
Chapter summary
IL-6 is a pleiotropic cytokine with a wide range of biological activities in immune regulation, hematopoiesis, inflammation, and oncogenesis. Its activities are shared by IL-6-related cytokines such as leukemia inhibitory factor and oncostatin M. The pleiotropy and redundancy of IL-6 functions have been identified by using a unique receptor system comprising two functional proteins: an IL-6 receptor (IL-6R) and gp130, the common signal transducer of cytokines related to IL-6. Signal transduction through gp130 is mediated by two pathways: the JAK–STAT (Janus family tyrosine kinase–signal transducer and activator of transcription) pathway and the Ras mitogen-activated protein kinase pathway. The negative regulators of IL-6 signaling have also been identified, although the physiological roles of the molecules are not yet fully understood. The pathological roles of IL-6 have also been clarified in various disease conditions, such as inflammatory, autoimmune, and malignant diseases. On the basis of the findings, a new therapeutic approach to block the IL-6 signal using humanized anti-IL-6R antibody for rheumatoid arthritis, Castleman's disease, and multiple myeloma has been attempted.
Similar content being viewed by others

Identification of IL-6 and its pleiotropic functions
IL-6 was originally identified as an antigen-nonspecific B-cell differentiation factor in the culture supernatants of mitogen- or antigen-stimulated peripheral blood mononuclear cells that induced B cells to produce immunoglobulins [1, 2], and was named B-cell stimulatory factor 2 (BSF-2). The cDNA encoding human BSF-2 was cloned in 1986 [3]. Simultaneously, IFN-β2 [4, 5] and a 26-kDa protein [6] in fibroblasts were independently cloned by different groups and found to be identical to BSF-2. Later, a hybridoma/plasmacytoma growth factor [7–10] and a hepatocyte-stimulating factor [11–13] were also proven to be the same molecule as BSF-2. Although various names have been used for this molecule because of its multiple biological activities, it is now known as IL-6.
A pleiotropic cytokine with a wide range of biological activities (Fig. 1), IL-6 is produced by various types of lymphoid and nonlymphoid cells, such as T cells, B cells, monocytes, fibroblasts, keratinocytes, endothelial cells, mesangial cells, and several tumor cells [14]. It induces growth of T cells and differentiation of cytotoxic T cells [15–19] by augmenting the expression of IL-2 receptor [15] and the production of IL-2 [20]. IL-6 acts synergistically with IL-3 to support the formation of multilineage blast cell colonies in hematopoiesis [21–25]. IL-6 also induces differentiation of macrophages [26], megakaryocytes [27–29], and osteoclasts [30]. In the acute-phase reaction, this cytokine stimulates hepatocytes to produce acute-phase proteins such as C-reactive protein (CRP), fibrinogen, α1-antitrypsin, and serum amyloid A [12, 13], and it simultaneously suppresses albumin production [11]. It causes leukocytosis and fever when administered in vivo[31] and also acts as a growth factor for renal mesangial cells [32], epidermal keratinocytes [33, 34], and various types of tumor cells, for example, in plasmacytoma [8], multiple myeloma [35], and renal cell carcinoma [36].

IL-6-producing cells and biological activities of IL-6. IL-6 is produced by lymphoid and nonlymphoid cells, such as T cells, B cells, monocytes, fibroblasts, keratinocytes, endothelial cells, mesangial cells, and several kinds of tumor cell (top of figure). IL-6 also has a wide range of biological activities on various target cells (bottom of figure).
Although IL-6 has pleiotropic effects on various target cells, some of the biological activities are also mediated by other cytokines, such as leukemia inhibitory factor (LIF) and oncostatin M (OSM). The pleiotropy and redundancy of IL-6 functions can be identified by using a unique receptor system of cytokines [14].
Identification and characterization of IL-6R as the specific receptor of IL-6, and of gp130 as the common signal transducer of the IL-6 superfamily
We and our colleagues identified the two components of IL-6 receptor (IL-6R), an 80-kDa IL-6-binding protein (α chain) and a 130-kDa signal transducer known as gp130 (β chain), in 1988 and 1990 [37–39], respectively. Although IL-6 cannot directly bind to gp130, it can bind to IL-6R to generate the high-affinity complex of IL-6/IL-6R/gp130. Furthermore, the complex of IL-6 and soluble IL-6R can generate IL-6-mediated signal transduction [38, 39]. Another feature of cytokines is the redundancy of their functions. For example, IL-6, LIF, and OSM all induce macrophage differentiation in the myeloid leukemia cell line M1 [40–43] and acute-phase protein synthesis in hepatocytes [11, 12, 44–46]. An important finding as regards cytokine receptors is that one constituent of a given cytokine receptor is shared by several other cytokine receptors [47]. For example, gp130 is in fact shared by the receptors for such cytokines of the IL-6 superfamily as ciliary neurotrophic factor (CNTF), LIF, OSM, IL-11, and cardiotrophin-1 [14, 48, 49]. Thus, the molecular mechanisms of redundancy in functions of cytokines of the IL-6 superfamily can be explained at least in part by the sharing of gp130 among their receptors.
Investigations of the IL-6R system have provided evidence that the combination of IL-6 and soluble IL-6R can act on cells that express gp130 but not IL-6R [48]. A complex consisting of a soluble cytokine receptor and its corresponding cytokine acquires different target specificity from the original cytokine and should therefore express different functions from those of the original cytokine. In fact, we found that doubly transgenic mice expressing human IL-6 and IL-6R showed myocardial hypertrophy [50], indicating that the combination of IL-6 and soluble IL-6R acts on heart muscle cells that express gp130, an action that IL-6 cannot exert by itself. The action leads to the induction of cardiac hypertrophy, so that the effect is similar to that of cardiotrophin-1. This combination of cytokine and its soluble receptor may contribute to the generation of the functional diversity of cytokines in a wide range of other receptor systems and may also play a pathological role in various diseases in which an increase in the serum-soluble form of various cytokine receptors has been reported.
Clarification of multiple signal cascades in IL-6 signal
As the cytoplasmic domain of most cytokine receptors, including gp130, does not have an intrinsic catalytic domain, one of the most controversial issues before 1993 was the identification of catalytic molecules that associate with cytokine receptors. This issue was resolved by the discovery of several Janus family tyrosine kinases (JAK1, JAK2, JAK3, Tyk-2), which are involved in the transduction of cytokine and hormone signals [51–53]. Furthermore, the signal transducer and activator of transcription (STAT) was found to play a central role in a variety of cytokine signal cascades. Our group and others found that JAK1, JAK2, and Tky-2 are activated and are tyrosine-phosphorylated in response to IL-6, CNTF, LIF, and OSM [14], and also identified and characterized STAT3 [54]. IL-6 activates STAT1 and STAT5 in addition to STAT3. In the absence of JAK1, the activation of transcriptional factor STATs following stimulation by IL-6 is not effective as long as both JAK2 and Tky-2 are activated. This finding suggests that there is a hierarchy among gp130-associated JAKs [55].
Several research groups, including ours, have identified two types of IL-6 response element (IL-6RE) in the genes encoding acute-phase proteins. The presence of type I IL-6RE, which is a binding site for NF-IL-6 (nuclear factor for IL-6 expression), IL-6DBP (IL-6 vitamin-D-binding protein), and C/EBPβ [56–59], has been confirmed in the genes for CRP, hemopexin A, and haptoglobin. The binding activity of NF-IL-6 is probably induced by IL-6 through the increased expression of the NF-IL-6 gene rather than through its post-translational modification. Type II IL-6RE is contained in the fibrinogen, α2-macroglobin, α1-acid glycoprotein, and haptoglobin genes. IL-6 triggers the rapid activation of a nuclear factor, known as the acute-phase response factor, which binds to type II IL-6RE [60]. Purification and molecular cloning of this factor revealed that it is identical to STAT3 [54, 61].
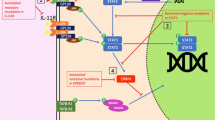
We clarified that human gp130 has 277 amino acid residues in its cytoplasmic domain, which contains two motifs, Box1 and Box2, conserved among the cytokine receptor family (Fig. 2) [39, 62, 63]. The membrane-proximal region containing Box1 and Box2 was found to be sufficient for the activation of JAK through gp130 [64]. Furthermore, human gp130 has six tyrosine residues in its cytoplasmic domain. Finally, the tyrosine phosphorylation of Src homology protein 2 tyrosine phosphatase-2 (SHP-2), a phosphotyrosine phosphatase, and that of STAT3 depend on the second tyrosine residue (Y2) from the membrane, and on any one of the four tyrosine residues (Y3, Y4, Y5, Y6) in the carboxy terminus that have a glutamine residue at the third position behind tyrosine (Y-X-X-Q) (see Fig. 2) [65, 66].

Schematic structure of gp130. Binding of IL-6 to IL-6R induces homodimerization of gp130, activating JAK associating with gp130 at Box1. This is followed by the tyrosine phosphorylation of the distal part of gp130 and recruitment of STAT3. STAT3 is then tyrosine-phosphorylated by JAK. SHP-2 on the second tyrosine (Y2) residue of gp130 activates the MAP kinase pathway. JAK, Janus family tyrosine kinase; SHP-2, Src homology protein 2 tyrosine phosphatase-2; STAT, signal transducer and activator of transcription; Y(2,3, etc.), (second, third, etc.) tyrosine residue (from the membrane).
It is known that IL-6 induces growth arrest and macrophage differentiation in the murine myeloid leukemic cell line M1. The essential role of STAT3 in the IL-6-induced macrophage differentiation of M1 cells was demonstrated by using dominant negative forms of STAT3 [67], which inhibit both IL-6-induced growth arrest and macrophage differentiation in M1 transformants. Blocking STAT3 activation inhibits IL-6-induced repression of c-Myb and c-Myc, but not EGR-1 induction [68], while IL-6 enhances the growth of M1 cells when STAT3 is suppressed. Thus, IL-6 simultaneously generates growth-enhancing signals as well as growth-arrest and differentiation-inducing signals, but the former are apparent only when STAT3 activation is suppressed. As for the growth signals, a 65-amino-acid region proximal to the transmembrane domain was found to be sufficient for generating a growth response by using gp130 transfectants of an IL-3-dependent proB-cell line BAF/BO3 [14, 63]. However, the membrane-proximal region of 68 amino acids is not sufficient for the induction of tritium thymidine (3H-Tdr) uptake when cells are starved of IL-3. For cell growth, the membrane-proximal region containing 133 amino acid residues is both required and sufficient [69]. Furthermore, at least two distinct signals are required for gp130-induced cell growth: a cell cycle progression signal dependent on the second tyrosine residue, Y2, and possibly mediated by SHP-2, and an antiapoptotic signal dependent on the third tyrosine residue, Y3, and mediated by STAT3 through induction of BCL-2. However, our recent study using mice with STAT3 deficiency in a T-cell-specific manner has revealed that STAT3 activation is involved in IL-6-dependent T cell proliferation through prevention of apoptosis without the need for BCL-2 induction [70]. Thus, STAT3 plays pivotal roles in gp130-mediated signal transduction regulating cell growth, differentiation, and survival. In addition to the JAK–STAT signal transduction pathway, it is known that the Ras mitogen-activated protein (MAP) kinase pathway is also activated through SHP-2 [69] or Shc [71]. Furthermore, nonreceptor tyrosine kinases, such as Btk, Tec, Fes, and Hck [72, 73] are activated through the IL-6 receptor, as well as through a variety of other cytokine receptors [74], although the biological significance of these signal transduction pathways remains to be clarified. Several distinct signal transduction pathways are generated through different regions of the cytoplasmic domain of gp130. The expression pattern of these signaling molecules determines which set of signaling pathways is activated in a given cell. Furthermore, these signaling pathways may interact with each other and contribute to a variety of biological activities. In fact, a recent study reported that knock-in mutation mice lacking SHP-2 signal showed sustained gp130-induced STAT3 activation; this finding indicates a negative regulatory role of SHP-2 for STAT3 activation [75]. These knock-in mice also displayed splenomegaly and lymphadenopathy and an acute-phase reaction. In contrast, all known mice deficient in the STAT3 binding site, such as the gp130-deficient mouse, died perinatally [75]. However, it has also been reported that mice deficient in STAT3 signal displayed a severe joint disease in association with mitogenic hyper-responsiveness of the synovial cells to the IL-6-family cytokines. This hyper-responsiveness was the result of sustained gp130-mediated SHP-2 activation due to a lack of the SHP-2 inhibitor induced by STAT3 [76].
Identification of new inhibitors of IL-6 signaling
Cytokine signaling, including that of IL-6, is negatively regulated with respect to both magnitude and duration. Recently, it has been found that at least two new families of inhibitors contribute to the negative regulation of cytokine signaling: the suppressor of cytokine signaling (SOCS) and the protein inhibitors of activated STATs (PIAS) (Fig. 3). In 1997, two other groups and ours identified SOCS-1, also known as SSI-1 (STAT-induced STAT inhibitor 1) or JAB-1 (JAK-binding protein 1), as a negative regulatory molecule of IL-6 signaling on the basis of its binding to JAK [77–79]. Subsequently, database searches have shown that the SOCS family now includes eight members (CIS and SOCS1–SOCS7), all of which are characterized by a central SH2 domain flanked by an N-terminal region containing a conserved motif known as the SOCS box [77, 80–82]. mRNA of SOCS-1, SOCS-2, and SOCS-3 is induced by cytokines such as IL-6, IFN-γ, IL-4, and granulocyte-colony-stimulating factor and several other members, and they inhibit cytokine-activated JAK–STAT signal pathways [83–85]. However, the factors that induce mRNA of the other SOCS families, such as SOCS-4-7, have not been clarified and their functions have not been thoroughly characterized.

Molecular mechanism of inhibition by new cytokine inhibitors. (Left) PIAS inhibits DNA-binding activity of STATs through association with activated them. (Center) SOCS-1 inhibits catalytic activity of JAKs by direct interaction with them. (Right) SOCS-3 inhibits catalytic activity of JAKs by binding to receptor complex. JAK, Janus family tyrosine kinases; P, phosphorylation; PIAS, protein inhibitors of activated STATs; SOCS, suppressor of cytokine signaling; STAT, signal transducer and activator of transcription; Y, tyrosine residue.
SOCS-1 and SOCS-3 are especially well known as inhibitors of cytokine signaling [86], acting through different mechanisms. SOCS-1 directly interacts with JAKs, and thus inhibits their catalytic activity. SOCS-3 also inhibits JAK activity (but only partially in comparison with SOCS-1) although the augmentation of its effect in the presence of receptors suggests that SOCS-3 inhibits cytokine signaling by binding to the receptor complex. In the IL-6 signal cascade, the SHP-2 interaction site of gp130 has also been shown to be a SOCS-3 contact site, so that SOCS-3 may compete for the SHP-2–gp130 interaction site [86, 87]. Gene-targeting mice of the SOCS family were used to show that SOCS-2 and SOCS-3 are critical molecules for, respectively, GH/IGF-1 and EPO signaling in vivo[88, 89]. In particular, mice deficient in SOCS-2 exhibit giantism, reduced production of major urinary proteins, increased local production of IGF-1, and accumulation of collagen in the dermis, while SOCS-3-deficient mice die at 12-16 days of age because of erythrocytosis by deregulation of fetal liver hematopoiesis. However, a recent study of SOCS-3-deficient mice showed that SOCS-3 was required for placental development but not for normal hematopoiesis in the mouse embryo [90].
Two groups of researchers, including ours, initially reported that SOCS-1-deficient mice are born healthy but with growth disclose various kinds of abnormalities, including stunted growth, fulminant hepatitis with serious fatty degradation, and mononuclear cell infiltration of several organs, and die within 3 weeks after birth [91, 92]. Subsequently, it was reported that SOCS-1 is a key molecule for IFN-γ actions in vivo as seen in SOCS-1-deficient mice that also lack the IFN-γ gene (SOCS-1/IFN-γ doubly deficient mice) [93, 94]. However, it was also found that SOCS-1 in vitro inhibits activation of STAT6 by IL-4 stimulation [92], and that SOCS-1 in vivo inhibits TNF-α and insulin signaling [95, 96]. In a recent study of SOCS-1/STAT1 and SOCS-1/STAT6 doubly deficient mice, we found that the physiological role of SOCS-1 is essential for inhibition of crosstalk in cytokine signaling, particularly for IFN-γ-induced inhibition of STAT6 [97]. SOCS-1-deficient mice feature an intact IL-6 signaling pathway, suggesting that SOCS-3 may act as a crucial inhibitor of IL-6 signaling in vivo.
Unlike the SOCS family, PIAS proteins constitute a family of constitutively expressed negative regulators of STATs. Five members of this family have been identified with the yeast two-hybrid method and by a search of the expressed sequence tag database: PIAS-1, PIAS-3, PIAS-Xα, PIAS-Xβ, and PIAS-Y [98, 99]. They all share homology and contain several highly conserved domains, including a putative zinc-binding motif and a highly acidic region. PIAS-1 and PIAS-3 have been identified as specific inhibitors of STAT signal pathways [98, 99]. Overexpression studies have shown that PIAS-1 associates only with activated STAT1 dimers and inhibits their DNA-binding activity, but that no monomeric forms of STAT1 are present [99]. Similarly, PIAS-3 associates specifically with activated STAT3 but not with STAT1, resulting in the blocking of all STAT3-mediated gene transcriptions, and is especially well known as an inhibitor of IL-6 signaling in M1 cell lines [98]. The constitutive expression of PIAS proteins implies that their physiological role differs from that of SOCS proteins, which are induced by cytokine stimulation. So far, however, the differences in the physiological roles of these two families of proteins are not well known.
Application of anti-IL-6R antibody to clinical medicine
Rheumatoid arthritis (RA) is a systemic inflammatory disease characterized by destructive changes in bone and cartilage of affected joints as well as the emergence of rheumatoid factors. Although the exact causes of RA remain unknown, immunological dysregulation by inflammatory cytokines has been shown to be involved in its development [100]. IL-6 is one of these cytokines and uncontrolled IL-6 overproduction appears to be responsible for the clinical symptoms and abnormal laboratory findings in RA [101]. Because of the B-cell differentiation factor activity of IL-6, overproduction of IL-6 is responsible for the increase in serum γ-globulin and the emergence of rheumatoid factors. IL-6 as a hepatocyte-stimulating factor causes an increase in CRP, serum amyloid A, and erythrocyte sedimentation rate and a decrease in serum albumin [11–13]. On the other hand, IL-6 as a megakaryocyte differentiation factor causes thrombocytosis [22, 27, 28]. Since IL-6 in the presence of soluble IL-6R activates osteoclasts to induce bone absorption [30], IL-6 may be involved in the osteoporosis [102] and destruction of bone and cartilage associated with RA. In fact, a large amount of IL-6 has been observed in both sera and synovial fluids from the affected joints of patients with RA [103–106]. Blockade of the IL-6 signal may thus constitute a new therapeutic strategy for RA.
Wendling et al. reported that the administration of mouse antihuman IL-6 monoclonal antibodies to patients with RA resulted in amelioration of RA symptoms and improvement of laboratory findings [107]. However, such therapeutic effects were transient, because murine antibodies were found to be highly immunogenic in humans, especially when they were administered repeatedly. To be effective as therapeutic agents administered to patients in repeated doses, mouse antibodies must therefore be engineered to look like human antibodies. A humanized anti-IL-6R antibody was constructed by grafting the complementarity-determining regions (CDRs) from mouse PM-1, a specific monoclonal antibody against human IL-6R, into human IgG to re-create a properly functioning antigen-binding site in a reshaped human antibody [108]. In vitro, humanized anti-IL-6R antibody is equivalent to both mouse and chimeric PM-1 in terms of antigen binding and growth inhibition of IL-6-dependent myeloma cells [108, 109]. Furthermore, it looks very much like a human antibody and can therefore be expected to be a poor immunogen in human patients [110].
The in vivo effect of humanized anti-IL-6R antibody on the development of collagen-induced arthritis was examined in cynomolgus monkeys because it cross-reacts with the monkey IL-6R [111]. Intravenous administration of humanized anti-IL-6R antibody (10 mg/kg once a week) significantly inhibited the onset of joint inflammation and the elevation of serum CRP and fibrinogen levels and erythrocyte sedimentation rate that were induced by immunization with bovine type II collagen with a complete adjuvant.
On the basis of the above findings, we administered humanized anti-IL-6R antibody to RA patients whose active disease was resistant to conventional therapy using methotrexate, various disease-modifying antirheumatic drugs, and corticosteroids, with the permission of the Ethical Committee of Osaka University Medical School. Low-grade fever and fatigue disappeared and CRP and fibrinogen levels were normalized within 2 weeks after the start of humanized anti-IL-6R antibody treatment (50 mg twice a week) (Fig. 4). This was followed by reduction of morning stiffness, improvement of the swollen-joint score and the pain and tenderness score, and reduction of anemia, thrombocytosis, and hypoalbuminemia. A score of ACR20 on the American College of Rheumatology scale was achieved in 7 of 8 patients after 8 weeks of treatment and ACR50 in 4 of 8 patients after 8 weeks. The treatment was well tolerated and no major side effects were observed. These data indicate that humanized anti-IL-6R antibody is useful for the treatment of RA. Phase I clinical trials in the United Kingdom and a phase I/II study in Japan also proved the safety and the efficacy of humanized anti-IL-6R antibody [112, 113]. Double-blind, randomized, placebo-controlled phase II studies for the use of the antibody to treat RA are now in the progress both in Europe and in Japan. In addition to RA, various other IL-6-related diseases such as Castleman's disease, multiple myeloma, mesangial proliferative glomerulonephritis, psoriasis, and Kaposi's sarcoma are possible targets of humanized anti-IL-6R antibody.

Representative clinical course of an RA patient treated with humanized anti-IL-6R antibody. A 51-year-old woman with RA was given humanized anti-IL-6R antibody intravenously (50 mg twice a week). Although she had active disease refractory to conventional treatment with drugs including methotrexate and prednisolone, treatment with humanized anti-IL-6R remarkably improved her condition. CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; RF, rheumatoid factor.
Conclusion
IL-6 participates in immune response, hematopoiesis, and acute-phase reactions. On the other hand, deregulation of IL-6 production has been implicated in the pathogenesis of a variety of diseases, including plasmacytoma/myeloma and several chronic inflammatory proliferative diseases. Future studies on the regulation of IL-6 expression and clarification of the molecular mechanisms of IL-6 functions, as well as of inhibitors of IL-6 signal, should provide information critical to a better understanding of the molecular mechanisms of these diseases and the development of new therapeutic methods such as antibody therapy.
Glossary of terms
BSF = B-cell stimulatory factor; CNTF = ciliary neurotrophic factor; IL-6RE = IL-6 response element; LIF = leukemia inhibitory factor; NF-IL-6 = nuclear factor for IL-6 expression; OSM = oncostatin M; PIAS = protein inhibitors of activated STATs; SHP-2 = SH2-containing protein tyrosine phosphatase-2; SOCS = suppressor of cytokine signaling; Y(2,3, etc.) = (second, third, etc.) tyrosine residue (from the membrane).
References
Muraguchi A, Kishimoto T, Miki T, Kuritani T, Kaieda T, Yoshizaki K, Yamamura Y: T cell-replacing factor (TRF)-induced IgG secretion in human B blastoid cell line and demonstration of acceptors for TRF. J Immunol. 1981, 127: 412-416.
Yoshizaki K, Nakagawa T, Kaieda T, Muraguchi A, Yamamura Y, Kishimoto T: Induction of proliferation and Ig production in human B leukemic cells by anti-immunoglobulins and T cell factors. J Immunol. 1982, 128: 1296-1301.
Hirano T, Yasukawa K, Harada H, Taga T, Watanabe Y, Matsuda T, Kashiwamura S, Nakajima K, Koyama K, Iwamatsu A, Tsuna-sawa S, Sakiyama F, Matsui H, Takahara Y, Taniguchi T, Kishimoto T: Complementary DNA for a novel human interleukin (BSF-2) that induces B cell lymphocytes to produce immunoglobulin. Nature. 1986, 324: 73-76.
Zilberstein A, Ruggieri R, Korn JH, Revel M: Structure and expression of cDNA and genes for human interferon-β2, a distinct species inducible by growth-stimulatory cytokines. EMBO J. 1986, 5: 2529-2537.
Sehgal PB, Walther Z, Tamm I: Rapid enhancement of b2-interferon/B-cell differentiation factor BSF-2 gene expression in human fibroblasts by diacylglycerols and calcium ionophore A23187. Proc Natl Acad Sci USA. 1987, 84: 3663-3667.
Haegeman G, Content J, Volckaert G, Derynck R, Taverneir J, Fires W: Structural analysis of the sequence encoding for an inducible 26-kDa protein in human fibroblasts. Eur J Biochem. 1986, 159: 625-632.
Van Damme J, Opdenakker G, Simpson RJ, Rubira MR, Cayphas S, Vink A, Billiau A, Van Snick JV: Identification of the human 26-kDa protein, interferon β2 (IFN-β2), as a B cell hybridoma/ plasmacytoma growth factor induced by interleukin-1 and tumor necrosis factor. J Exp Med. 1987, 165: 914-919.
Nordan RP, Pumphrey JG, Rudikoff S: Purification and NH2-terminal sequence of a plasmacytoma growth factor derived from the murine macrophage cell line P388D1. J Immunol. 1987, 139: 813-817.
Uyttenhove C, Coulie PG, Van Snick JV: T cell growth and differentiation induced by interleukin-HP1/IL-6, the murine hybridoma/plasmacytoma growth factor. J Exp Med. 1988, 167: 1417-1427.
Van Snick JV, Cayphas S, Szikora J-P, Renauld J-C, Van Roost E, Boon T, Simpson RJ: cDNA cloning of murine interleukin-HP1: homology with human interleukin 6. Eur J Immunol. 1988, 18: 193-197.
Andus T, Geiger T, Hirano T, Northoff H, Ganter U, Bauer J, Kishimoto T, Heinrich PC: Recombinant human B cell stimulatory factor 2 (BSF-2/ IFNβ2) regulates β-fibrinogen and albumin mRNA levels in Fao-9 cells. FEBS Lett. 1987, 221: 18-22. 10.1016/0014-5793(87)80344-7.
Gauldie J, Richards C, Harnish D, Landsdorp P, Baumann H: Interferon-β2/ B cell-stimulatory factor type 2 shares identity with monocyte-derived hepatocyte-stimulating factor and regulates the major acute phase protein response in liver cells. Proc Natl Acad Sci USA. 1987, 84: 7251-7255.
Castell JV, Gomez-Lechon MJ, David M, Hirano T, Kishimoto T, Heinrich PC: Recombinant human interleukin-6 (IL-6/BSF-2/ HSF) regulates the synthesis of acute phase proteins in human hepatocytes. FEBS Lett. 1988, 232: 347-350. 10.1016/0014-5793(88)80766-X.
Kishimoto T, Akira S, Narazaki M, Taga T: Interleukin-6 family of cytokines and gp130. Blood. 1995, 86: 1243-1254.
Noma T, Mizuta T, Rosen A, Hirano T, Kishimoto T, Honjo T: Enhancement of the interleukin-2 receptor expression on T cells by multiple B-lymphotropic lymphokines. Immunol Lett. 1987, 15: 249-253. 10.1016/0165-2478(87)90032-0.
Okada M, Kitahara M, Kishimoto S, Matsuda T, Hirano T, Kishimoto T: BSF-2/IL-6 functions as killer helper factor in the in vitro induction of cytotoxic T cells. J Immunol. 1988, 141: 1543-1549.
Ceuppens JL, Baroja ML, Lorre K, Van Damme J, Billiau A: Human T cell activation with phytohemagglutinin: the function of IL-6 as an accessory signal. J Immunol. 1988, 141: 3868-3874.
Le J, Fredrickson G, Reis L, Diamantsein T, Hirano T, Kishimoto T, Vilcek J: Interleukin-2-dependent and interleukin-2-independent pathways of regulation of thymocyte function by interleukin-6. Proc Natl Acad Sci USA. 1988, 85: 8643-8647.
Lotz M, Jirik F, Kabouridis R, Tsoukas C, Hirano T, Kishimoto T, Carson DA: BSF-2/IL-6 is costimulant for human thymocytes and T lymphocytes. J Exp Med. 1988, 167: 1253-1258.
Garman RD, Jacobs KA, Clark SC, Raulet DH: B cell-stimulatory factor 2 (β2 interferon) functions as a second signal for interleukin 2 production by mature murine T cells. Proc Natl Acad Sci USA. 1987, 84: 7629-7633.
Ikebuchi K, Wong GG, Clark. SC, Ihle JN, Hirai Y, Ogawa M: Interleukin-6 enhancement of interleukin-3-dependent proliferation of multipotential hemopoietic progenitors. Proc Natl Acad Sci USA. 1987, 84: 9035-9039.
Koike K, Nakahata T, Takagi M, Kobayashi T, Ishiguro A, Tsujii K, Naganuma K, Okano A, Akiyama Y, Akabane T: Synergism of BSF2/interleukin-6 and interleukin-3 on development of multipotential hemopoietic progenitors in serum free culture. J Exp Med. 1988, 168: 879-890.
Leary A, Ikebuchi K, Hirai Y, Wong G, Yang Y-C, Clark SC, Ogawa M: Synergism between interleukin-6 and interleukin-3 in supporting proliferation of human hematopoietic stem cells: comparison with interleukin-1a. Blood. 1988, 71: 1759-1763.
Ogawa M: Differentiation and proliferation of hematopoietic stem cells. Blood. 1993, 81: 2844-2853.
Stanley ER, Bartocci A, Patinkin D, Rosendaal M, Bradley TR: Regulation of very primitive, multipotent, hemopoietic cells by hemopoietin-1. Cell. 1986, 45: 667-674.
Nicola NA, Metcalf D, Matsumoto M, Johnson GR: Purification of a factor inducing differentiation in murine myelomonocytic leukemia cells. Identification as granulocyte colony-stimulating factor. J Biol Chem. 1983, 258: 9017-9023.
Ishibashi T, Kimura H, Uchida T, Kariyone S, Friese P, Burstein SA: Human interleukin6 is a direct promoter of maturation of megakaryocytes in vitro. Proc Natl Acad Sci USA. 1987, 86: 5953-5957.
Ishibashi T, Kimura H, Shikama Y, Uchida T, Kariyone S, Hirano T, Kishimoto T, Takatsuki F, Akiyama Y: Interleukin-6 is a potent thrombopoietic factor in vivo in mice. Blood. 1989, 74: 1241-1244.
Koike K, Nakahata T, Kubo T, Kikuchi T, Takagi M, Ishiguro A, Tsuji K, Naganuma K, Okano A, Akiyama Y, Akabane T: Interleukin-6 enhances murine megakariocytopoiesis in serum-free culture. Blood. 1990, 75: 2286-2291.
Tamura T, Udagawa N, Takahashi N, Miyaura C, Tanaka S, Yamada Y, Koishihara Y, Ohsugi Y, Kumaki K, Taga T, Kishimoto T, Suda T: Soluble interleukin-6 receptor triggers osteoclast formation by interleukin 6. Proc Natl Acad Sci USA. 1993, 90: 11924-11928.
Ulich TR, del Castillo J, Guo KZ: In vivo hematologic effects of recombinant interleukin-6 on hematopoiesis and circulating numbers of RBCs and WBCs. Blood. 1989, 73: 108-110.
Horii Y, Muraguchi A, Iwano M, Matsuda T, Hirayama T, Yamada H, Fujii Y, Dohi K, Ishikawa H, Ohmoto Y, Yoshizaki K, Hirano T, Kishimoto T: Involvement of interleukin-6 in mesangial proliferation of glomerulonephritis. J Immunol. 1989, 143: 3949-3955.
Grossman RM, Krueger J, Yourish D, Granelli-Piperno A, Murphy DP, May LT, Kupper TS, Sehgal PB, Gottlieb AB: Interleukin 6 is expressed in high levels in psoriasis skin and stimulates proliferation of cultured human keratinocytes. Proc Natl Acad Sci USA. 1989, 86: 6367-6371.
Yoshizaki K, Nishimoto N, Matsumoto K, Tagoh H, Taga T, Deguchi Y, Kuritani T, Hirano T, Kishimoto T: Interleukin-6 and its receptor expression on the epidermal keratinocytes. Cytokine. 1990, 2: 381-387.
Kawano M, Hirano T, Matsuda T, Taga T, Horii Y, Iwato K, Asaoku H, Tang B, Tanabe O, Tanaka H, Kuramoto A, Kishimoto T: Autocrine generation and requirement of BSF-2/IL-6 for human multiple myelomas. Nature. 1988, 332: 83-85. 10.1038/332083a0.
Miki S, Iwano M, Miki Y, Yamamoto M, Tang B, Yokokawa K, Sonoda T, Hirano T, Kishimoto T: Interleukin-6 (IL-6) functions as an in vitro autocrine growth factor in renal cell carcinomas. FEBS Lett. 1989, 250: 607-610. 10.1016/0014-5793(89)80805-1.
Yamasaki K, Taga T, Hirata Y, Yawata H, Kawanishi Y, Seed B, Taniguchi T, Hirano T, Kishimoto T: Cloning and expression of the human interleukin-6 (BSF-2/INF b2) receptor. Science. 1988, 241: 825-828.
Taga T, Hibi M, Hirata Y, Yamasaki K, Yasukawa K, Matsuda T, Hirano T, Kishimoto T: Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell. 1989, 58: 573-581.
Hibi M, Murakami M, Saito M, Hirano T, Taga T, Kishimoto T: Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell. 1990, 63: 1149-1157.
Miyaura C, Onozaki K, Akiyama Y, Taniyama T, Hirano T, Kishimoto T, Suda T: Recombinant human interleukin 6 (B-cell stimulatory factor 2) is a potent inducer of differentiation of mouse myeloid leukemia cells (M1). FEBS Lett. 1988, 234: 17-21. 10.1016/0014-5793(88)81293-6.
Shabo Y, Lotem J, Rubinstein M, Revel M, Clark SC, Wolf SF, Kamen R, Sachs L: The myeloid blood cell differentiation-inducing protein MGI-2A is interleukin-6. Blood. 1988, 72: 2070-2073.
Metcalf D: Actions and interactions of G-CSF, LIF, and IL-6 on normal and leukemic murine cells. Leukemia. 1989, 3: 349-355.
Rose TM, Bruce AG: Oncostatin M is a member of a cytokine family which includes leukemia inhibitory factor, granulocyte colony-stimulatory factor and interleukin-6. Proc Natl Acad Sci USA. 1991, 88: 8641-8645.
Baumann H, Onorato V, Gauldie J, Jahreis GP: Distinct sets of acute phase plasma proteins are stimulated by separate human hepatocyte-stimulating factors and monokines in rat hepatoma cells. J Biol Chem. 1987, 262: 9756-9768.
Baumann H, Wong GG: Hepatocyte-stimulatory factor III shares structural and functional identity with leukemia inhibitory factor. J Immunol. 1989, 143: 1163-1167.
Richards CD, Brown TJ, Shoyab M, Baumann H, Gauldie J: Recombinant oncostatin M stimulates the production of acute phase protein in HepG2 cells and rat primary hepatocytes in vitro. J Immunol. 1992, 148: 1731-1736.
Miyajima A, Kitamura T, Harada N, Yokota T, Arai K: Cytokine receptors and signal transduction. Annu Rev Immunol. 1992, 10: 295-331. 10.1146/annurev.iy.10.040192.001455.
Hirano T, Matsuda T, Nakajima K: Signal transduction through gp130 that is shared among the receptors for the interleukin 6 related cytokine subfamily. Stem Cells. 1994, 12: 262-277.
Hibi M, Nakajima K, Hirano H: IL-6 cytokine family and signal transduction: a model of the cytokine system. J Mol Med. 1996, 74: 1-12.
Hirota H, Yoshida K, Kishimoto T, Taga T: Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc Natl Acad Sci USA. 1995, 92: 4862-4866.
Darnell JE, Kerr IM, Stark GR: Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994, 264: 1415-1421.
Ihle JN, Witthuhn BA, Quelle FW, Yamamoto K, Thierfelder WE, Kreider B, Silvennoinen O: Signaling by the cytokine receptor superfamily: JAKs and STATs. Trends Biochem Sci. 1994, 19: 222-227. 10.1016/0968-0004(94)90026-4.
Schindler C, Darnell JE: Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu Rev Biochem. 1995, 64: 621-651. 10.1146/annurev.bi.64.070195.003201.
Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, Yoshida K, Sudo T, Naruto M, Kishimoto T: Molecular cloning of APRF, a novel IFN-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell. 1994, 77: 63-71.
Guschin D, Rogers N, Briscoe J, Witthuhn BA, Wathing D, Horn F, Pellegrini S, Yasukawa K, Heinrich P, Stark GR, Ihle JN, Kerr IM: A major role for the protein kinase JAK1 in the JAK/STAT signal transduction pathway in response to the interleukin-6. EMBO J. 1995, 14: 1421-1429.
Akira S, Isshiki H, Sugita T, Tanabe O, Kinoshita S, Nishio Y, Nakajima T, Hirano T, Kishimoto T: A nuclear factor for IL-6 expression (NF-IL6) is a member of a C/EBP family. EMBO J. 1990, 9: 1897-1906.
Poli V, Mancini FP, Cortese R: IL-6DBP, a nuclear protein involved in interleukin-6 signal transduction, defines a new family of leucine zipper proteins related to C/EBP. Cell. 1990, 63: 643-653.
Cao Z, Umkek RM, McKnight SL: Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev. 1991, 5: 1538-1552.
De Groot RP, Auwerx J, Karperien M, Staels B, Kruijer W: Activation of junB by PKC and PKA signal transduction through a novel cis-acting element. Nucleic Acids Res. 1991, 19: 775-781.
Wegenka UM, Buschmann J, Lutticken C, Heinrich PC, Horn F: Acute-phase response factor, a nuclear factor binding to acute-phase response elements, is rapidly activated by interleukin-6 at the posttranslational level. Mol Cell Biol. 1993, 13: 276-288.
Zhong Z, Wen Z, Darnell JE: Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994, 264: 95-98.
Fukunaga R, Ishizaka Ikeda E, Pan CX, Seto Y, Nagata S: Functional domains of the granulocyte colony-stimulating factor receptor. EMBO J. 1991, 10: 2855-2865.
Murakami M, Narazaki M, Hibi M, Yawata H, Yasukawa K, Hamaguchi M, Taga T, Kishimoto T: Critical cytoplasmic region of the interleukin 6 signal transducer gp130 is conserved in the cytokine receptor family. Proc Natl Acad Sci USA. 1991, 88: 11349-11353.
Narazaki M, Witthuhn BA, Yoshida K, Silvennoinen O, Yasukawa K, Ihle JN, Kishimoto T, Taga T: Activation of JAK2 kinase mediated by the interleukin 6 signal transducer gp130. Proc Natl Acad Sci USA. 1994, 91: 2285-2289.
Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JJ, Yan-copoulos GD: Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science. 1995, 267: 1349-1353.
Yamanaka Y, Nakajima K, Fukada T, Hibi M, Hirano T: Differentiation and growth arrest signals are generated through the cytoplamic region of gp130 that is essential for Stat3 activation. EMBO J. 1996, 15: 1557-1565.
Minami M, Inoue M, Wei S, Takeda K, Matsumoto M, Kishimoto T, Akira S: STAT3 activation is a critical step in gp130-mediated terminal differentiation and growth arrest of a myeloid cell line. Proc Natl Acad Sci USA. 1996, 93: 3963-3966. 10.1073/pnas.93.9.3963.
Nakajima K, Yamanaka Y, Nakae K, Kojima H, Kiuchi N, Ichiba M, Kitaoka T, Fukada T, Hibi M, Hirano T: A central role for Stat3 in IL-6-induced regulation of growth and differentiation in M1 leukemia cells. EMBO J. 1996, 15: 3651-3658.
Fukada T, Hibi M, Yamanaka Y, Takahashi-Tezuka M, Fijitani Y, Yamaguchi T, Nakajima K, Hirano T: Two signals are necessary for cell proliferation induced by a cytokine receptor gp130: involvement of STAT3 in anti-apoptosis. Immunity. 1996, 5: 449-460.
Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S: Stat3 activation is irresponsible for IL-6-dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell-specific Stat3-deficient mice. J Immunol. 1998, 161: 4652-4660.
Kumar G, Gupta S, Wang S, Nel AE: Involvement of Janus kinases, p52shc, Raf-1, and MEK-1 in the IL-6-induced mitogen-activated protein kinase cascade of a growth-responsive B cell line. J Immunol. 1994, 153: 4436-4447.
Ernst M, Gearing DP, Dunn AR: Functional and biochemical association of Hck with the LIF/IL-6 receptor signal transduct-ing subunit gp130 in embryonic stem cells. EMBO J. 1994, 13: 1574-1584.
Matsuda T, Fukada T, Takahashi-Tezuka M, Okuyama Y, Fujitani Y, Hanazono Y, Hirai H, Hirano T: Association and activation of Btk and Tec tyrosine kinases by gp130, a signal transducer of the interleukin-6 family of cytokines. J Biol Chem. 1995, 270: 11037-11039. 10.1074/jbc.270.19.11037.
Taniguchi T: Cytokine signaling through nonreceptor protein tyrosine kinase. Science. 1995, 268: 251-255.
Ohtani T, Ishihara K, Atsumi T, Nishida K, Kaneko Y, Miyata H, Itoh T, Narimatsu M, Maeda H, Fukada T, Itoh M, Okano H, Hibi M, Hirano T: Dissection of signaling cascades through gp130 in vivo: reciprocal roles for STAT3- and SHP2-mediated signals in immune responses. Immunity. 2000, 12: 95-105.
Ernst M, Inglese M, Waring P, Cambell IK, Bao S, Clay FJ, Alexander WS, Wicks IP, Tarlinton DM, Novak U, Heath JK, Dunn AR: Defective gp130-mediated signal transducer and activator of transcription (STAT) signaling results in degenerative joint disease, gastrointestinal ulceration, and failure of uterine implantation. J Exp Med. 2001, 194: 189-203. 10.1084/jem.194.2.189.
Starr R, Willson TA, Viney EM, Murray LJL, Rayner JR, Jenkis BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, Hilton DJ: A family of cytokine-inducible inhibitors of signaling. Nature. 1997, 387: 917-921. 10.1038/43206.
Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, Nishimoto N, Kajita T, Taga T, Yoshizaki K, Akira S, Kishimoto T: Structure and function of a new STAT-induced STAT inhibitor. Nature. 1997, 387: 924-929. 10.1038/43219.
Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, Matsumoto A, Tanimura S, Ohtsubo M, Misawa H, Miyazaki T, Leonor N, Taniguchi T, Fujita T, Kanakura Y, Komiya S, Yoshimura A: A new protein containing an SH2 domain that inhibits JAK kinase. Nature. 1997, 387: 921-924. 10.1038/43213.
Minamoto S, Ikegame K, Ueno K, Narazaki M, Naka T, Yamamoto H, Matsumoto T, Saito H, Hosoe S, Kishimoto T: Cloning and functional analysis of new members of STAT induced STAT inhibitor (SSI) family: SSI-2 and SSI-3. Biochem Biophys Res Commun. 1997, 237: 79-83. 10.1006/bbrc.1997.7080.
Masuhara M, Sakamoto H, Matsumoto A, Suzuki R, Yasukawa H, Mitsui K, Wakioka T, Tanimura S, Sasaki A, Misawa H, Yokouchi M, Ohtsubo M, Yoshimura A: Cloning and characterization of novel CIS family genes. Biochem Biophys Res Commun. 1997, 239: 436-446. 10.1006/bbrc.1997.7484.
Hilton DJ, Richardson RT, Alexander WS, Viney EM, Sprigg NS, Nicholson SE, Metcalf D, Nicola NA: Twenty proteins containing C-terminal SOCS-box form five structural classes. Proc Natl Acad Sci USA. 1998, 95: 114-119. 10.1073/pnas.95.1.114.
Naka T, Fujimoto M, Kishimoto T: Negative regulation of cytokine signaling: STAT-induced STAT inhibitor. Trends Biochem Sci. 1999, 24: 394-398. 10.1016/S0968-0004(99)01454-1.
Yasukawa H, Sasaki A, Yoshimura A: Negative regulation of cytokine signaling pathways. Annu Rev Immunol. 2000, 18: 143-164. 10.1146/annurev.immunol.18.1.143.
Krebs DL, Hilton DJ: SOCS proteins: negative regulators of cytokine signaling. Stem Cells. 2001, 19: 378-387.
Nicholson SE, Willson TA, Farley A, Starr R, Zhang JG, Baca M, Alexander WS, Metcalf D, Hilton DJ, Nicola NA: Mutational analyses of the SOCS proteins suggest a dual domain requirement but distinct mechanisms for inhibition of LIF and IL-6 signal transduction. EMBO J. 1999, 18: 375-385. 10.1093/emboj/18.2.375.
Schmitz J, Weissenbach M, Haan S, Heinrich PC, Schaper F: SOCS-3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP-2 recruitment site of gp130. J Biol Chem. 2000, 275: 12848-12856. 10.1074/jbc.275.17.12848.
Metcalf D, Greenhalgh CJ, Viney E, Willson TA, Starr R, Nicola NA, Hilton DJ, Alexander WS: Giantism in mice lacking suppressor of cytokine signal-2. Nature. 2000, 405: 1069-1074. 10.1038/35016611.
Marine JC, Topham DJ, McKay C, Wang D, Parganas E, Nakajima H, Pendeville H, Yasukawa H, Sasaki A, Yoshimura A, Ihle JN: SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell. 1999, 98: 617-627.
Robert AW, Robb L, Rakar S, Hartley L, Nicola NA, Metcalf D, Hilton DJ, Alexander WS: Placental defects and embryonic lethality in mice lacking suppressor of cytokine signaling 3. Proc Natl Acad Sci USA. 2001, 98: 9324-9329. 10.1073/pnas.161271798.
Starr R, Metcalf D, Elefanty AG, Brysha M, Willson TA, Nicola NA, Hilton DJ, Alexander WS: Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc Natl Acad Sci USA. 1998, 95: 14395-14399. 10.1073/pnas.95.24.14395.
Naka T, Matsumoto T, Narazaki M, Fujimoto M, Morita Y, Ohsawa Y, Saito H, Nagasawa T, Uchiyama Y, Kishimoto T: Accelerated apoptosis of lymphocytes by augmented induction of Bax in SSI-1(STAT-induced STAT inhibitor) deficient mice. Proc Natl Acad Sci USA. 1998, 95: 15575-15582. 10.1073/pnas.95.26.15577.
Alexander WA, Starr R, Fenner JE, Scott CL, Handman E, Springg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, Kay TWH, Nicola NA, Hertzog PJ, Metcalf D, Hilton DJ: SOCS1 is critical inhibitor of interferon γ signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999, 98: 597-608.
Marine JC, Topham DJ, McKay C, Wang D, Parganas E, Stravopodis D, Yoshimura A, Ihle JN: SOCS1 deficiency causes a lymphocyte-dependent perinatal lethality. Cell. 1999, 98: 609-616.
Morita Y, Naka T, Kawazoe Y, Fujimoto M, Narazaki M, Nakagawa R, Fukuyama H, Nagata S, Kishimoto T: SSI/SOCS-1 suppresses TNF α-induced cell death by regulation of p38 MAP kinase signal in fibroblast. Proc Natl Acad Sci USA. 2000, 97: 5405-5410. 10.1073/pnas.090084797.
Kawazoe Y, Naka T, Fujimoto M, Kohzaki H, Morita Y, Narazaki M, Okumura K, Saitoh H, Nakagawa R, Uchiyama Y, Akira S, Kishimoto T: SSI-1/SOCS-1 family proteins inhibit insulin signal transduction pathway through modulating IRS-1 phosphorylation. J Exp Med. 2001, 193: 263-269. 10.1084/jem.193.2.263.
Naka T, Tsutsui H, Fujimoto M, Kawazoe Y, Kohzaki H, Morita Y, Nakagawa R, Narazaki M, Adachi K, Yoshimoto T, Nakanishi K, Kishimoto T: SOCS-1/SSI-1-deficient NKT cells participate in severe hepatitis through dysregulated cross-talk inhibition of IFN-γ and IL-4 signaling in vivo. Immunity. 2001, 14: 535-545. 10.1016/S1074-7613(01)00132-7.
Chung CD, Liao J, Lui B, Rao X, Jay P, Berta P, Shuai K: Specific inhibiton of Stat3 signal transduction by PIAS3. Science. 1997, 278: 1803-1805. 10.1126/science.278.5344.1803.
Liu B, Liao J, Rao X, Kushner SA, Chung CD, Chang DD, Shuai K: Inhibition of Stat-1 mediated gene activation by PIAS1. Proc Natl Acad Sci USA. 1998, 95: 10626-10631. 10.1073/pnas.95.18.10626.
Feldmann M, Elliott MJ, Woody JN, Maini RN: Anti-tumor necrosis factor-α therapy of rheumatoid arthritis. Adv Immunol. 1997, 64: 283-350.
Yoshizaki K, Nishimoto N, Mihara M, Kishimoto T: Therapy of RA by blocking IL-6 signal transduction with humanized anti-IL-6 receptor antibody. Springer Semin Immunopathol. 1998, 20: 247-259. 10.1007/s002810050033.
Jilka RH, Hangoc G, Girasole G, Passeri G, Williams DC, Abrams JS, Boice B, Broxmeyer H, Manolagas SC: Increased osteoclast development after estrogen loss: Mediation by interleukin-6. Science. 1992, 257: 88-91.
Hirano T, Matsuda T, Turner M, Miyasaka N, Buchan G, Tang B, Sato K, Shimizu M, Maini R, Feldman M, Kishimoto T: Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. Eur J Immunol. 1988, 18: 1797-1801.
Houssiau FA, Devogelaer JP, Van Damme J, De Deuxchaisies CN, Van Snick J: Interleukin-6 in synovial fluid and serum of patients with rheumatoid arthritis and other inflammatory arthritis. Arthritis Rheum. 1988, 31: 784-788.
Sack U, Kinne R, Marx T, Heppt P, Bender S, Emmrich F: Interleukin-6 in synovial fluid is closely associated with chronic synovitis in rheumatoid arthritis. Rheumatol Int. 1993, 13: 45-51.
Madhok R, Crilly A, Watson J, Capell HA: Serum interleukin 6 levels in rheumatoid arthritis: correlation with clinical and laboratory indices of disease activity. Ann Rheum Dis. 1993, 52: 232-234.
Wendling D, Racadot E, Wijdenes J: Treatment of severe rheumatoid arthritis by anti-interleukin 6 monoclonal antibody. J Rheumatol. 1993, 20: 259-262.
Sato K, Tsuchiya M, Saldanha J, Koishihara Y, Ohsugi Y, Kishimoto T, Bendig MM: Reshaping a human antibody to inhibit the interleukin-6-dependent tumor cell growth. Cancer Res. 1993, 53: 851-856.
Nishimoto N, Sasai M, Shima Y, Nakagawa M, Matsumoto T, Shirai T, Kishimoto T, Yoshizaki Y: Improvement in Castleman's disease by humanized anti-IL-6 receptor antibody therapy. Blood. 2000, 95: 56-61.
Nishimoto N, Maeda K, Kuritani T, Deguchi H, Sato B, Imai N, Kakehi T, Suemura M, Kishimoto T, Yoshizaki K: Safety and efficacy of repetitive treatment with humanized anti-interleukin-6 receptor antibody (MRA) in rheumatoid arthritis (RA) [abstract]. Arthritis Rheum. 2001, 44: S84-
Mihara M, Kotoh M, Nishimoto N, Oda Y, Kumagai E, Takagi N, Tsunemi K, Ohsugi Y, Kishimoto T, Yoshizaki Y, Takeda Y: Humanized antibody to human interleukin-6 receptor inhibits the development of collagen arthritis in cynomolgus monkeys. Clin Immunol. 2001, 98: 319-326. 10.1006/clim.2000.4989.
Choy EH, Isenberg DA, Farrow S, Garrood T, Ioannou Y, Bird H, Cheung N, Williams BD, Hazlemau B, Price R, Kishimoto T, Panayi GS: A double-blind, randomized, placebo-controlled trial of anti-interleukin-6 (IL-6) receptor monoclonal antibody in rheumatoid arthritis (RA) [abstract]. Arthritis Rheum. 2001, 44: S84-10.1002/1529-0131(200109)44:9<1993::AID-ART347>3.0.CO;2-A.
Nishimoto N, Ogata A, Shima Y, Tani Y, Ogawa H, Nakagawa M, Sugiyama H, Yoshizaki K, Kishimoto T: Oncostatin M, leukemia inhibitory factor, and interleukin 6 induce the proliferation of human plasmacytoma cells via the common signal transducer, gp130 [abstract]. J Exp Med. 1994, 179: 1343-1347.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Naka, T., Nishimoto, N. & Kishimoto, T. The paradigm of IL-6: from basic science to medicine. Arthritis Res Ther 4 (Suppl 3), S233 (2002). https://doi.org/10.1186/ar565
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar565