27 and CD70 expressed by B cells only [77] was also reported to be sufficient for B cell differentiation, raising the possibility that B cells might be able to regulate themselves by CD27–CD70 interactions. In another recent study [78] it was shown that CD27- B cells can be differentiated into IgG-producing or IgE-producing plasma cells in vitro. It needs emphasis that class switching, but not somatic hypermutation, could be induced, although the recently discovered activation-induced deaminase (AID) [79] has been found to be expressed in these cells.
On the basis of the available data on the distribution of B cell subsets in SLE versus pSS, there is increasing evidence that diseases associated with immunologic activity can be characterized by unique features of B cell distribution [33, 72, 80–83]. Whereas patients with active SLE [33] revealed increased circulating CD27+ memory B cells, reduced naive CD27- B cells and markedly increased CD27high plasma cells that seemed to be related to lupus disease activity, analysis in pSS [40] showed a clear predominance of CD27- naive B cells (Fig. 1) that also lacked expression of CD5 compared with normal donors as well as patients with SLE (P < 0.001) and a significant decrease in the frequency of memory CD27+ B cells that were predominantly CD5+[40]. The reduced frequency of CD27+ B cells in pSS was significant when compared with either normal controls (P < 0.05) or patients with SLE (P < 0.0002). Another recent study [57] characterized peripheral B cells in 11 patients with pSS as well as patients with RA and normal controls. These investigators also found a predominance of naive B cells that were CD27- and a reduced frequency of memory B cells in patients with pSS.

Analysis of the distribution of peripheral CD19+ B cell subsets demonstrates that patients with primary Sjögren's syndrome (pSS) have reduced frequencies of CD27+ memory B cells in the peripheral blood compared with normal donors. In addition, patients with pSS with secondary non-Hodgkin lymphoma exhibited an increase in CD27+ B cells in the blood.
The difference between pSS and SLE (Fig. 2) is noteworthy because of the many common clinical and serologic similarities (hyperimmunoglobulinemia, positive anti-Ro and anti-La autoantibodies, rheumatoid factor) between patients with pSS and those with SLE. Another difference between patients with pSS and with SLE was the normal peripheral B cell count in the former, whereas patients with SLE exhibited significant decreases in peripheral B cell frequencies [33, 40]. In one study [57], patients with RA also manifested an increased frequency of CD27+ memory B cells and a normal frequency of CD27- naive B cells. The pattern of B cell subpopulations in pSS, RA and SLE defined by CD27 expression therefore seemed to be unique.

Schematic distribution of B cell subsets in peripheral blood of normals compared with patients with systemic lupus erythematosus and Sjögren's syndrome.
Previous data have shown that the frequency of CD27+ B cells reflects the accumulation of antigen experience of an individual that is, at least in part, related to age [73, 82]. Cord blood and blood from hyper-IgM patients normally do not contain CD27+ B cells [73]. Because of the usually more advanced age of patients with pSS than of those with SLE, the actual differences identified between the SLE and pSS groups might have been underestimated. In contrast, the peripheral status of B cell distributions looks very similar in patients with pSS and in those with HIV with predominantly naive B cells [40, 80]. Because CD4+ T cells are depleted in HIV but not in pSS, one interpretation of these observations may be that T cell dependent priming of B cells might be less in patients with pSS than in normals and patients with SLE.
Remarkably, the CD27- B cells could be further subdivided by the mutational status of their productive VH rearrangements into a majority of naive cells (35 of 39 with no mutations; mutational frequency less than 0.1%) and a minority of memory-type cells (4 of 39 with mutations; mutational frequency 4.6%), whereas all but one of the CD27+ B cells (31 of 32) analyzed expressed mutated IgV genes [40]. Currently, the finding of the small population of CD27- B cells expressing mutated VH rearrangements remains unclear. It is noteworthy that this population had a significantly lower mutational frequency than CD27+ B cells (4.6% versus 7.8%; P = 0.0009). Possible explanations might be transient or low-level CD27 expression, shedding of CD27, or stimulation that results in mutations but fails to upregulate CD27. Notably, however, there are no striking differences in the frequency of this population between SS patients and normals [40], and, very recently, this population has also been detected in other normal and abnormal conditions by studies on single cells. Importantly, the regulation of CD27 and its association with the acquisition of IgVH mutations seems to be normal in patients with pSS.
Previous molecular analysis documented that CD27 can be taken as a reliable marker for memory B cells in healthy normals [68, 74] as well as in patients with SLE [33]. The analysis of a patient with SLE revealed a mutational frequency of 0.4% in the CD27- B cells and 6.1% in the CD27+ B cells. Overall, there was no major difference in the frequency of mutations in VH rearrangements of CD27- and CD27+ B cells, respectively, obtained from patients with pSS or SLE and from normals, which is consistent with the conclusion that expression of CD27 indicates previous antigen contact by the respective B cell.
Several studies have identified an enhancement of CD5-expressing B cells in the periphery of patients with pSS [84–87], although to the best of our knowledge no study has analyzed in detail the proportions of CD5+ B cells among naive and memory B cells. In contrast, a few reports did not identify an enhanced frequency of CD5+ B cells in pSS [88]. Earlier studies [89] found enhanced frequencies of CD5+ B cells in about half of patients with pSS, as well as in about half of patients with RA and about a quarter of patients with SLE and normals. By contrast, there was an increase only of CD5-/CD27- naive B cells in patients with pSS [40], whereas there was no significant increase in the overall CD5+ B cell population. However, a subgroup of seven patients with pSS with the highest frequencies of naive B cells (86–94%) also had larger numbers of CD5+/CD27- naive B cells (14.2–37.2%). In the patients analyzed, there were no clinical features that distinguished these seven patients with enhanced CD5+ B cells from the remainder. These data indicate that the previously known enhancement in CD5+ B cells in SS stems preferentially from an increase in the immature B cell pool. It should be noted that B cells infiltrating the parotids frequently express CD5, further supporting the hypothesis of homing and activation of specific B cells in the glands.
B cell malignancies and Sjögren's syndrome
In contrast to the focal sialadenitis of the minor (labial) salivary glands, the lymphocytic lesions of the major salivary glands often contain secondary lymph follicles. B cells have been shown to infiltrate the glandular duct epithelium and thereby to contribute to the characteristic pattern of chronic lymphocytic inflammation called myoepithelial sialadenitis (MESA) or benign lymphoepithelial lesion [90]. These lesions are thought to form the substrate for the development of extranodal non-Hodgkin lymphomas (NHLs) [91, 92]. In this context, it is well known that patients with pSS have an increased risk of developing such lymphomas compared with normals. Extranodal lymphomas in pSS are almost exclusively of B cell origin and are frequently identified in the major salivary glands. Recently, the suggested linkage between autoimmunity, autoantibody-producing cells and lymphoma [66, 93, 94] has been emphasized by the demonstration of two cases of parotid gland lymphomas in pSS producing mono-specific rheumatoid factors [66].
A remarkably biased usage of individual VH segments (in particular the VH1–69/DP-10 and VH3–07/DP-54 segments) has been shown in both benign and malignant clonal B cell expansions in the salivary glands of patients with pSS, exhibiting some evidence for (auto)antigen selection, for example by rheumatoid factor activity [4, 58, 66, 95, 96]. Moreover, a previous anti-idiotypic study has suggested that B cells expressing VH1–69/DP10 cross-reactive idiotypes G6, G8 and H1 are increased in infiltrates in the minor salivary glands of patients with pSS [65].
Support for a role of B cell activation in the development of lymphoma comes from phenotypic analyses of peripheral B cells in patients with pSS that demonstrated an enhanced frequency of CD27+ memory B cells in their peripheral blood, contrasting with patients with pSS but no lymphoma.
Because the expression of CD27 as well as its ligand, CD70, is strictly regulated on normal lymphocytes, it is striking that neoplastic B cells at different stages of B cell differentiation strongly express CD27 [97, 98]. Notably, this included B cell malignancies with a putative origin from antigen-inexperienced B cells, such as mantle-zone lymphomas [98]. In addition, a recent study reported that 7 of 10 high-grade lymphomas from HIV-positive patients and 6 of 10 HIV-negative patients with different lymphomas expressed CD27 [81]. The extent to which these findings indicate a loss of regulation of CD27 expression by the malignant cells and the nature of these abnormalities remain unknown. Potential explanations for the different expression of CD27 by lymphoma might be alterations in the circulation or stimulation of these cells as well as a loss of normal regulatory activity. Importantly, co-expression of both CD27 and CD70 by several tumors indicate that this receptor–ligand pair promote autocrine growth regulation of these lymphomas [98].
It is notable that exceptionally high frequencies of CD27+ (including CD27high) B cells were seen in two patients with pSS and secondary NHL, in contrast with all other patients with pSS (Fig. 1). Although in both cases these lymphomas were putatively derived from later stages of B cell differentiation (immunocytoma and plasmocytoid lymphoma), CD27 expression has been shown in almost all types of B cell NHL potentially associated with pSS [97, 98]. Thus, the observations suggest that significantly enhanced expression of CD27 might serve as an early indicator of the development of an NHL in pSS, a disease with a well-known increased risk for secondary NHL but for which there are no reliable early laboratory parameters. A recent study of a patient with cold agglutinin disease subsequently developing NHL demonstrated that almost all peripheral B cells were CD27+, although not all B cells belonged to the lymphoma [99]. Whether the B cells expressing CD27 in the patients with pSS without lymphoma differ from the cells found in patients with NHL or merely reflect a higher overall activation of B cells in these patients needs to be further examined.
Conclusions
Characteristic disturbances of peripheral B cell homeostasis with depletion of memory B cells in the peripheral blood, and evidence for the accumulation and retention of these antigen-experienced B cells in the parotids, together with new findings of the role of chemokines and chemokine receptors, permitted new insight into the immunopathogenesis of pSS. Although most the current data indicate that there is no major molecular abnormality in generating the IgV heavy and light chain repertoire in patients with pSS, influences of disordered selection apparently lead to remarkable differences in V gene usage by B cells in these patients. Most notably, selective influences after encountering (auto)antigen lead to preferential changes in VL gene usage and the length of the CDR3 of VH rearrangements in patients with pSS. One possible explanation is that fine tuning of the antigen-binding pocket is preferentially active on the VH CDR3 and IgVL chains. Overall, concentration and maintenance of B cell activation in the salivary glands of patients with pSS leads to a significant depletion of memory B cells in the peripheral blood, probably resulting in autoantibody production and potential malignant transformation of B lymphocytes in the glands. It will be important to identify factors directing the migration and accumulation of B lymphocytes in order to interrupt the apparent immunopathology in patients with SS.
Abbreviations
- BCA-1:
-
= B-cell-attracting chemokine 1
- CDR:
-
= complementarity-determining region
- GC:
-
= germinal center
- IFN:
-
= interferon
- IgV:
-
= Ig variable region
- IL:
-
= interleukin
- LT:
-
= lymphotoxin
- NHL:
-
= non-Hodgkin lymphoma
- pSS:
-
= primary Sjögren's syndrome
- RA:
-
= rheumatoid arthritis
- RAG:
-
= recombination activating gene
- SDF-1:
-
= stromal cell-derived factor 1
- SLE:
-
= systemic lupus erythematosus
- TGF:
-
= transforming growth factor
- Th:
-
= T helper
- TNF:
-
= tumor necrosis factor.
References
Fox RI, Stern M, Michelson P: Update in Sjögren syndrome. Curr Opin Rheumatol. 2000, 12: 391-398. 10.1097/00002281-200009000-00007.
Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, Daniels TE, Fox PC, Fox RI, Kassan SS, Pillemer SR, Talal N, Weisman MH: European Study Group on Classification Criteria for Sjögren's Syndrome. Assessment of the European classification criteria for Sjögren's syndrome in a series of clinically defined cases: results of a prospective multicentre study. The European Study Group on Diagnostic Criteria for Sjögren's Syndrome. Ann Rheum Dis. 1996, 55: 116-121.
Xanthou G, Tapinos NI, Polihronis M, Nezis IP, Margaritis LH, Moutsopoulos HM: CD4 cytotoxic and dendritic cells in the immunopathologic lesion of Sjögren's syndrome. Clin Exp Immunol. 1999, 118: 154-10.1046/j.1365-2249.1999.01037.x.
Stott DI, Hiepe F, Hummel M, Steinhauser G, Berek C: Antigen-driven clonal proliferation of B cells within the target tissue of an autoimmune disease. The salivary glands of patients with Sjögren's syndrome. J Clin Invest. 1998, 1102: 938-946.
Bodeutsch C, deWilde PC, Kater L, van den Hoogen FH, Hene RJ, van Houwelingen JC, van de Putte LB, Vooijs GP: Monotypic plasma cells in labial salivary glands of patients with Sjögren's syndrome: prognosticator for systemic lymphoproliferative disease. J Clin Pathol. 1993, 46: 123-128.
Horsfall AC, Venables PJ, Allard SA, Maini RN: Co-existent anti-La antibodies and rheumatoid factors bear distinct idiotypic markers. Scand J Rheumatol. 1988, 75 (Suppl): 84-88.
Fox RI, Konttinen Y, Fisher A: Use of muscarinic agonists in the treatment of Sjögren's syndrome. Clin Immunol. 2001, 101: 249-263. 10.1006/clim.2001.5128.
Schröder AE, Greiner A, Seyfert C, Berek C: Differentiation of B cells in the nonlymphoid tissue of the synovial membrane of patients with rheumatoid arthritis. Proc Natl Acad Sci USA. 1996, 93: 221-225. 10.1073/pnas.93.1.221.
Meffre E, Davis E, Schiff C, Cunningham-Rundles C, Ivashkiv LB, Staudt LM, Young JW, Nussenzweig MC: Circulating human B cells that express surrogate light chains and edited receptors. Nat Immunol. 2000, 1: 207-213. 10.1038/79739.
Amft N, Bowman SJ: Chemokines and cell trafficking in Sjögren's syndrome. Scand J Immunol. 2001, 54: 62-69. 10.1046/j.1365-3083.2001.00970.x.
Amft N, Curnow SJ, Scheel-Toellner D, Devadas A, Oates J, Crocker J, Hamburger J, Ainsworth J, Mathews J, Salmon M, Bowman SJ, Buckley CD: Ectopic expression of the B cell-attracting chemokine BCA-1 (CXCL13) on endothelial cells and within lymphoid follicles contributes to the establishment of germinal center-like structures in Sjögren's syndrome. Arthritis Rheum. 2001, 44: 2633-2641. 10.1002/1529-0131(200111)44:11<2633::AID-ART443>3.0.CO;2-9.
Shi K, Hayashida K, Kaneko M, Hashimoto J, Tomita T, Lipsky PE, Yoshikawa H, Ochi T: Lymphoid chemokine B cell-attracting chemokine-1 (CXCL13) is expressed in germinal center of ectopic lymphoid follicles within the synovium of chronic arthritis patients. J Immunol. 2001, 166: 650-655.
Halse A, Tengner P, Wahren-Herlenius M, Haga H, Jonsson R: Increased frequency of cells secreting interleukin-6 and interleukin-10 in peripheral blood of patients with primary Sjögrens syndrome. Scand J Immunol. 1999, 49: 533-528. 10.1046/j.1365-3083.1999.00533.x.
Moutsopoulos HM, Kordossis T: Sjögren's syndrome revisited: autoimmune epithelitis. Br J Rheumatol. 1996, 35: 204-206.
Taga K, Cherney B, Tosato G: IL-10 inhibits apoptotic cell death in human T cells starved of IL-2. Int Immunol. 1993, 5: 1599-1608.
Fox R, Kang HI, Ando D, Abrams J, Pisa E: Cytokine mRNA expression in salivary gland biopsies of Sjögrens syndrome. J Immunol. 1994, 152: 5532-5539.
Ngo VN, Korner H, Gunn MD, Schmidt KN, Riminton DS, Cooper MD, Browning JL, Sedgwick JD, Cyster JG: Lymphotoxin-αβ and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J Exp Med. 1999, 189: 403-412. 10.1084/jem.189.2.403.
Gunn MD, Kyuwa S, Tam C, Kakiuchi T, Matsuzawa A, Williams LT, Nakano H: Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med. 1999, 189: 451-460. 10.1084/jem.189.3.451.
Cyster JG: Chemokines and cell migration in secondary lymphoid organs. Science. 1999, 286: 2098-2102. 10.1126/science.286.5447.2098.
Forster R, Mattis AE, Kremmer E, Wolf E, Brem G, Lipp M: A putative chemokines receptor, BLR1, directs B cell migration to defined lymphoid organs and specific anatomic compartments of the spleen. Cell. 1996, 87: 1037-1047.
Luther SA, Lopez T, Bai W, Hanahan D, Cyster JG: BCA-1 expression in pancreatic islets causes B cell recruitment and lymphotoxin-dependent lymphoid neogenesis. Immunity. 2000, 12: 471-481.
Nanki T, Hayashida K, El-Gabalawy HS, Suson S, Shi K, Girschick HJ, Yavuz S, Lipsky PE: Stromal cell-derived factor-1-CXC chemokine receptor 4 interactions play a central role in CD4+ T cell accumulation in rheumatoid arthritis synovium. J Immunol. 2000, 165: 6590-6598.
Groom J, Kalled SL, Cutler AH, Olson C, Woodcock SA, Schneider P, Tschopp J, Cachero TG, Batten M, Wheway J, Mauri D, Cavill D, Gordon TP, Mackay CR, Mackay F: Association of BAFF/BLyS overexpression and altered B cell differentiation with Sjögren's syndrome. J Clin Invest. 2002, 109: 59-68. 10.1172/JCI200214121.
Zhang J, Roschke V, Baker KP, Wang Z, Alarcon GS, Fessler BJ, Bastian H, Kimberly RP, Zhou T: Cutting edge: a role for B lymphocyte stimulator in systemic lupus erythematosus. J Immunol. 2001, 166: 6-10.
Dörner T, Lipsky PE: Immunoglobulin variable-region gene usage in systemic autoimmune diseases. Arthritis Rheum. 2001, 44: 2715-2727. 10.1002/1529-0131(200112)44:12<2715::AID-ART458>3.0.CO;2-L.
Brezinschek HP, Foster SJ, Dörner T, Brezinschek RI, Lipsky PE: Pairing of variable heavy and variable kappa chains in individual naive and memory B cells. J Immunol. 1998, 160: 4762-4767.
Brezinschek HP, Brezinschek RI, Lipsky PE: Analysis of the heavy chain repertoire of human peripheral blood B cells using single-cell polymerase chain reaction. J Immunol. 1995, 155: 190-202.
Brezinschek HP, Foster SJ, Brezinschek RI, Dörner T, Domiati-Saad R, Lipsky PE: Analysis of the human VH gene repertoire. Differential effects of selection and somatic hypermutation on peripheral CD5+/IgM+ and CD5-/IgM+ B cells. J Clin Invest. 1997, 99: 2488-2501.
Farner NL, Dörner T, Lipsky PE: Molecular mechanisms and selection influence the generation of the human VλJλ repertoire. J Immunol. 1999, 162: 2137-2145.
Foster SJ, Brezinschek HP, Brezinschek RI, Lipsky PE: Molecular mechanisms and selective influences that shape the kappa gene repertoire of IgM+ B cells. J Clin Invest. 1997, 99: 1614-1627.
Dörner T, Brezinschek HP, Brezinschek RI, Foster SJ, Domiati-Saad R, Lipsky PE: Analysis of the frequency and pattern of somatic mutations within non-productively rearranged human VH genes. J Immunol. 1997, 158: 2779-2789.
Dörner T, Brezinschek HP, Foster SJ, Brezinschek RI, Farner NL, Lipsky PE: Comparable impact of mutational and selective influences in shaping the expressed repertoire of peripheral IgM+/CD5- and IgM+/CD5+ B cells. Eur J Immunol. 1998, 28: 657-668. 10.1002/(SICI)1521-4141(199810)28:10<3384::AID-IMMU3384>3.3.CO;2-K.
Odendahl M, Jacobi A, Hansen A, Feist E, Hiepe F, Burmester GR, Lipsky PE, Radbruch A, Dörner T: Disturbed peripheral B lymphocyte homeostasis. J Immunol. 2000, 165: 5970-5979.
Dörner T, Foster SJ, Farner NL, Lipsky PE: Immunoglobulin kappa chain receptor editing in systemic lupus erythematosus. J Clin Invest. 1998, 102: 688-694.
Dörner T, Farner NL, Lipsky PE: Immunoglobulin lambda and heavy chain gene usage in early untreated systemic lupus erythematosus suggests intensive B cell stimulation. J Immunol. 1999, 163: 1027-1036.
Heimbächer C, Hansen A, Pruss A, Jacobi A, Reiter K, Lipsky PE, Dörner T: Immunoglobulin Vκ light chain analysis in patients with Sjögren's syndrome. Arthritis Rheum. 2001, 44: 626-637. 10.1002/1529-0131(200103)44:3<626::AID-ANR111>3.3.CO;2-K.
Kaschner S, Hansen A, Jacobi A, Reiter K, Monson NL, Odendahl M, Burmester GR, Lipsky PE, Dörner T: Immunoglobulin Vλ light chain gene usage in patients with Sjögren's syndrome. Arthritis Rheum. 2001, 44: 2620-2632. 10.1002/1529-0131(200111)44:11<2620::AID-ART442>3.0.CO;2-M.
Jacobi AM, Hansen A, Kaufmann O, Burmester GR, Lipsky PE, Dörner T: Analysis of immunoglobulin light chain rearrangements in the salivary gland and blood of a patient with Sjögren's syndrome. Arthritis Res. 2002, 4: R4-10.1186/ar423.
Hansen A, Jacobi AM, Burmester GR, Lipsky PE, Dörner T: Comparison of heavy chain rearrangements in the blood and parotid gland of a patient with Sjögren's syndrome. Scand J Immunol. 2002,
Hansen A, Odendahl M, Reiter K, Jacobi AM, Feist E, Scholze J, Burmester GR, Lipsky PE, Dörner T: Evidence for the migration and accumulation of memory B cells in the salivary glands of patients with Sjögren's syndrome. Arthritis Rheum. 2002, 46: 2160-2171. 10.1002/art.10445.
Dörner T, Kaschner S, Hansen A, Pruss A, Lipsky PE: Perturbations in the impact of mutational activity on Vλ genes in systemic lupus erythematosus. Arthritis Res. 2001, 3: 368-374. 10.1186/ar329.
de Wildt RM, Hoet RM, van Venrooij WJ, Tomlinson IM, Winter G: Analysis of heavy and light chain pairings indicates that receptor editing shapes the human antibody repertoire. Mol Biol. 1999, 285: 895-901. 10.1006/jmbi.1998.2396.
Silberstein LE, Jefferies LC, Goldman J, Friedman D, Moore JS, Nowell PC, Roelcke D, Pruzanski W, Roudier J, Silverman GJ: Variable region gene analysis of pathologic human autoantibodies to the related i and I red blood cell antigens. Blood. 1991, 78: 2372-2386.
Dörner T, Heimbacher C, Farner NL, Lipsky PE: Enhanced mutational activity of Vκ gene rearrangements in systemic lupus erythematosus. Clin Immunol. 1999, 92: 188-196. 10.1006/clim.1999.4740.
Pugh-Bernard AE, Silverman GJ, Cappione AJ, Villano ME, Ryan DH, Insel RA, Sanz I: Regulation of inherently autoreactive VH4-34 B cells in the maintenance of human B cell tolerance. J Clin Invest. 2001, 108: 1061-1070. 10.1172/JCI200112462.
Yavuz S, Grammer AC, Yavuz AS, Nanki T, Lipsky PE: Comparative characteristics of mu chain and alpha chain transcripts expressed by individual tonsil plasma cells. Mol Immunol. 2001, 38: 19-34. 10.1016/S0161-5890(01)00036-0.
Kelsoe G: Life and death in germinal centers (Redux). Immunity. 1996, 4: 107-111.
Nemazee D, Weigert M: Revising B cell receptors. J Exp Med. 2000, 191: 1813-1817. 10.1084/jem.191.11.1813.
Girschick HJ, Grammer AC, Nanki T, Mayo M, Lipsky PE: RAG1 and RAG2 expression by B cell subsets from human tonsil and peripheral blood. J Immunol. 2001, 166: 377-386.
Lee J, Monson NL, Lipsky PE: The VλJλ repertoire in human fetal spleen: evidence for positive selection and extensive receptor editing. J Immunol. 2000, 165: 6322-6333.
Radic MZ, Zouali M: Receptor editing, immune diversification and self-tolerance. Immunity. 1996, 5: 505-511.
Bensimon C, Chastagner P, Zouali M: Human lupus anti-DNA autoantibodies undergo essentially primary Vκ gene rearrangements. EMBO J. 1994, 13: 2951-2962.
Chen C, Luning-Prak E, Weigert M: Editing disease-associated autoantibodies. Immunity. 1997, 6: 97-105.
Itoh K, Meffre E, Albesiano E, Farber A, Dines D, Stein P, Asnis SE, Furie RA, Jain RI, Chiorazzi N: Immunoglobulin heavy chain variable region gene replacement as a mechanism for receptor revision in rheumatoid arthritis synovial tissue B lymphocytes. J Exp Med. 2000, 192: 1151-1164. 10.1084/jem.192.8.1151.
Zhang Z, Wu X, Limbaugh BH, Bridges SL: Expression of recombination-activating genes and terminal deoxynucleotidyl transferase and secondary rearrangement of immunoglobulin kappa light chains in rheumatoid arthritis synovial tissue. Arthritis Rheum. 2001, 44: 2275-2284. 10.1002/1529-0131(200110)44:10<2275::AID-ART390>3.0.CO;2-K.
Elagib KE, Borretzen M, Thompson KM, Natvig JB: Light chain variable (VL) sequences of rheumatoid factors (RF) in patients with primary Sjögren's syndrome (pSS): moderate contribution of somatic hypermutation. Scand J Immunol. 1999, 50: 492-498. 10.1046/j.1365-3083.1999.00624.x.
Bohnhorst J, Bjorgan MB, Thoen JE, Natvig JB, Thompson KM: Bm1–bm5 classification of peripheral blood B cells reveals circulating germinal center founder cells in healthy individuals and disturbance in the B cell subpopulations in patients with primary Sjögren's syndrome. J Immunol. 2001, 167: 3610-3618.
Bahler DW, Swerdlow SH: Clonal salivary gland infiltrates associated with myoepithelial sialadenitis (Sjögren's syndrome) begin as nonmalignant antigen-selected expansion. Blood. 1998, 91: 1864-1872.
Kipps TJ, Tomhave E, Chen PP, Fox RI: Molecular characterization of a major autoantibody-associated cross-reactive idiotype in Sjögren's syndrome. J Immunol. 1989, 142: 4261-4268.
Gellrich S, Rutz S, Borkowski A, Golembowski S, Gromnica-Ihle E, Sterry W, Jahn S: Analysis of V(H)-D-J(H) gene transcripts in B cells infiltrating the salivary glands and lymph node tissues of patients with Sjögren's syndrome. Arthritis Rheum. 1999, 42: 240-247. 10.1002/1529-0131(199902)42:2<240::AID-ANR5>3.0.CO;2-I.
Tomlinson IM, Cook GP, Walter G, Carter NP, Riethman H, Buluwela L, Rabbitts TH, Winter G: A complete map of the human immunoglobulin VH locus. Ann NY Acad Sci. 1995, 764: 43-46.
Insel RA, Adderson EE, Carroll WL: The repertoire of human antibody to the Haemophilus influenzae type b capsular polysaccharide. Int Rev Immunol. 1992, 9: 25-43.
Sun Y, Park MK, Kim J, Diamond B, Solomon A, Nahm MH: Repertoire of human antibodies against the polysaccharide capsule of Streptococcus pneumoniae serotype 6B. Infect Immun. 1999, 67: 1172-1179.
Ikematsu W, Kobarg J, Ikematsu H, Ichiyoshi Y, Casali P: Clonal analysis of a human antibody response. III. Nucleotide sequences of monoclonal IgM, IgG, and IgA to rabies virus reveal restricted Vκ gene utilization, junctional VκJκ and VλJλ diversity, and somatic hypermutation. J Immunol. 1998, 161: 2895-2905.
Deacon EM, Matthews JB, Potts AJ, Hamburger J, Mageed RA, Jefferis R: Expression of rheumatoid factor associated cross-reactive idiotypes by glandular B cells in Sjögren's syndrome. Clin Exp Immunol. 1991, 83: 280-285.
Martin T, Weber JC, Levallois H, Labouret N, Soley A, Koenig S, Korganow AS, Pasquali JC: Salivary gland lymphomas in patients with Sjögren's syndrome may frequently develop from rheumatoid factor B cells. Arthritis Rheum. 2000, 43: 908-916. 10.1002/1529-0131(200004)43:4<908::AID-ANR24>3.0.CO;2-K.
Bahler DW, Miklos JA, Swerdlow SH: Ongoing Ig gene hypermutation in salivary gland mucosa-associated lymphoid tissue-type lymphomas. Blood. 1997, 89: 3335-3344.
Klein U, Rajewsky K, Küppers R: Human immunoglobulin (Ig)M+IgD+ peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J Exp Med. 1998, 188: 1679-1689. 10.1084/jem.188.9.1679.
Miklos JA, Swerdlow SH, Bahler DW: Salivary gland mucosa associated lymphoid tissue lymphoma immunoglobulin VH genes show frequent use of V1-69 with distinctive CDR3 features. Blood. 2000, 95: 3878-3884.
Rosner K, Winter DB, Tarone RE, Skovgaard GL, Bohr VA, Gearhart PJ: Third complementarity-determining region of mutated VH immunoglobulin genes contains shorter V, D, J, P, and N components than non-mutated genes. Immunology. 2001, 103: 179-187. 10.1046/j.1365-2567.2001.01220.x.
Küppers R, Zhao M, Hansmann ML, Rajewsky K: Tracing B cell development in human germinal centres by molecular analysis of single cells picked from histological sections. EMBO J. 1993, 12: 4955-4967.
Arce E, Jackson DG, Gill MA, Bennett LB, Banchereau J, Pascual V: Increased frequency of pre-germinal center B cells and plasma cell precursors in the blood of children with systemic lupus erythematosus. J Immunol. 2001, 167: 2361-2369.
Agematsu K, Nagumo H, Yang FC, Nakazawa T, Fukushima K, Ito S, Sugita K, Mori T, Kobata T, Morimoto C: B cell subpopulations separated by CD27 and crucial collaboration of CD27+ B cells and helper T cells in immunoglobulin production. Eur J Immunol. 1997, 27: 2073-2079.
Tangye SG, Liu YJ, Aversa G, Phillips JH, de Vries JE: Identification of functional human splenic memory B cells by expression of CD148 and CD27. J Exp Med. 1998, 188: 1691-1703. 10.1084/jem.188.9.1691.
Agematsu K, Nagumo H, Oguchi Y, Nakazawa T, Fukushima K, Yasui K, Ito S, Kobata T, Morimoto C, Komiyama A: Generation of plasma cells from peripheral blood memory B cells: synergistic effect of interleukin-10 and CD27/CD70 interaction. Blood. 1998, 91: 173-180.
Nagumo H, Agematsu K, Shinozaki K, Hokibara S, Ito S, Takamoto M, Nikaido T, Yasui K, Uehara Y, Yachie A: CD27/CD70 interaction augments IgE secretion by promoting the differentiation of memory B cells into plasma cells. J Immunol. 1998, 161: 6496-6502.
Shinozaki K, Yasui K, Agematsu K: Direct B/B-cell interactions in immunoglobulin synthesis. Clin Exp Immunol. 2001, 124: 386-391. 10.1046/j.1365-2249.2001.01518.x.
Nagumo H, Agematsu K, Kobayashi N, Shinozaki K, Hokibara S, Nagase H, Takamoto M, Yasui K, Sugane K, Komiyama A: The different process of class switching and somatic hypermutation; a novel analysis by CD27- naive B cells. Blood. 2002, 99: 567-575. 10.1182/blood.V99.2.567.
Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T: Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000, 102: 553-563.
Moir S, Malaspina A, Ogwaro KM, Donoghue ET, Hallahan CW, Ehler LA, Liu S, Adelsberger J, Lapointe R, Hwu P: HIV-1 induces phenotypic and functional perturbations of B cells in chronically infected individuals. Proc Natl Acad Sci USA. 2001, 98: 10362-10367. 10.1073/pnas.181347898.
Widney D, Gundapp G, Said JW, van der Meijden M, Bonavida B, Demidem A, Trevisan C, Taylor J, Detels R, Martinez-Maza O: Aberrant expression of CD27 and soluble CD27 (sCD27) in HIV infection and in AIDS-associated lymphoma. Clin Immunol. 1999, 93: 114-123. 10.1006/clim.1999.4782.
Agematsu K, Nagumo H, Shinozaki K, Hokibara S, Yasui K, Terada K, Kawamura N, Toba T, Nonoyama S, Ochs HD: Absence of IgD-CD27+ memory B cell population in X-linked hyper-IgM syndrome. J Clin Invest. 1998, 102: 853-860.
Denz A, Eibel H, Illges H, Kienzle G, Schlesier M, Peter HH: Impaired up-regulation of CD86 in B cells of 'type A' common variable immunodeficiency patients. Eur J Immunol. 2000, 30: 1069-1077. 10.1002/(SICI)1521-4141(200004)30:4<1069::AID-IMMU1069>3.0.CO;2-M.
Ebo D, DeClerck LS, Bridts CH, Stevens WJ: Expression of CD5 and CD23 on B cells of patients with rheumatoid arthritis, systemic lupus erythematosus and Sjögren's syndrome. Relationship with disease activity and treatment. In Vivo. 1994, 8: 577-580.
Zeher M, Suranyi P, Nagy G, Szegedi G: B cells expressing CD5 in minor labial salivary glands of patients with primary Sjögren's syndrome. Arthritis Rheum. 1990, 33: 453-
Brennan F, Plater-Zyberk C, Maini RN, Feldmann M: Coordinate expansion of 'fetal type' lymphocytes (TCRγΔ+T and CD5+B) in rheumatoid arthritis and primary Sjögren's syndrome. Clin Exp Immunol. 1989, 77: 175-178.
Plater-Zyberk C, Brennan FM, Feldmann M, Maini RN: 'Fetal-type' B and T lymphocytes in rheumatoid arthritis and primary Sjögren's syndrome. J Autoimmun. 1989, 2: 233-241.
Foster HE, Calvert JE, Kelly CA, Griffiths ID: Levels of CD5+ B cells are not increased in probands or relatives in a family study of primary Sjögren's syndrome. Autoimmunity. 1992, 12: 207-214.
Dauphinee M, Tovar Z, Talal N: B cells expressing CD5 are increased in Sjögren's syndrome. Arthritis Rheum. 1988, 31: 642-647.
DiGiuseppe JA, Corio RL, Westra WH: Lymphoid infiltrates of the salivary glands: pathology, biology, and clinical significance. Curr Opin Oncol. 1996, 8: 232-237.
De Vita S, Boiocchi M, Sorrentino D, Carbone A, Avellini C: Characterization of prelymphomatous stages of B cell lymphoproliferation in Sjögren's syndrome. Arthritis Rheum. 1997, 40: 318-331.
Kassan SS, Thomas TL, Moutsoloulos HM, Hoover R, Kimberly RP, Budman DR, Costa J, Decker JL, Cused TM: Increased risk of lymphoma in sicca syndrome. Ann Intern Med. 1978, 89: 888-892.
Chen PP, Olsen NJ, Yang PM, Soto-Gil RW, Olee T, Siminovitch KA, Carson DA: From human autoantibodies to the fetal antibody repertoire to B cell malignancy: it's a small world after all. Intern Rev Immunol. 1990, 5: 239-251.
Mackay IR, Rose NR: Autoimmunity and lymphoma: tribulations of B cells. Nat Immunol. 2001, 2: 793-795. 10.1038/ni0901-793.
Bahler DW, Miklos JA, Swerdlow SH: Ongoing Ig gene hypermutation in salivary gland mucosa-associated lymphoid tissue type lymphomas. Blood. 1997, 89: 3335-3344.
Miklos JA, Swerdlow SH, Bahler DW: Salivary gland mucosa-associated lymphoid tissue lymphoma immunoglobulin VH genes show frequent use of V1-69 with distinctive CDR3 features. Blood. 2000, 95: 3878-3884.
van Oers MH, Pals ST, Evers LM, van der Schoot CE, Koopman G, Bonfrer JM, Hintzen RQ, von dem Borne AE, van Lier RA: Expression and release of CD27 in human B-cell malignancies. Blood. 1993, 82: 3430-3406.
Lens SM, Tesselaar K, van Oers MH, van Lier RA: Control of lymphocyte function through CD27-CD70 interactions. Semin Immunol. 1998, 10: 491-499. 10.1006/smim.1998.0154.
Ruzickova S, Pruss A, Odendahl M, Wolbart K, Burmester GR, Dörner T, Hansen A: Chronic lymphocytic leukemia preceeded by cold agglutinin disease: intraclonal Ig light chain diversity in VH4-34 expressing single B cells. Blood. 2002,
Acknowledgements
This work was supported by Deutsche Forschungsgemeinschaft Grants Sonderforschungsbereich 421/TP C7, Do 491/4–1, 4–3 and 5–1, and by National Institutes of Health Grant AI 31229.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dörner, T., Lipsky, P.E. Abnormalities of B cell phenotype, immunoglobulin gene expression and the emergence of autoimmunity in Sjögren's syndrome. Arthritis Res Ther 4, 360 (2002). https://doi.org/10.1186/ar603
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar603