
Abstract
Introduction
Chondrocalcinosis (CC) most commonly results from calcium pyrophosphate crystal deposition (CPPD). The objective of this study is to examine the association between candidate single-nucleotide polymorphisms (SNPs) and radiographic CC.
Methods
SNPs in ankylosis human (ANKH), high ferritin (HFE), tissue non-specific alkaline phosphatase (TNAP), ecto-neucleotide pyrophosphatase 1 (ENPP1), and transferrin (TE) genes were genotyped in participants of the Genetics of Osteoarthritis and Lifestyle (GOAL) and Nottingham Osteoarthritis Case-Control studies. Adjusted genotype odds ratio (aORGENOTYPE), the OR for association between one additional minor allele and CC, was calculated and adjusted for age, gender, body mass index (BMI), and osteoarthritis (OA) by using binary logistic regression. Statistical significance was set at P ≤0.003 after Bonferroni correction for multiple tests.
Results
The -4bpG > A polymorphism in the 5′ untranslated region (5′ UTR) of ANKH associated with CC after Bonferroni correction. This was independent of age, gender, OA, and BMI; aORGENOTYPE (95% confidence interval, or CI) was 1.39 (1.14-1.69) (P = 0.001). rs3045 and rs875525, two other SNPs in ANKH, associated with CC; aORGENOTYPE (95% CI) values were 1.31 (1.09-1.58) (P = 0.005) and 1.18 (1.03-1.35) (P = 0.015), respectively; however, this was non-significant after Bonferroni correction.
Conclusions
This study validates the association between a functional polymorphism in the 5′ UTR of ANKH and CC and shows for the first time that this is independent of age and OA – the two key risk factors for CC. It shows that other SNPs in ANKH may also associate with CC. This supports the role of extracellular inorganic pyrophosphate in the pathogenesis of CC. The findings of this hospital-based study require replication in a community-based population.
Similar content being viewed by others


Introduction
Chondrocalcinosis (CC) most commonly results from calcium pyrophosphate (CPP) crystal deposition (CPPD) [1]. CPPD may present as acute CCP crystal arthritis, CPPD with osteoarthritis (OA), chronic CPP crystal inflammatory arthritis, or asymptomatic CC [1]. Age, OA, diuretic use, and joint injury are recognized risk factors for CC [1, 2]. Additionally, metabolic diseases that elevate extracellular pyrophosphate (ePPi) levels (hyperparathyroidism, hypomagnesemia, and hypophosphatasia), hemochromatosis, and familial predisposition are uncommon risk factors [1, 2]. Though rare, familial predisposition is reported from several countries and different ethnic groups [3–9]. The pattern of inheritance is usually autosomal dominant. The main clinical phenotype is characterized by early onset (in the 20s or 30s) of acute CPP crystal arthritis with florid polyarticular CC and variable severity of accompanying structural arthritis/OA. However, a second phenotype with later onset in the sixth to seventh decades and oligo-articular CC that more closely resembles sporadic CPPD has also been reported [10]. This latter familial form may be more common than is recognized, the late onset of disease expression and geographic dispersal of families tending to mask such predisposition. An association with benign childhood fits appears unique to one UK family with early-onset polyarticular CC, and the responsible gene—CC gene 2 (CCAL2) on chromosome 5p15—was first identified in this family [11]. Other kindreds with CC due to mutations at this locus [3] have been reported, and the responsible gene was subsequently identified as the ankylosis human (ANKH) gene [12]. The other reported locus in an American family with premature OA and associated CPPD (CCAL1) is on chromosome 8q [13], and a specific gene predisposing to CPPD at this site has not been identified.
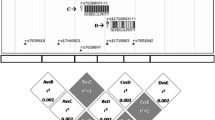
Although several kindreds with familial CPPD are reported, the evidence for genetic contribution to sporadic CC is conflicting [3]. For example, the -4bpG > A transition in the 5′ untranslated region (5′ UTR) of ANKH (which encodes the trans-membrane PPi transport protein ANKH) associated with CC in a study of 128 CC cases and 475 healthy controls [14], whereas hereditary contribution to CC was not detected in a larger sibling study (n = 1,841) [15]. Therefore, the objectives of this study were to validate the reported association between -4bpG > A transition in the 5′ UTR of ANKH and CC, to examine whether this is independent of other risk factors of CC such as age and OA, and to investigate whether other candidate single-nucleotide polymorphisms (SNPs) in genes involved in PPi metabolism—ANKH, tissue non-specific alkaline phosphatase (TNAP), and ectonucleotide pyrophosphatase 1 (ENPP1) ( Figure 1)—or iron overload (for example, high ferritin and transferrin) associate with CC.

Extracellular pyrophosphate metabolism. ANKH, (progressive) ankylosis human; Mg++, magnesium; NTP, nucleotide tri-phosphate; PC1, plasma cell glycoprotein 1; Pi, phosphate; PPi, pyrophosphate; TNAP, tissue non-specific alkaline phosphatase.
Methods
Study design and participants
The case-control study used data from the Genetics of Osteoarthritis and Lifestyle (GOAL) and Nottingham Osteoarthritis Case-Control (NOAC) studies [16, 17] (Table 1). The GOAL study was approved by the Nottinghamshire Research Ethics Committee (UK). The NOAC study was approved by the North Nottinghamshire Research Ethics Committee and the Leicestershire, Northamptonshire, and Rutland Research Ethics Committee. All study participants gave written informed consent. These studies, originally set up to examine the genetics of large-joint OA, recruited Caucasians with (a) hip or knee OA severe enough to require joint replacement surgery or (b) no radiographic or clinical features of hip or knee OA [16, 17]. All participants were residents of Nottinghamshire, UK. Information about disease and demographic variables were collected at the study visit. Genomic DNA was extracted from the peripheral blood leukocytes by using standard protocols. GOAL study participants had radiographs of hands, knees, and pelvis; and the NOAC study participants had radiographs of the index knee or hip joint awaiting replacement. These radiographs were scored for CC at the knees, hips, wrists, and symphysis pubis and for metacarpophalangeal joint (MCPJ) calcification by a single trained observer (Sally Doherty).
Cases and controls
Cases were participants with CC at any joint, whereas controls did not have CC at any joint x-rayed.
Single-nucleotide polymorphism selection
Seventeen SNPs were selected on the basis of published associations with various phenotypes (Table 2).
Genotyping
Genotyping was carried out at AstraZeneca laboratories in Macclesfield, UK, by using the Taqman method and at Kbioscience Ltd (Hertfordshire, UK) by using the Kompetitive Allele Specific PCR (KASPar) chemistry. All 17 SNPs were genotyped in the GOAL study. All selected SNPs in any gene which contained at least one SNP that associated with CC with an uncorrected P ≤0.10 in the GOAL study were genotyped in the NOAC study participants.
Covariates
Data about age (years), height (centimeters), and weight (kilograms) were collected at the study visit. Height and weight were used to calculate body mass index (BMI) (kg/m2). OA was defined as knee or hip OA clinically severe enough to warrant consideration of joint replacement surgery.
Statistical analysis
Mean and standard deviation (SD) and number (percentage) were used for descriptive purposes. Chi-square test and student t test were used to compare categorical and continuous variables. Cases with CC were compared with controls without CC. All SNPs were checked for Hardy-Weinberg equilibrium (HWE). Data from the GOAL and NOAC studies were pooled together for analyzing genetic risk. Genotype odds ratio (ORGENOTYPE)—the OR for association between increasing number of minor alleles of an SNP and CC—was calculated. Binary logistic regression was used to calculate aORGENOTYPE (95% confidence interval, or CI) adjusting for age (tertiles), gender, BMI (tertiles), and OA. Additionally, ORGENOTYPE was meta-analyzed with published studies by using fixed effects analysis given the lack of heterogeneity between studies. Meta-analyses were performed by using R V.2.13.1 [26]. Other analyses were carried out by using SPSSv14. Statistical significance for genetic association was set at P ≤0.003 after application of Bonferroni correction for multiple tests. Linkage disequilibrium for the four ANKH SNPs studied was estimated from unphased genotype data by using the Haploview 4.2 version [27].
Results and discussion
The descriptive characteristics of study participants are presented in Table 1. Three thousand one hundred forty-one GOAL and 1,800 NOAC study participants were included. The mean age (SD), number of females (percentage), and mean BMI (SD) of the GOAL study and NOAC study participants were 66.6 (7.9) and 70.5 (9.2) years, 1,520 (48.4%) and 1,045 (58.1%) women, and 29.3 (5.3) and 29.5 (5.6) kg/m2, respectively. GOAL study participants were significantly younger (P <0.001), were less likely to be female (P <0.001), and had similar BMI (P = 0.22) compared with the NOAC study participants.
Six hundred fifty-eight (13.2%) participants had CC at any site. All genotyped SNPs were in HWE. As TNAP571 [28] was a rare mutation in the GOAL study population (minor allele frequency = 0.01), it was excluded from further analysis. Three ANKH and one HFE SNPs associated with CC with an uncorrected P ≤0.10 in the GOAL study population (Table 2). Thus, all ANKH (-4bpG > A 5′ UTR, rs3045, rs39968, and rs875525) and HFE (rs1800562 and rs1799945) SNPs genotyped in the GOAL study were further genotyped in the NOAC study participants. The four ANKH SNPs were not in linkage disequilibrium (Table 3). Among the six SNPs genotyped in both the GOAL and NOAC studies, the -4bpG > A 5′ UTR of ANKH associated with CC after adjustment for age, gender, BMI, and OA; aORGENOTYPE was 1.39 (1.14-1.69) (P = 0.001) (Table 4). There was a trend toward an association between rs3045 and CC; aORGENOTYPE was 1.31 (1.09-1.58) (P = 0.005); however, it was not statistically significant after application of Bonferroni correction (Table 4). The frequency of rare and common allelic variants in the ANKH and HFE SNPs genotyped in both the GOAL and NOAC studies is shown in an additional file (Additional file 1: Table S1).
Comparison with published studies
Two studies have reported on the association between CC and HFE SNPs [20, 21]. Of these, one study reported genotype data about rs1800562, and rs1799945 without reporting full data on compound heterozygote numbers [20], whereas the other did not report detailed genotype data [21]. Therefore, the latter study was not included in the meta-analysis. In a meta-analysis of the data of the present study and of the study by Alizadeh et al. [20], the values for pooled ORGENOTYPE (95% CI) for association between rs1799945 and rs1800562 and CC were 1.20 (1.04-1.39) (P = 0.015) and 1.08 (0.88-1.33) (P = 0.445), respectively. Similarly, the results of the present study and the previous study that reported an association between CC and -4bpG > A transition in the 5′ UTR of ANKH were meta-analyzed [14]. The pooled ORGENOTYPE (95% CI) was 1.36 (1.13-1.61) (P = 0.001).
The present study confirms the association between CC and -4bpG > A transition in the 5′ UTR of ANKH. It is the first study to show that this association is independent of age and OA, which are the two major established risk factors of CC. This study also raises the possibility that other SNPs in ANKH (for example, rs3045) may also associate with CC. However, in keeping with previous reports, there was no association between SNPs in TNAP or ENPP1 and CC [29].
ANKH encodes a multipass transmembrane protein (ANKH) in joints and other tissues and participates in the export of intracellular PPi [30, 31]. PPi cannot diffuse across cell membranes passively, and ANKH is the principal way in which intracellular PPi reaches the extracellular environment. ANKH-mediated control of PPi levels regulates tissue calcification and susceptibility to arthritis [30, 31]. The autosomal dominant mutations in ANKH are thought to confer a gain in PPi transport function leading to increased extracellular PPi levels [14, 32]. Functional assays show that the -4bpG > A transition in the 5′ UTR of ANKH reduces intracellular PPi (a surrogate for increased transcellular PPi export) and increases ANKH expression in vitro[14, 18]. The minor alleles of rs3045 also result in lower intracellular PPi levels in vitro, providing external validity to our finding of a possible association between this polymorphism and CC [18]. These two SNPs are not in linkage disequilibrium. The minor allele frequency for the -4bpG > A transition in 5′ UTR of ANKH and rs3045 is higher in this study than that in the multi-ethnic 1000 Genomes Project. (See Additional file 2: Table S2 for genotype frequencies of the selected SNPs in the 1000 Genomes Project). This may explain, in part, why CPPD is more common in Caucasians than in other ethnicities.
Though not statistically significant, results from the data suggest that rs1799945 (HFE SNP associated with smaller iron overload), but not rs1800562 (HFE SNP associated with greater iron overload), may associate with CC. Similar findings have been reported previously [20]. The lack of association between homozygosity for minor allele at rs1800562 and CC may be due to a channeling bias (that is, patients with hemochromatosis are excluded from these studies).
This is the largest reported study of genetic risk factors for CC. This study has several strengths. First, the analysis of genetic risk was adjusted for factors that associate with CC, specifically age and OA, and also for gender, which associates with iron overload. Moreover, correction for multiple testing was applied to reduce the chances of a type I error. However, this study has several caveats. First, this is a hospital-based study carried out by reconstituting cases and controls within cohorts assembled primarily to examine risk factors for knee or hip OA within the East Midlands region of the UK. Cases with mild to moderate large-joint OA were not included. The study sample therefore does not resemble a community-based population and is restricted to one area of the country. Moreover, as more than 78% of participants had severe large-joint OA, the results may be confounded by their OA status. However, to minimize any confounding and to improve the generalizability of these findings, we have adjusted for OA at the knee and hip. Second, participants of the NOAC study did not have the same extensive radiographic phenotyping of CC as participants of the GOAL study. As a result, some NOAC study participants who may have CC at distant joints without CC at the joint to be replaced will be misclassified as ‘CC negative’ controls. This misclassification is likely to minimize the genetic association and does not invalidate the genetic associations observed in this study.
Conclusions
This study validates an established genetic association with CC and shows that this is independent of age and OA. This study also raises the possibility that other SNPs in ANKH associate with CC. Larger studies with greater power are required to confirm these findings. Finally, the findings of this study derived from a hospital-based cohort warrant confirmation in a community-based population including cases with mild to moderate disease and in other countries.
Authors’ information
AMV and MD are joint senior authors.
Abbreviations
- 5′ UTR:
-
5′ untranslated region
- ANKH:
-
ankylosis human
- BMI:
-
body mass index
- CC:
-
chondrocalcinosis
- CCAL (1 or 2):
-
chondrocalcinosis (1 or 2) gene
- CI:
-
confidence interval
- CPP:
-
calcium pyrophosphate crystal
- CPPD:
-
calcium pyrophosphate crystal deposition
- ENPP1:
-
ectonucleotide pyrophosphatase 1
- GOAL:
-
Genetics of Osteoarthritis and Lifestyle
- HFE:
-
high ferritin
- HWE:
-
Hardy-Weinberg equilibrium
- MCPJ:
-
metacarpophalangeal joint
- NOAC:
-
Nottingham Osteoarthritis Case-Control
- OA:
-
osteoarthritis
- ORGENOTYPE:
-
genotype odds ratio
- PPi:
-
pyrophosphate
- SD:
-
standard deviation
- SNP:
-
single-nucleotide polymorphism
- TNAP:
-
tissue non-specific alkaline phosphatase.
References
Zhang W, Doherty M, Bardin T, Barskova V, Guerne PA, Jansen TL, Leeb BF, Perez-Ruiz F, Pimentao J, Punzi L: European league against rheumatism recommendations for calcium pyrophosphate deposition. Part I: terminology and diagnosis. Ann Rheum Dis. 2011, 70: 563-570. 10.1136/ard.2010.139105.
Richette P, Bardin T, Doherty M: An update on the epidemiology of calcium pyrophosphate dihydrate crystal deposition disease. Rheumatology (Oxford). 2009, 48: 711-715. 10.1093/rheumatology/kep081.
Abhishek A, Doherty M: Pathophysiology of articular chondrocalcinosis–role of ANKH. Nat Rev Rheumatol. 2011, 7: 96-104. 10.1038/nrrheum.2010.182.
Andrew LJ, Brancolini V, de la Pena LS, Devoto M, Caeiro F, Marchegiani R, Reginato A, Gaucher A, Netter P, Gillet P, Loeuille D, Prockop DJ, Carr A, Wordsworth BF, Lathrop M, Butcher S, Considine E, Everts K, Nicod A, Walsh S, Williams CJ: Refinement of the chromosome 5p locus for familial calcium pyrophosphate dihydrate deposition disease. Am J Hum Genet. 1999, 64: 136-145. 10.1086/302186.
Béjia I, Rtibi I, Touzi M, Zrour S, Younes M, Naceur B: Familial calcium pyrophosphate dihydrate deposition disease. A Tunisian kindred. Joint Bone Spine. 2004, 71: 401-408. 10.1016/j.jbspin.2003.10.012.
Gaudreau A, Camerlain M, Pibarot ML, Beauregard G, Lebrun A, Petitclerc C: Familial articular chondrocalcinosis in Quebec. Arthritis Rheum. 1981, 24: 611-615. 10.1002/art.1780240407.
Hamza M, Meddeb N, Bardin T: Hereditary chondrocalcinosis in a Tunisian family. Clin Exp Rheumatol. 1992, 10: 43-49.
Hamza M, Ayed K, Bardi R, Gebuhrer L, Betuel H, Bardin T, Plaetke R, Lathrop M: HLA-antigens in a Tunisian familial chondrocalcinosis. Dis Markers. 1990, 8: 109-112.
Reginato AJ, Hollander JL, Martinez V, Valenzuela F, Schiapachasse V, Covarrubias E, Jacobelli S, Arinoviche R, Silcox D, Ruiz F: Familial chondrocalcinosis in the Chiloe Islands, Chile. Ann Rheum Dis. 1975, 34: 260-268. 10.1136/ard.34.3.260.
Riestra JL, Sanchez A, Rodriguez-Valverde V, Alonso JL, de la Hera M, Merino J: Radiographic features of hereditary articular chondrocalcinosis. A comparative study with the sporadic type. Clin Exp Rheumatol. 1988, 6: 369-372.
Doherty M, Hamilton E, Henderson J, Misra H, Dixey J: Familial chondrocalcinosis due to calcium pyrophosphate dihydrate crystal deposition in English families. Br J Rheumatol. 1991, 30: 10-15. 10.1093/rheumatology/30.1.10.
Williams CJ, Zhang Y, Timms A, Bonavita G, Caeiro F, Broxholme J, Cuthbertson J, Jones Y, Marchegiani R, Reginato A, Russell RG, Wordsworth BP, Carr AJ, Brown MA: Autosomal dominant familial calcium pyrophosphate dihydrate deposition disease is caused by mutation in the transmembrane protein ANKH. Am J Hum Genet. 2002, 71: 985-991. 10.1086/343053.
Baldwin CT, Farrer LA, Adair R, Dharmavaram R, Jimenez S, Anderson L: Linkage of early-onset osteoarthritis and chondrocalcinosis to human chromosome 8q. Am J Hum Genet. 1995, 56: 692-697.
Zhang Y, Johnson K, Russell RG, Wordsworth BP, Carr AJ, Terkeltaub RA, Brown MA: Association of sporadic chondrocalcinosis with a -4-basepair G-to-A transition in the 5’-untranslated region of ANKH that promotes enhanced expression of ANKH protein and excess generation of extracellular inorganic pyrophosphate. Arthritis Rheum. 2005, 52: 1110-1117. 10.1002/art.20978.
Zhang W, Neame R, Doherty S, Doherty M: Relative risk of knee chondrocalcinosis in siblings of index cases with pyrophosphate arthropathy. Ann Rheum Dis. 2004, 63: 969-973. 10.1136/ard.2003.015206.
Valdes AM, De Wilde G, Doherty SA, Lories RJ, Vaughn FL, Laslett LL, Maciewicz RA, Soni A, Hart DJ, Zhang W: The Ile585Val TRPV1 variant is involved in risk of painful knee osteoarthritis. Ann Rheum Dis. 2011, 70: 1556-1561. 10.1136/ard.2010.148122.
Robertson J, Zhang W, Liu JJ, Muir KR, Maciewicz RA, Doherty M: Radiographic assessment of the index to ring finger ratio (2D:4D) in adults. J Anat. 2008, 212: 42-48.
Peach CA, Zhang Y, Dunford JE, Brown MA, Carr AJ: Cuff tear arthropathy: evidence of functional variation in pyrophosphate metabolism genes. Clin Orthop Relat Res. 2007, 462: 67-72.
Vistoropsky Y, Keter M, Malkin I, Trofimov S, Kobyliansky E, Livshits G: Contribution of the putative genetic factors and ANKH gene polymorphisms to variation of circulating calciotropic molecules, PTH and BGP. Hum Mol Genet. 2007, 16: 1233-1240. 10.1093/hmg/ddm071.
Alizadeh BZ, Njajou OT, Hazes JM, Hofman A, Slagboom PE, Pols HA, van Duijn CM: The H63D variant in the HFE gene predisposes to arthralgia, chondrocalcinosis and osteoarthritis. Ann Rheum Dis. 2007, 66: 1436-1442. 10.1136/ard.2006.063099.
Timms AE, Sathananthan R, Bradbury L, Athanasou NA, Wordsworth BP, Brown MA: Genetic testing for haemochromatosis in patients with chondrocalcinosis. Ann Rheum Dis. 2002, 61: 745-747. 10.1136/ard.61.8.745.
Goseki-Sone M, Sogabe N, Fukushi-Irie M, Mizoi L, Orimo H, Suzuki T, Nakamura H, Orimo H, Hosoi T: Functional analysis of the single nucleotide polymorphism (787T>C) in the tissue-nonspecific alkaline phosphatase gene associated with BMD. J Bone Miner Res. 2005, 20: 773-782.
Suk EK, Malkin I, Dahm S, Kalichman L, Ruf N, Kobyliansky E, Toliat M, Rutsch F, Nürnberg P, Livshits G: Association of ENPP1 gene polymorphisms with hand osteoarthritis in a Chuvasha population. Arthritis Res Ther. 2005, 7: R1082-R1090. 10.1186/ar1786.
Valli-Jaakola K, Suviolahti E, Schalin-Jäntti C, Ripatti S, Silander K, Oksanen L, Salomaa V, Peltonen L, Kontula K: Further evidence for the role of ENPP1 in obesity: association with morbid obesity in Finns. Obesity (Silver Spring). 2008, 16: 2113-2119. 10.1038/oby.2008.313.
Benyamin B, McRae AF, Zhu G, Gordon S, Henders AK, Palotie A, Peltonen L, Martin NG, Montgomery GW, Whitfield JB, Visscher PM: Variants in TF and HFE explain approximately 40% of genetic variation in serum-transferrin levels. Am J Hum Genet. 2009, 84: 60-65. 10.1016/j.ajhg.2008.11.011.
R Development Core Team: R: A Language and Environment for Statistical Computing. 2010, Vienna, Austria: R Foundation for Statistical Computing, Retrieved from http://www.R-project.org
Barrett JC, Fry B, Maller J, Daly MJ: Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005, 21: 263-265. 10.1093/bioinformatics/bth457.
Fauvert D, Brun-Heath I, Lia-Baldini AS, Bellazi L, Taillandier A, Serre JL, de Mazancourt P, Mornet E: Mild forms of hypophosphatasia mostly result from dominant negative effect of severe alleles or from compound heterozygosity for severe and moderate alleles. BMC Med Genet. 2009, 10: 51.
Zhang Y, Brown MA, Peach C, Russell G, Wordsworth BP: Investigation of the role of ENPP1 and TNAP genes in chondrocalcinosis. Rheumatology (Oxford). 2007, 46: 586-589.
Ho AM, Johnson MD, Kingsley DM: Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000, 289: 265-270. 10.1126/science.289.5477.265.
Gurley KA, Reimer RJ, Kingsley DM: Biochemical and genetic analysis of ANK in arthritis and bone disease. Am J Hum Genet. 2006, 79: 1017-1029. 10.1086/509881.
Pendleton A, Johnson MD, Hughes A, Gurley KA, Ho AM, Doherty M, Dixey J, Gillet P, Loeuille D, McGrath R, Reginato A, Shiang R, Wright G, Netter P, Williams C, Kingsley DM: Mutations in ANKH cause chondrocalcinosis. Am J Hum Genet. 2002, 71: 933-940. 10.1086/343054.
Acknowledgments
AstraZeneca UK funded the GOAL study sample and data collection. The Arthritis Research Council provided infrastructure support during the GOAL study (grant 14581) and for part of the NOAC study. Genotyping for the NOAC study was funded by the EU FP7 large collaborative project grant 200800 TREAT-OA.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
RM is an employee of AstraZeneca (London, UK), owns shares in that company, and was involved as a scientific collaborator in the GOAL study. She is named on a patent application for a gene associated with osteoarthritis. The other authors declare that they have no competing interests. AstraZeneca had no role in the study design, data analysis, or drafting of the manuscript.
Authors’ contributions
AA, RM, and WZ were involved in study conception and design and in analysis and interpretation of the data. KM and MD were involved in study conception and design and in acquisition, analysis, and interpretation of the data. AMV was involved in study conception and design and in acquisition, analysis, and interpretation of the data and carried out the meta-analysis. SD was involved in study conception, data acquisition, and radiographic scoring. All authors were involved in drafting the article or revising it for important intellectual content. All authors read and approved the final version of the manuscript.
Electronic supplementary material
13075_2013_4109_MOESM1_ESM.doc
Additional file 1: Table S1: Genotypes frequencies of ankylosis human (ANKH) and high ferritin (HFE) single-nucleotide polymorphisms (SNPs) in Genetics of Osteoarthritis and Lifestyle (GOAL) and Nottingham Osteoarthritis Case-Control (NOAC) studies. (DOC 72 KB)
13075_2013_4109_MOESM2_ESM.doc
Additional file 2: Table S2: Published minor allele frequency (MAF) of selected single-nucleotide polymorphisms (SNPs) from the 1000 Genomes Project phase 1. Genotype data from 1,094 individuals from across the world (http://www.1000genomes.org/node/506). (DOC 74 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Abhishek, A., Doherty, S., Maciewicz, R. et al. The association between ANKH promoter polymorphism and chondrocalcinosis is independent of age and osteoarthritis: results of a case–control study. Arthritis Res Ther 16, R25 (2014). https://doi.org/10.1186/ar4453
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar4453