
Chapter summary
Signal transduction induced by tumor necrosis factor (TNF) family members and their receptors has been an intensive area of research for several years. The major impact of these studies has been the delineation of apoptotic and cell survival signaling pathways. These discoveries, coupled with major advances in the study of mammalian apoptotic machinery, constitute a promising blueprint of the molecular network governing the fate of all living cells. In this review, we concentrate on the fate of cells in the immune system, where regulation of cell death and cell survival is a frequent and important exercise. A small imbalance in favor of either fate can result in disastrous pathological outcomes, such as cancer, autoimmunity or immune deficiency. It is an insurmountable task to discuss all molecules reported in the literature that are implicated in lymphocyte death or survival. We have therefore focused on discoveries made by mouse gene targeting, as these studies provide the most physiologically relevant information on each molecule. We begin with a description of signaling channels initiated by TNF receptor type 1 engagement, which can lead to either cell survival or to cell death. The point of bifurcation of this pathway and the decision-making molecules FADD, TRAF2 and RIP are discussed. We then follow apoptotic and survival pathways from upstream to downstream, describing many important players involved in signal transduction. Molecules important for NF-κB and JNK/stress-activated protein kinase activation such as IKKβ, NEMO, MAP3K and TRAF6 are discussed, as is the impact of BAFF and its receptors on B-cell survival. Mouse mutants that have helped to define the mammalian apoptosis execution machinery, including animals lacking Apaf-1, caspase-3 and caspase-9, are also described. We conclude with a brief analysis of the potential therapeutic options arising from this body of work.
Similar content being viewed by others

Introduction
Homeostasis is an essential aspect of the mammalian immune system [1, 2]. The majority of lymphocytes that expand and proliferate in response to antigens in vivo has to subsequently die to maintain a constant number of these cells between immune responses. During the initial expansion phase of an immune response, T cells and B cells are induced to divide by antigens, growth factors and cytokines. These agents trigger cellular signals that sustain lymphocyte growth and survival. During the eclipse phase of an immune response, however, apoptosis (programmed cell death [PCD]) must occur to reduce the number of accumulated cells and to restore homeostasis. These survival and apoptotic signals are highly regulated [3, 4], and the understanding of the processes mediating these cellular decisions should aid in the design of rational therapeutics for autoimmune diseases, inflammation and infections.
For the immune system, many apoptotic signals and survival signals are mediated via receptors present on the surfaces of lymphocytes and other hemopoietic cells. The most prominent of these receptors belong to a group of structurally related signaling proteins called the TNF receptor (TNFR) superfamily [5–7]. Over the past decade, many TNFRs and their ligands have been molecularly cloned and characterized.
Even more exciting is the identification of the myriad of downstream effector proteins involved in coordinating and controlling various signaling cascades. Both apoptotic signals and survival signals in lymphocytes are primarily mediated by various members of the TNFR superfamily. Intriguingly, in some cases, signals associated with PCD or survival can be transmitted through the same receptor in response to the same ligand. Understanding how such signal transduction is executed is a key goal for the many laboratories studying immune and inflammatory responses.
In the present review, we shall concentrate on information gained from studies of gene-targeted mutant mice deficient in the expression of proteins involved in apoptotic and survival signaling pathways in lymphocytes. We shall focus particularly on signal mediators and effectors that act in the TNFR pathways and on the common apoptosis execution machinery. For a more in-depth survey of work in this area, please refer to other recent comprehensive reviews [2–11].
TNFR superfamily
The most important members of the TNFR superfamily involved in lymphocyte signal transduction are TNF receptor-associated factor type 1 (TNFR1), TNF receptor-associated factor type 2 (TNFR2) and Fas (CD95). TNFR1 and TNFR2, which both bind to the cytokine TNF, were the first members of the TNFR family to be identified.
Studies of gene-targeted mice lacking expression of TNFR1 demonstrated that this receptor is involved in signaling leading to inflammation and cell survival [12]. Paradoxically, however, PCD was also found to be impaired in TNFR1-/- mutants [13]. In response to a single ligand (TNF), TNFR1 thus mediates not only survival and inflammatory signaling, but also apoptotic signaling. In contrast, in response to TNF binding, TNFR2 transduces signals principally for survival and inflammation [8]. The Fas (CD95) receptor, which was originally discovered as the product of the mutated gene in MRL/lpr mice [14], mediates mainly signals for apoptosis in response to the binding of its ligand FasL. Mice deficient for Fas or FasL show massive lymphoadenopathy and disruption of lymphocyte homeostasis due to a lack of PCD.
How is the signaling initiated by engagement of TNFR superfamily members transduced within the cell? In response to ligand binding, various proteins are recruited to specific domains in the cytoplasmic tails of the receptors. It is these downstream effectors, which vary in structure and function, that determine signaling outcomes. Some effectors are enzymes, such as kinases, phosphatases and proteases, some are adaptor proteins that serve to recruit still more signaling intermediaries, and some are regulatory proteins. Understanding the nature and functions of these downstream effectors will allow us to explain how particular cellular outcomes result from signaling through a limited number of receptors.
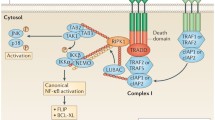
TNFR superfamily members are categorized into two classes based on the effectors recruited to their cytoplasmic domains. In the first class, which includes TNFR1, Fas, death receptor (DR)3, DR4, DR5 and DR6, the cytoplasmic tail contains a 'death domain' (DD) [6, 15]. The DDs may vary slightly in sequence between receptors but all are highly homologous and provide the capacity for protein–protein interaction. The DD in a receptor tail recruits intracellular adaptor proteins that also contain a DD. For example, TNFR1 contains a DD that binds to the TNF receptor-associated death domain protein (TRADD), and Fas contains a DD that binds to both Fas-associated death domain protein (FADD) and receptor-interacting protein (RIP) [5, 9] (Fig. 1).

Intracellular signaling pathways downstream of tumor necrosis factor (TNF) receptor superfamily receptors. The Fas, tumor necrosis factor receptor type 1 (TNFR1) and tumor necrosis factor receptor type 2 (TNFR2) receptors are shown extending through the cell membrane with their extracellular domains projecting into the extracellular space. Various adaptor proteins and signal transducing molecules that convey signals initiated by the binding of the ligands Fas ligand (FasL) to Fas and TNF to TNFR1 and TNFR2, respectively, are shown, as is the crosstalk between various molecules and pathways. Functional domains within a protein are shown as colored blocks. Recruitment of one protein to another is indicated by the juxtaposition of like-colored and like-shaped domain blocks. BID, beta interaction domain; cIAPs, cellular inhibitors of apoptosis; FADD, Fas-associated death domain protein; IKK, IκB kinase; JNK, c-Jun N-terminal kinase; MAP3K, mitogen-activated protein 3 kinase; NEMO, NF-κB essential modulator protein; RIP, receptor-interacting protein; T2K, TRAF2-associated kinase; TANK, TRAF family member associated NF-κB activator; TRADD, TNF receptor-associated death domain protein; TRAF, TNF receptor-associated factor.
The second class of TNFR superfamily receptors includes TNFR2, CD30, CD40, lymphotoxin receptor, the osteoprotegerin ligand (OPGL) receptor activator of NF-κB (RANK), and B-lymphocyte activating factor (BAFF) receptors [6, 16]. The cytoplasmic tails of these receptors do not contain DDs. Instead, they have sequences allowing them to associate with a different set of intracellular adaptors called TNF receptor-associated factors (TRAFs) [17, 18]. Although six TRAF proteins (TRAF1–TRAF6) have been identified to date, only five (TRAF1, TRAF2, TRAF3, TRAF5 and TRAF6) actually bind to the cytoplasmic tails of the second class of TNFR superfamily receptors.
Other groups and ourselves have generated gene-targeted 'knockout mice' for several signaling proteins downstream of these receptors, and have been able to gain insights into their functions and regulation from examining the phenotypes of these animals and their cells. In the present review, we discuss the receptor-proximal signaling adaptors and their key functions in deciding whether a cell will survive or will undergo apoptosis. We also examine the successive molecular events that make up the initial phase of the cascade of apoptosis, and its terminal execution machinery.
FADD, TRAF2 and RIP
It is now well established that TNFR1 can mediate signals for either apoptosis or cell survival. Which signaling path is triggered following receptor aggregation appears to be a function of the recruitment of the different adaptor proteins appropriate for each path. Indeed, Hsu et al. showed that when TRADD binds to TNFR1, TRADD can then associate with either FADD or TRAF2 and with RIP [19] (Fig. 1). This observation led to the hypothesis that recruitment of FADD to the TNFR1 complex would lead to PCD, while recruitment of TRAF2 and RIP would signal for survival. We tested this hypothesis by generating gene-targeted mutant mice deficient for expression of either FADD or TRAF2.
FADD-deficient mice do not survive beyond day 11.5 of embryogenesis [20, 21], indicating that FADD is essential for embryonic development. In situ hybridization studies confirmed that FADD is expressed widely during embryogenesis, consistent with its role in fundamental developmental processes. Most FADD-/- embryos have thin ventricular myocardium, poorly developed inner trabeculation, and abdominal hemorrhage.
The precise function of FADD in embryonic development is not clearly understood at this time. Using cells derived from FADD-/- embryos, however, we have demonstrated the importance of FADD in apoptotic signaling initiated by TNFR superfamily receptors. Following engagement of Fas, TNFR1 or DR3, apoptosis was dramatically reduced in FADD-/- cells compared with in controls. However, levels of PCD in response to viral oncogene overexpression or chemotherapeutic drug administration were normal in FADD-/- cells [20], indicating that not all agents inducing apoptosis transduce their signals through FADD. Interestingly, FADD-deficient T cells, in addition to being resistant to Fas-induced apoptosis, showed a defect in activation-induced proliferation [21].
In contrast, TRAF2-/- cells showed increased levels of PCD on stimulation by TNF [22], consistent with a scenario in which cell survival depends on the recruitment of TRAF2 to TNFR1. Surprisingly, however, TRAF2-/- cells were still able to activate the transcription factor NF-κB following engagement of TNFR1. The activation of NF-κB had previously been thought to depend on signaling through TRAF2, in that a dominant negative TRAF2 protein blocked TNF-mediated activation of NF-κB. Rather than effects on NF-κB, we found that TNF-induced activation of the Jun kinase (JNK) pathway was greatly impaired in the absence of TRAF2. Interestingly, cells deficient in RIP, another adaptor protein associated with TRADD, show a reduction in TNF-mediated NF-κB activation [23]. Taken together, these results indicate that cell survival signaling mediated by TRAF2 does more than just activate NF-κB. The JNK pathway and perhaps other proteins, such as the cellular inhibitors of apoptosis, may also contribute to TNF-induced cell survival [24].
Caspase-8 and c-FLICE (caspase-8) inhibitory protein
Caspase-8 is the key initiator caspase downstream of the apoptosis pathway induced by TNFR1 and other death receptors (Fig. 1). Caspase-8 interacts with FADD through homologous death effector domains. Aggregation and close proximity result in the activation of caspase-8 such that its caspase domain is processed into two active subunits.
c-FLICE (caspase-8) inhibitory protein (c-FLIP), a cellular homolog of viral FLICE (caspase-8) inhibitory protein, is structurally similar to caspase-8 in that it possesses death effector domains and a caspase-like domain. However, c-FLIP lacks caspase enzymatic activity. It is thought that c-FLIP recruited by FADD to the receptor complex in the place of caspase-8 impedes the progression of apoptotic signals. Caspase-8 and c-FLIP thus appear to play critical, but opposite, roles in regulating DR signaling.
Mice lacking caspase-8 are phenotypically similar to FADD-deficient mice [25]. Caspase-8-/- mice die at approximately day 12.5 of embryogenesis with apparent hyperemia and heart defects. Caspase-8-/- cells, like FADD-/- cells, are specifically resistant to DR-induced apoptosis. Surprisingly, c-FLIP-/- mice show a very similar embryonic phenotype to FADD-/- mice and caspase-8-/- mice [26]. The ventricular myocardium and trabeculae are poorly developed in day 10.5 of embryogenesis c-FLIP-/- embryos.
Like FADD-deficient embryos, embryos lacking c-FLIP do not show signs of increased or decreased cell death, suggesting that the developmental defects in these mutant mice may be independent of apoptosis. In contrast, cells lacking c-FLIP are highly sensitive to apoptotic stimuli that trigger death receptors [26]. This result is consistent with the predicted role of c-FLIP to counteract FADD and caspase-8 in the regulation of DR-induced apoptosis.
Bid and Bim
Bid is a proapoptotic Bcl-2 family member that is cleaved and activated by caspase-8 in response to DR signaling. Bid then translocates to the mitochondria and mediates the release of cytochrome c that leads to apoptotic changes (Fig. 2). Bid-deficient mice are resistant to the anti-Fas antibody-induced apoptosis of hepatocytes that kills wild-type mice. However, a milder defect of FasL-induced or TNF-induced apoptosis was observed in Bid-deficient thymocytes and mouse embryonic fibroblasts (MEF) [27]. These results suggest that, depending on tissue type, DR-induced apoptosis does not necessarily have to go through Bid and the mitochondrial pathway.

Intrinsic and extrinsic pathways of apoptosis. The extrinsic pathway is triggered by death receptor engagement, which initiates a signaling cascade mediated by caspase-8 activation. Caspase-8 both feeds directly into caspase-3 activation and stimulates the release of cytochrome c by the mitochondria. Caspase-3 activation leads to the degradation of cellular proteins necessary to maintain cell survival and integrity. The intrinsic pathway occurs when various apoptotic stimuli trigger the release of cytochrome c from the mitochondria (independently of caspase-8 activation). Cytochrome c interacts with Apaf-1 and caspase-9 to promote the activation of caspase-3. Various intermediary signaling molecules (many of whose functions are not completely defined) and proteins inhibiting the apoptotic cascade are also shown. Apaf-1, apoptosis-activating factor 1; Bak, bacille Calmette–Guérin; Bax, BCL-2-associated × protein; Bid, proapoptotic Bcl-2 family member; Bim, proapoptotic Bcl-2 family member; DIABLO, direct IAP binding protein with low PI; FADD, Fas-associated death domain protein; IAP, inhibitors of apoptosis; Smac, second mitochondria-derived activator of caspase; tBid, truncated beta interaction domain.
Bim is another proapoptotic Bcl-2 family member that is not involved in the DR pathway but is critical for thymocyte negative selection. Bim-deficient thymocytes are refractory to apoptosis induced by TCR stimulation, and autoreactive thymocytes accumulate in TCR transgenic Bim-/- mice [28]. Moreover, Bim-deficient lymphocytes accumulate in peripheral blood and in the spleen, and these cells are resistant to apoptotic stimuli such as cytokine deprivation and microtubule perturbation [29].
Mitogen-activated protein 3 kinases
During the induction of cell survival by TNFR1 engagement, a number of mitogen-activated protein 3 kinase (MAP3K) family members has been shown to associate with TRAF2 or RIP (Fig. 1). NF-κB inducing kinase (NIK) was initially proposed to be the downstream target of TRAF2 in mediating TNF-induced NF-κB activation leading to survival [30]. However, NIK-deficient mice show a specific defect in lymph node development and in lymphotoxin-β-receptor signaling [31, 32], and cells lacking NIK respond normally to TNF in activating NF-κB.
Instead, the mitogen-activated protein extracellular signal regulated kinase kinase (MEKK)-1 was implicated in this NF-κB activation pathway [33]. In addition, MEKK-1 and apoptosis signal-regulating kinase 1 (ASK-1) were reported to mediate TRAF2-triggered JNK activation [34–36]. From studies of knockout mice, however, it seems that MEKK-1 is required for TNF-induced JNK activation only in embryonic stem cells and not in fibroblasts or T cells [37–39]. Similarly, ASK-1 is not required for early phase TNF-induced JNK activation, and ASK-1-/- cells exhibit only a partial defect in sustained stress kinase activation [40]. A new member of the MEKK family (MEKK-3) has recently been found to associate with RIP, meaning that it could potentially play a role in downstream survival signaling. Indeed, disruption of MEKK-3 severely impairs the activation of NF-κB induced by TNF, and MEKK-3-/- cells are highly sensitive to TNF-induced apoptosis [41].
The IκB kinase complex
NF-κB is a key transcription factor whose activation generally promotes cell survival. Mice lacking RelA (p65), a principal subunit of NF-κB, die during embryogenesis due to massive liver apoptosis [42]. NF-κB is normally held inactive in the cytoplasm by its association with the inhibitor of NF-κB protein, IκB. To activate NF-κB, IκB must be removed via phosphorylation followed by ubiquitination and proteasomal degradation. Phosphorylation of IκB is mediated primarily by the IκB kinase (IKK) complex containing the proteins IKKα, IKKβ and NF-κB essential modulator (NEMO) (also known as IKKγ) [10]. IKKα and IKKβ are active kinases, while NEMO is a regulatory protein that binds tightly to both kinases.
Deficiency for IKKβ is embryonic lethal, with the mice displaying a liver apoptosis phenotype similar to that of the RelA knockout [43–45]. In addition, the activation of NF-κB by TNF or IL-1 is defective in IKKβ-/- cells. Reconstitution of lethally irradiated hosts with IKKβ-deficient fetal liver cells revealed a defect in T-lymphocyte survival [46]. The absence of T cells in the IKKβ-deficient fetal liver chimerae was due to the activity of circulating TNF, and normal development of T cells was restored in mice lacking both IKKβ and TNFR1.
NEMO-deficient mice also display a phenotype of fetal liver apoptosis and embryonic lethality, consistent with an essential role for NEMO in the central pathway mediating NF-κB activation [47]. Like RelA-/- cells and IKKβ-/- cells, NEMO-/- cells show an increased susceptibility to TNF-induced apoptosis. NEMO is an X-linked gene, and female NEMO+/- mice develop a self-limiting inflammatory skin disorder characterized by hyperkeratosis and increased apoptosis. This phenotype is presumably dependent on X-chromosome inactivation [48, 49]. Importantly, these symptoms are reminiscent of incontinentia pigmenti, an X-linked dominant hereditary disease in humans. Indeed, genetic studies of incontinentia pigmenti patients have revealed mutations in the NEMO gene and defects in NF-κB activation in the majority of cases [50].
Interestingly, IKKα-deficient mice do not exhibit a fetal liver defect like IKKβ-/- mice. Instead, IKKα-/- mice display abnormal limb and skeletal patterns and display defective epidermal differentiation [51, 52]. The defect in skin formation seems to be independent of NF-κB activity and is due instead to failed secretion of a keratinocyte differentiation-inducing factor [53]. IKKα is not required for IKK or NF-κB activation in response to inflammatory cytokines. Further studies have revealed that IKKα is required for B-cell maturation and secondary lymphoid organogenesis. Most recently, it has been found that IKKα does play a role in NF-κB activation by mediating the processing of the NF-κB2 (p100) precursor protein [54, 55].
TRAF2-associated kinase, glycogen synthase kinase 3β and atypical protein kinase C
NF-κB activation can occur via signaling pathways that are independent of the IKK complex. TRAF2-associated kinase (T2K) (also called TBK and NAK) associates with TRAF2 through an intermediary protein, TRAF family member associated NF-κB activator (TANK) [56–58]. TANK is a serine threonine kinase that is distantly related to IKKα and IKKβ. TANK phosphorylates serine 36 on the IκBα subunit of IκB, but this partial phosphorylation is not sufficient to trigger degradation of IκB. T2K-/- cells show normal IκB phosphorylation and degradation, normal NF-κB translocation into the nucleus, and normal NF-κB binding to target DNA sequences in response to TNF and IL-1. However, NF-κB transactivation activity is affected in cells lacking T2K [58]. This phenomenon is validated by the phenotype of T2K-/- mice, which show liver apoptosis and embryonic lethality similar to that in mice lacking RelA, IKKβ or NEMO. Furthermore, elimination of TNFR1 rescues T2K-deficient mice from embryonic lethality, and the double-knockout animals survive for extended periods with no gross abnormalities [58].
The kinases glycogen synthase kinase (GSK3)α and GSK3β act as inhibitory components of the Wnt signaling pathway important for embryonic development. In frogs and zebrafish, GSK3 is required for the definition of the embryonic axes [59]. However, analysis of GSK3β-deficient mice revealed a surprisingly specific and limited phenotype of fetal liver apoptosis similar to that in mice with NF-κB activation defects [60]. GSK3β-/- MEF were highly susceptible to TNF killing, much like wild-type fibroblasts treated with TNF in the presence of the documented GSK3 inhibitor lithium. Interestingly, TNF-induced IκB phosphorylation and degradation were normal in GSK3β-/- cells, but NF-κB activation was diminished due to reduced DNA binding activity and due to severely impaired transcriptional activation of a target reporter gene [60].
Another molecule implicated in TNF-induced NF-κB activation is atypical protein kinase C (ζPKC). ζPKC associates with RIP through the adapter protein p62 [61]. A lack of ζPKC severely impairs cellular responses to TNF that depend on the transcriptional activity of NF-κB [62]. However, IKK activation is normal in ζPKC-deficient MEF, and NF-κB DNA binding is only mildly reduced in response to TNF. Intriguingly, RelA phosphorylation is defective in ζPKC-/- cells, suggesting that an alternative pathway may exist to activate NF-κB. While ζPKC-/- cells are hypersensitive to TNF-induced apoptosis, ζPKC-/- mice do not exhibit fetal liver apoptosis or embryonic lethality [62]. These mutant mice are viable but display a reduced number of Peyer's patches.
TNF receptor-associated factor 6
TRAF6 is emerging as one of the key players in survival signaling. TRAF6 binds to the cytoplasmic tails of both the B-cell costimulatory molecule CD40 and the inflammatory IL-1 receptor [17]. In response to the binding of CD40 ligand and IL-1 to CD40 and IL-1 receptor, respectively, TRAF6 mediates the activation of NF-κB and JNK/stress-activated protein kinase. Study of TRAF6-/- cells confirmed the importance of TRAF6 in NF-κB activation in response to CD40 or IL-1 signaling [63].
Interestingly, lipopolysaccharide signaling was also impaired in TRAF6-/- cells, suggesting that TRAF6 may be involved in signal transduction downstream of the Toll-like receptors, particularly Toll-like receptor 4. A most unexpected finding was that TRAF6-/- mice suffered from severe osteopetrosis, showing defects in tooth eruption and bone remodeling due to impaired osteoclast function [63]. Interestingly, mice lacking expression of OPGL [64], the ligand for RANK, also exhibited osteopetrosis, strongly suggesting that TRAF6 may act downstream of RANK. OPGL-/- mice also show defects in T-cell and B-cell homeostasis [64].
BAFF and its receptors
BAFF (also called BlyS, TALL-1 or zTNF4) is a new TNF family member that has been implicated in promoting B-cell survival [16]. Mice overexpressing BAFF display mature B-cell hyperplasia and systemic lupus erythematosus-like symptoms. Conversely, BAFF-deficient mice show a complete loss of follicular and marginal-zone B lymphocytes [65]. Moreover, both T-cell-dependent and T-cell-independent antibody responses are impaired in mice lacking BAFF.
BAFF can bind to three receptors on the cell surface, B-cell maturation antigen (BCMA), transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI) and BAFF-R. Studies of knockout mice have shown that these molecules are not directly equivalent in function [66, 67]. Mice lacking BCMA show normal B-cell development and antibody responses [68], while TACI-deficient mice are deficient only in T-cell-independent antibody responses [69, 70]. Paradoxically, mice lacking TACI show increased B-cell proliferation and accumulation, suggesting an inhibitory role for TACI in B-cell homeostasis. Gene-targeted mice lacking BAFF-R have yet to be reported, but the natural mouse mutant A/WySnJ has a disruption of the intracellular domain of BAFF-R. A/WySnJ mice display a phenotype that is similar to BAFF-/- mice, although follicular and marginal-zone B cells are not completely abolished [65]. In addition, A/WySnJ mice are impaired only in T-cell-dependent antibody responses, unlike the more comprehensive defect observed in BAFF-deficient mice. These results suggest that, while BAFF-R may be the major receptor relaying BAFF-mediated signals for B-cell survival, some redundancy in function may be provided by the other two receptors, particularly TACI.
Apoptosis execution machinery
In mammals, apoptosis depends on the actions of a family of cysteine proteases called caspases. Two classes of caspases exist: the executioner caspases, which initiate the apoptotic cascade in response to extracellular signals transduced by cell surface receptors such as TNFR1; and the effector caspases, which are activated by the executioner caspases.
In a resting cell, caspases are present as proenzymes containing prodomains. The prodomains must be cleaved off to activate caspase activity. Cleavage of caspases is an element of the two pathways of apoptosis operating in mammalian cells: the 'intrinsic' pathway, which depends on mitochondrial involvement; and the 'extrinsic' pathway, which is independent of the mitochondria. The extrinsic pathway is initiated by the binding of a ligand to a DR as already described. In the intrinsic pathway, the mitochondria in a stimulated cell make the critical decision whether to initiate PCD [71]. In so doing, the mitochondria evaluate the balance of apoptotic signals with other signals promoting survival. If the net result favors the triggering of apoptosis, cytochrome c is released from the mitochondria at a level sufficient to initiate the activation of executioner caspases.
Specifically, cytochrome c in conjunction with dATP promotes the association of procaspase-9 with apoptosis-activating factor 1 (Apaf-1) [72]. This recruitment activates caspase-9 through an unknown mechanism. Activated caspase-9 in turn removes the prodomain of procaspase-3, resulting in the activation of this critical effector caspase (Fig. 2). Activated caspase-3 drives the apoptotic process by degrading a panel of cellular proteins crucial for cell survival
We have taken a genetic approach to dissecting apoptotic pathways in various cell types. The evidence shows, perhaps surprisingly, that not all elements are required in all cell types or in response to all apoptotic agents.
Apoptosis-activating factor 1
Deficiency for Apaf-1 is embryonic lethal [73, 74]. The mutant embryos show dramatically reduced levels of apoptosis in the brain, and hyperproliferation of neuronal cells that leads to striking craniofacial deformities [73]. Analysis of various cell types lacking Apaf-1 confirmed the vital role of this protein in the induction of apoptosis by a range of stimuli. However, we were surprised to observe that Fas-induced cell death of thymocytes and T cells was normal in the absence of Apaf-1. This was the first clue that, at least in some cell types, Fas-mediated apoptosis occurs by mechanism that can bypass Apaf-1.
Caspase-3
Most caspase 3-/- mice die at or before birth [75, 76]. The few survivors are smaller than their littermates and have visible masses in their heads. These masses are ectopic protrusions of supernumerary cells that accumulate in place of the pyknotic clusters usually derived from apoptosis during normal brain development [75, 76]. Caspase-3 is thus crucial for normal embryonic development.
Deficiency for caspase-3 also dramatically reduces the ability of many different cell types to undergo apoptosis in different settings. For example, PCD of oncogenically transformed MEF induced by a chemotherapeutic agent is greatly compromised in the absence of caspase-3. Activation-induced cell death of peripheral T cells is also dramatically reduced. However, while caspase-3 is necessary for efficient apoptosis of embryonic stem cells following UV irradiation, it is not required for PCD induced by γ-irradiation [76]. In addition, the requirement for caspase-3 in PCD induced by a given stimulus can be tissue specific. For example, while TNF induces the apoptosis of caspase-3-/- thymocytes, transformed caspase-3-/- MEFs are resistant to this stimulus.
Finally, caspase-3 is required for certain events transpiring during PCD but is not required for other events. In response to certain stimuli, caspase-3-/- cells fail to display DNA degradation or chromatin condensation but still exhibit other physical signs characteristic of apoptosis [76]. Thus, while caspase-3 is an important player in many instances of PCD, it is dispensable in certain apoptotic settings.
Caspase-9
Caspase-9 deficiency also results in embryonic lethality due to defective brain development [77, 78]. Analysis of neuronal tissues of these mutant embryos again revealed decreased apoptosis. Determination of the PCD of several caspase-9-/- cell types showed that, like caspase-3, caspase-9 is not required in all cell types for all apoptotic events. Caspase-9 is necessary for the PCD of embryonic stem cells and MEFs induced by UV irradiation or γ-irradiation, and for PCD of thymocytes exposed to dexamethasone or γ-irradiation. However, caspase-9-/- thymocytes readily undergo apoptosis in response to UV irradiation or engagement of Fas [78].
Future prospects and concluding remarks
Over the past decade, the understanding of the molecular pathways leading to activation of apoptosis and cell survival has increased substantially. This knowledge is an essential prerequisite to our contemplating the design of therapeutics to combat diseases. It is now clear that in addition to autoimmune diseases, this understanding will help in the development of drugs that may have an impact on the treatment of cancer and degenerative diseases.
Glossary of terms
Apaf-1 = apoptosis-activating factor 1; ASK-1 = apoptosis signal-regulating kinase 1; BAFF = B-lymphocyte activating factor; BAFF-R = BAFF receptor; BCMA = B-cell maturation antigen; Bid = proapoptotic Bcl-2 family member; Bim = proapoptotic Bcl-2 family member; DD = death domain; DR = death receptor; FADD = Fas-associated death domain protein; c-FLIP = cellular homologue of FLICE (caspase-8) inhibitory protein; GSK3 = glycogen synthase kinase 3; IKK = IκB kinase; JNK = Jun kinase; MAP3K = mitogen-activated protein 3 kinase; MEF = mouse embryonic fibroblasts; MEKK = mitogen-activated protein extracellular signal regulated kinase; NEMO = NF-κB essential modulator protein; NIK = NF-κB inducing kinase; OPGL = osteoproteregin ligand; PCD = programmed cell death; ζPKC = atypical protein kinase C; RANK = receptor activator of NF-κB; RIP = receptor-interacting protein; T2K = TRAF2-associated kinase; TACI = transmembrane activator and calcium modulator and cyclophilin ligand interactor; TANK = TRAF family member associated NF-κB activator; TRADD = TNF receptor-associated death domain protein; TRAF = TNF receptor-associated factor; TNFR1 = TNF receptor-associated factor type 1; TNFR2 = TNF receptor-associated factor type 2.
Funds for research
Funds for research in this field may be obtained from CANVAC (contact: Rafic Sekaly, 500 Sherbrooke Street West, Suite 800, Mon-treal, Quebec, Canada H3A 3C6), by the National Cancer Institute Of Canada (10 Alcorn Avenue, Suite 200, Toronto, Ontario, Canada M4V 3B1), and by Canadian Institutes of Health Research (formerly the Medical Research Council of Canada; 410 Laurier Avenue West, 9th Floor, Address Locator 4209A, Ottawa, Ontario, Canada K1A 0W9).
References
Debatin KM: Cell death in T- and B-cell development. Ann Hematol. 2001, 80: B29-B31.
Pinkoski MJ, Green DR: Lymphocyte apoptosis: refining the paths to perdition. Curr Opin Hematol. 2002, 9: 43-49. 10.1097/00062752-200201000-00008.
Kaufmann SH, Hengartner MO: Programmed cell death: alive and well in the new millennium. Trends Cell Biol. 2001, 11: 526-534. 10.1016/S0962-8924(01)02173-0.
Meier P, Finch A, Evan G: Apoptosis in development. Nature. 2000, 407: 796-801. 10.1038/35037734.
Baud V, Karin M: Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001, 11: 372-377. 10.1016/S0962-8924(01)02064-5.
Locksley RM, Killeen N, Lenardo MJ: The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001, 104: 487-501. 10.1016/S0092-8674(01)00237-9.
Chan KF, Siegel MR, Lenardo JM: Signaling by the TNF receptor superfamily and T cell homeostasis. Immunity. 2000, 13: 419-422. 10.1016/S1074-7613(00)00041-8.
Yeh WC, Hakem R, Woo M, Mak TW: Gene targeting in the analysis of mammalian apoptosis and TNF receptor superfamily signaling. Immunol Rev. 1999, 169: 283-302.
Strasser A, O'Connor L, Dixit VM: Apoptosis signaling. Annu Rev Biochem. 2000, 69: 217-245. 10.1146/annurev.biochem.69.1.217.
Senftleben U, Karin M: The IKK/NF-kappa B pathway. Crit Care Med. 2002, 30: S18-S26. 10.1097/00003246-200201001-00003.
Karin M, Ben-Neriah Y: Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000, 18: 621-663. 10.1146/annurev.immunol.18.1.621.
Pfeffer K, Matsuyama T, Kundig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Kronke M, Mak TW: Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993, 73: 457-467. 10.1016/0092-8674(93)90134-C.
Zheng L, Fisher G, Miller RE, Peschon J, Lynch DH, Lenardo MJ: Induction of apoptosis in mature T cells by tumour necrosis factor. Nature. 1995, 377: 348-351. 10.1038/377348a0.
Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S: Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992, 356: 314-317. 10.1038/356314a0.
Tartaglia LA, Ayres TM, Wong GH, Goeddel DV: A novel domain within the 55 kd TNF receptor signals cell death. Cell. 1993, 74: 845-853. 10.1016/0092-8674(93)90464-2.
Laabi Y, Egle A, Strasser A: TNF cytokine family: More BAFFling complexities. Curr Biol. 2001, 11: R1013-R1016. 10.1016/S0960-9822(01)00613-3.
Bradley JR, Pober JS: Tumor necrosis factor receptor-associated factors (TRAFs). Oncogene. 2001, 20: 6482-6491. 10.1038/sj.onc.1204788.
Wajant H, Henkler F, Scheurich P: The TNF-receptor-associated factor family: scaffold molecules for cytokine receptors, kinases and their regulators. Cell Signal. 2001, 13: 389-400. 10.1016/S0898-6568(01)00160-7.
Hsu H, Shu HB, Pan MG, Goeddel DV: TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996, 84: 299-308. 10.1016/S0092-8674(00)80984-8.
Yeh WC, de la Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, El-Deiry WS, Lowe SW, Goeddel DV, Mak TW: FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science. 1998, 279: 1954-1958. 10.1126/science.279.5358.1954.
Zhang J, Cado D, Chen A, Kabra NH, Winoto A: Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature. 1998, 392: 296-300. 10.1038/32681.
Yeh WC, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A, de la Pompa JL, Ferrick D, Hum B, Iscove N, Ohashi P, Rothe M, Goeddel DV, Mak TW: Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997, 7: 715-725. 10.1016/S1074-7613(00)80391-X.
Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P: The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998, 8: 297-303. 10.1016/S1074-7613(00)80535-X.
Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS: NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998, 281: 1680-1683. 10.1126/science.281.5383.1680.
Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P, Wallach D: Targeted disruption of the mouse caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998, 9: 267-276. 10.1016/S1074-7613(00)80609-3.
Yeh WC, Itie A, Elia AJ, Ng M, Shu HB, Wakeham A, Mirtsos C, Suzuki N, Bonnard M, Goeddel DV, Mak TW: Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000, 12: 633-642. 10.1016/S1074-7613(00)80214-9.
Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Roth KA, Korsmeyer SJ: Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999, 400: 886-891. 10.1038/23730.
Bouillet P, Purton JF, Godfrey DI, Zhang LC, Coultas L, Puthalakath H, Pellegrini M, Cory S, Adams JM, Strasser A: BH3-only Bcl-2 family member Bim is required for apoptosis of autore-active thymocytes. Nature. 2002, 415: 922-926. 10.1038/415922a.
Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM, Strasser A: Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999, 286: 1735-1738. 10.1126/science.286.5445.1735.
Malinin NL, Boldin MP, Kovalenko AV, Wallach D: MAP3K-related kinase involved in NF-kappaB induction by TNF, CD95 and IL-1. Nature. 1997, 385: 540-544. 10.1038/385540a0.
Shinkura R, Kitada K, Matsuda F, Tashiro K, Ikuta K, Suzuki M, Kogishi K, Serikawa T, Honjo T: Alymphoplasia is caused by a point mutation in the mouse gene encoding Nf-kappa b-inducing kinase. Nat Genet. 1999, 22: 74-77. 10.1038/8780.
Yin L, Wu L, Wesche H, Arthur CD, White JM, Goeddel DV, Schreiber RD: Defective lymphotoxin-beta receptor-induced NF-kappaB transcriptional activity in NIK-deficient mice. Science. 2001, 291: 2162-2165. 10.1126/science.1058453.
Lee FS, Hagler J, Chen ZJ, Maniatis T: Activation of the IkappaB alpha kinase complex by MEKK1, a kinase of the JNK pathway. Cell. 1997, 88: 213-222. 10.1016/S0092-8674(00)81842-5.
Baud V, Liu ZG, Bennett B, Suzuki N, Xia Y, Karin M: Signaling by proinflammatory cytokines: oligomerization of TRAF2 and TRAF6 is sufficient for JNK and IKK activation and target gene induction via an amino-terminal effector domain. Genes Dev. 1999, 13: 1297-1308.
Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H: ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell. 1998, 2: 389-395. 10.1016/S1097-2765(00)80283-X.
Hoeflich KP, Yeh WC, Yao Z, Mak TW, Woodgett JR: Mediation of TNF receptor-associated factor effector functions by apoptosis signal-regulating kinase-1 (ASK1). Oncogene. 1999, 18: 5814-5820. 10.1038/sj/onc/1202975.
Yujiri T, Ware M, Widmann C, Oyer R, Russell D, Chan E, Zaitsu Y, Clarke P, Tyler K, Oka Y, Fanger GR, Henson P, Johnson GL: MEK kinase 1 gene disruption alters cell migration and c-Jun NH2-terminal kinase regulation but does not cause a measurable defect in NF-kappa B activation. Proc Natl Acad Sci USA. 2000, 97: 7272-7277. 10.1073/pnas.130176697.
Xia Y, Makris C, Su B, Li E, Yang J, Nemerow GR, Karin M: MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proc Natl Acad Sci USA. 2000, 97: 5243-5248. 10.1073/pnas.97.10.5243.
Yujiri T, Sather S, Fanger GR, Johnson GL: Role of MEKK1 in cell survival and activation of JNK and ERK pathways defined by targeted gene disruption. Science. 1998, 282: 1911-1914. 10.1126/science.282.5395.1911.
Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H: ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001, 2: 222-228. 10.1093/embo-reports/kve046.
Yang J, Lin Y, Guo Z, Cheng J, Huang J, Deng L, Liao W, Chen Z, Liu Z, Su B: The essential role of MEKK3 in TNF-induced NF-kappaB activation. Nat Immunol. 2001, 2: 620-624. 10.1038/89769.
Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D: Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kB. Nature. 1995, 376: 167-170. 10.1038/376167a0.
Li Q, Van Antwerp D, Mercurio F, Lee KF, Verma IM: Severe liver degeneration in mice lacking the IkappaB kinase 2 gene. Science. 1999, 284: 321-325. 10.1126/science.284.5412.321.
Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL, Goeddel DV: Embryonic lethality, liver degeneration, and impaired NF-kappa B activation in IKK-beta-deficient mice. Immunity. 1999, 10: 421-429. 10.1016/S1074-7613(00)80042-4.
Li ZW, Chu W, Hu Y, Delhase M, Deerinck T, Ellisman M, Johnson R, Karin M: The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med. 1999, 189: 1839-1845. 10.1084/jem.189.11.1839.
Senftleben U, Li ZW, Baud V, Karin M: IKKbeta is essential for protecting T cells from TNFalpha-induced apoptosis. Immunity. 2001, 14: 217-230. 10.1016/S1074-7613(01)00104-2.
Rudolph D, Yeh WC, Wakeham A, Rudolph B, Nallainathan D, Potter J, Elia AJ, Mak TW: Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes Dev. 2000, 14: 854-862.
Makris C, Godfrey VL, Krahn-Senftleben G, Takahashi T, Roberts JL, Schwarz T, Feng L, Johnson RS, Karin M: Female mice heterozygous for IKK gamma/NEMO deficiencies develop a dermatopathy similar to the human X-linked disorder incontinentia pigmenti. Mol Cell. 2000, 5: 969-979. 10.1016/S1097-2765(00)80262-2.
Schmidt-Supprian M, Bloch W, Courtois G, Addicks K, Israel A, Rajewsky K, Pasparakis M: NEMO/IKK gamma-deficient mice model incontinentia pigmenti. Mol Cell. 2000, 5: 981-992. 10.1016/S1097-2765(00)80263-4.
Smahi A, Courtois G, Vabres P, Yamaoka S, Heuertz S, Munnich A, Israel A, Heiss NS, Klauck SM, Kioschis P, Wiemann S, Poustka A, Esposito T, Bardaro T, Gianfrancesco F, Ciccodicola A, D'Urso M, Woffendin H, Jakins T, Donnai D, Stewart H, Ken-wrick SJ, Aradhya S, Yamagata T, Levy M, Lewis RA, Nelson DL: Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature. 2000, 405: 466-472. 10.1016/S0168-9002(97)86454-6.
Hu Y, Baud V, Delhase M, Zhang P, Deerinck T, Ellisman M, Johnson R, Karin M: Abnormal morphogenesis but intact IKK activation in mice lacking the IKKalpha subunit of IkappaB kinase. Science. 1999, 284: 316-320. 10.1006/abio.2000.4698.
Li Q, Lu Q, Hwang JY, Buscher D, Lee KF, Izpisua-Belmonte JC, Verma IM: IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev. 1999, 13: 1322-1328.
Hu Y, Baud V, Oga T, Kim KI, Yoshida K, Karin M: IKKalpha controls formation of the epidermis independently of NF-kappaB. Nature. 2001, 410: 710-714. 10.1038/35070605.
Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M: Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001, 293: 1495-1499. 10.1126/science.1062677.
Kaisho T, Takeda K, Tsujimura T, Kawai T, Nomura F, Terada N, Akira S: IkappaB kinase alpha is essential for mature B cell development and function. J Exp Med. 2001, 193: 417-426. 10.1084/jem.193.4.417.
Pomerantz JL, Baltimore D: NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. Embo J. 1999, 18: 6694-6704. 10.1093/emboj/18.23.6694.
Tojima Y, Fujimoto A, Delhase M, Chen Y, Hatakeyama S, Nakayama K, Kaneko Y, Nimura Y, Motoyama N, Ikeda K, Karin M, Nakanishi M: NAK is an IkappaB kinase-activating kinase. Nature. 2000, 404: 778-782. 10.1038/35008109.
Bonnard M, Mirtsos C, Suzuki S, Graham K, Huang J, Ng M, Itie A, Wakeham A, Shahinian A, Henzel WJ, Elia AJ, Shillinglaw W, Mak TW, Cao Z, Yeh W-C: Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. Embo J. 2000, 19: 4976-4985. 10.1093/emboj/19.18.4976.
He X, Saint-Jeannet JP, Woodgett JR, Varmus HE, Dawid IB: Glycogen synthase kinase-3 and dorsoventral patterning in Xenopus embryos. Nature. 1995, 374: 617-622. 10.1038/374617a0.
Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR: Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000, 406: 86-90. 10.1038/35017574.
Sanz L, Sanchez P, Lallena MJ, Diaz-Meco MT, Moscat J: The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. Embo J. 1999, 18: 3044-3053. 10.1093/emboj/18.11.3044.
Leitges M, Sanz L, Martin P, Duran A, Braun U, Garcia JF, Camacho F, Diaz-Meco MT, Rennert PD, Moscat J: Targeted disruption of the zetaPKC gene results in the impairment of the NF-kappaB pathway. Mol Cell. 2001, 8: 771-780. 10.1016/S1097-2765(01)00361-6.
Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, van der Heiden A, Itie A, Wakeham A, Khoo W, Sasaki T, Cao Z, Penninger JM, Paige CJ, Lacey DL, Dunstan CR, Boyle WJ, Goeddel DV, Mak TW: TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999, 13: 1015-1024.
Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Boyle WJ, Penninger JM: OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999, 397: 315-323. 10.1038/16852.
Schiemann B, Gommerman JL, Vora K, Cachero TG, Shulga-Morskaya S, Dobles M, Frew E, Scott ML: An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science. 2001, 293: 2111-2114. 10.1126/science.1061964.
Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, Xu W, Parrish-Novak J, Foster D, Lofton-Day C, Moore M, Littau A, Grossman A, Haugen H, Foley K, Blumberg H, Harrison K, Kindsvogel W, Clegg CH: TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000, 404: 995-999. 10.1038/35010115.
Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, Hession C, Schneider P, Sizing ID, Mullen C, Strauch K, Zafari M, Benjamin CD, Tschopp J, Browning JL, Ambrose C: BAFF-R, a newly identified TNF receptor that specifically interacts with BAFF. Science. 2001, 293: 2108-2111. 10.1126/science.1061965.
Xu S, Lam KP: B-cell maturation protein, which binds the tumor necrosis factor family members BAFF and APRIL, is dispensable for humoral immune responses. Mol Cell Biol. 2001, 21: 4067-4074. 10.1128/MCB.21.12.4067-4074.2001.
Yan M, Wang H, Chan B, Roose-Girma M, Erickson S, Baker T, Tumas D, Grewal IS, Dixit VM: Activation and accumulation of B cells in TACI-deficient mice. Nat Immunol. 2001, 2: 638-643. 10.1038/89790.
von Bulow GU, van Deursen JM, Bram RJ: Regulation of the T-independent humoral response by TACI. Immunity. 2001, 14: 573-582. 10.1016/S1074-7613(01)00130-3.
Green DR, Reed JC: Mitochondria and apoptosis. Science. 1998, 281: 1309-1312. 10.1126/science.281.5381.1309.
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X: Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997, 91: 479-489. 10.1016/S0092-8674(00)80434-1.
Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW: Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998, 94: 739-750. 10.1016/S0092-8674(00)81733-X.
Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P: Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998, 94: 727-737. 10.1016/S0092-8674(00)81732-8.
Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA: Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996, 384: 368-372. 10.1038/384368a0.
Woo M, Hakem R, Soengas MS, Duncan GS, Shahinian A, Kagi D, Hakem A, McCurrach M, Khoo W, Kaufman SA, Senaldi G, Howard T, Lowe SW, Mak TW: Essential contribution of caspase 3/CPP32 to apoptosis and its associated nuclear changes. Genes Dev. 1998, 12: 806-819.
Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MSS, Rakic P, Flavell RA: Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998, 94: 325-337. 10.1016/S0092-8674(00)81476-2.
Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW: Differential requirement for caspase 9 in apoptotic pathways in vivo. Cell. 1998, 94: 339-352. 10.1016/S0092-8674(00)81477-4.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mak, T.W., Yeh, WC. Signaling for survival and apoptosis in the immune system. Arthritis Res Ther 4 (Suppl 3), S243 (2002). https://doi.org/10.1186/ar569
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar569