
Abstract
Osteoarthritis (OA) is a major cause of disability in the adult population. As a progressive degenerative joint disorder, OA is characterized by cartilage damage, changes in the subchondral bone, osteophyte formation, muscle weakness, and inflammation of the synovium tissue and tendon. Although OA has long been viewed as a primary disorder of articular cartilage, subchondral bone is attracting increasing attention. It is commonly reported to play a vital role in the pathogenesis of OA. Subchondral bone sclerosis, together with progressive cartilage degradation, is widely considered as a hallmark of OA. Despite the increase in bone volume fraction, subchondral bone is hypomineralized, due to abnormal bone remodeling. Some histopathological changes in the subchondral bone have also been detected, including microdamage, bone marrow edema-like lesions and bone cysts. This review summarizes basic features of the osteochondral junction, which comprises subchondral bone and articular cartilage. Importantly, we discuss risk factors influencing subchondral bone integrity. We also focus on the microarchitectural and histopathological changes of subchondral bone in OA, and provide an overview of their potential contribution to the progression of OA. A hypothetical model for the pathogenesis of OA is proposed.
Similar content being viewed by others


Introduction
Osteoarthritis (OA) is a common leading cause of pain and disability in the aging population. As a slowly progressive degenerative joint disorder, OA is characterized by cartilage damage, changes in the subchondral bone, osteophyte formation, muscle weakness, and inflammation of the synovium tissue and tendon [1].
Although OA has long been considered as a primary disorder of articular cartilage, the contribution of subchondral bone to the physiopathology of OA is arousing interest [2]. Subchondral bone deterioration is commonly associated with articular cartilage defects [3], and subchondral bone sclerosis, together with progressive cartilage degradation, is widely considered as a hallmark of OA [4, 5]. Despite the increase in the number of trabeculae and bone volume, subchondral bone is hypomineralized and of inferior quality, as a consequence of abnormal local high bone turnover [6]. Some histopathological changes in the subchondral bone have also been detected, including microdamage, bone marrow edema-like lesions and bone cysts [7–9].
In this review, we summarize basic features of a functional joint unit comprised of subchondral bone and articular cartilage. We also discuss factors that influence the integrity of subchondral bone. Importantly, we focus on the microarchitectural and histopathological changes of subchondral bone in OA, and provide an overview of their potential contribution to the progression of OA.
The structure and function of subchondral bone
OA is considered as an organ disease that affects the whole joint. Subchondral bone plays a crucial role in the initiation and progression of OA [10]. Although ‘subchondral bone’ has been defined in a number of ways, the term most often refers to the bony components lying distal to calcified cartilage [3, 4]. Subchondral bone can be separated into two distinct anatomic entities: subchondral bone plate and subchondral trabecular bone [11] (Figure 1).

The structure of articular cartilage and subchondral bone in a normal human joint. CC, calcified cartilage; NCC, non-calcified cartilage; SBP, subchondral bone plate; STB, subchondral trabecular bone. Arrows denote the tidemark; the dotted line indicates the cement line.
Subchondral bone plate is a thin cortical lamella, lying immediately beneath the calcified cartilage [12]. This cortical endplate is not an impenetrable structure, but possesses a marked porosity. It is invaded by channels that provide a direct link between articular cartilage and subchondral trabecular bone. A surprisingly high number of arterial and venous vessels, as well as nerves, penetrate through the channels and send tiny branches into calcified cartilage [3, 13]. The distribution and intensity of the channels depend not only on aging, but also on the magnitude of the compressive forces transmitting through cartilage and subchondral bone within and between joints [12]. These channels are preferentially concentrated in the heavily stressed areas of the joint. Channel shape and diameter also differs with the thickness of the cortical plate. Channels are narrower and form a tree-like mesh in regions where the subchondral plate is thicker, while they tend to be wider and resemble ampullae where the plate is thinner [12].
Arising from subchondral bone plate is the supporting trabeculae, which comprises subchondral trabecular bone, together with deeper bone structure [3]. Subchondral trabecular bone exerts important shock-absorbing and supportive functions in normal joints, and may also be important for cartilage nutrient supply and metabolism [10]. Relative to the subchondral bone plate, subchondral trabecular bone is more porous and metabolically active, containing blood vessels, sensory nerves, and bone marrow [2]. Subchondral trabecular bone has an inhomogeneous structure that varies with the distance from the articular surface. It exhibits significant structural and mechanical anisotropy; that is, the bone trabeculae shows preferential spatial orientation and parallelism [14].
Subchondral bone is a very dynamic structure and is uniquely adapted to the mechanical forces imposed across the joint. In addition to bone density patterns and mechanical properties, subchondral bone also dynamically adjusts trabecular orientation and scale parameters in a precise relationship with principal stress [15]. Mechanical stress also modifies the contour and shape of subchondral bone by means of bone modeling and remodeling [16].
The interaction between subchondral bone and articular cartilage
Articular cartilage overlies subchondral bone, and provides a vital function of maintaining homeostasis of the joint environment. It encompasses superficial non-calcified cartilage and deeper calcified cartilage. Calcified cartilage is permeable to small molecule transport, and plays an important role in the biochemical interaction between non-calcified cartilage and subchondral bone [17]. It is separated from non-calcified cartilage by a boundary called the ‘tidemark’, a dynamic structure that appears as a basophilic line in histological sections. The tidemark represents the mineralization front of calcified cartilage, and provides a gradual transition between the two dissimilar cartilage regions [18]. Continuous collagen fibrils cross the tidemark, indicating the strong link between non-calcified and calcified cartilage [3]. There is also a sharp borderline between calcified cartilage and subchondral bone, called the ‘cement line’ [19]. Unlike the tidemark, however, no continuous collagen fibrils cross the cement line.
Given the intimate contact between articular cartilage and subchondral bone, they form a closely composited functional unit called the ‘osteochondral junction’ [2] (Figure 1). The osteochondral junction is peculiarly complex, and consists of a deeper layer of non-calcified cartilage, the tidemark, calcified cartilage, the cement line and subchondral bone [18]. Alterations of either tissue will modulate the properties and functions of other parts of the osteochondral junction [20]. There is intensive biomechanical and biochemical cross-talk across this region that may play a role in maintenance and degeneration of the joint.
Subchondral bone and cartilage are dynamic stress-bearing structures that play complementary roles in load-bearing of joints [17]. Subchondral bone supports overlying articular cartilage and distributes mechanical loads across joint surfaces with a gradual transition in stress and strain. Stiffened and less pliable subchondral bone could transmit increased loads to overlying cartilage, leading to secondary cartilage damage and degeneration [11]. The load transmitted to underlying bone will also be substantially increased after articular cartilage damage or loss [21].
Although biomechanical coupling between subchondral bone and cartilage is well established, the biochemical interaction remains comparatively undefined. The permeability of calcified cartilage and subchondral bone plates allows crossover communication, and provides connecting channels between subchondral bone and cartilage [17]. In vivo studies showed that prostaglandins, leukotrienes and various growth factors released by osteoblasts during subchondral bone remodeling could reach overlying articular cartilage [22]. Accordingly, inflammatory and osteoclast stimulation factors released by articular cartilage may also lead to subchondral bone deterioration through increased bone remodeling in OA [5, 23].
Factors affecting subchondral bone integrity
OA is a progressive degenerative joint disease with different etiologies, and multifaceted risk factors have been suggested for the onset of OA, which include genetic predisposition, gender, aging, obesity, physical activity, previous joint injury, joint malalignment and abnormal joint shape [24]. These factors are reported to specifically influence subchondral bone.
Genetic predisposition
OA has been widely considered to be a polygenic disease, having an important hereditary component [25]. Inheritance studies involving family groups and twin pairs have revealed a considerable genetic contribution to the development of OA, with heritability estimates ranging from 39% to 78% at different joints [26–28].
Most of the candidate OA-associated genes reported to date relate to joint development and structural components of the joint [29]. Malformations in joint structure (for example, developmental dysplasia of the hip (DDH) and femoroacetabular impingement) caused by relative genetic mutations could lead to abnormal subchondral bone and contribute substantially to the susceptibility to OA by altering joint biomechanics [30–32].
In a transgenic Del1 mouse harboring a mutated Col2a1 gene (the type II collagen gene), augmentation of matrix metalloproteinase-13, cysts and sclerosis were detected in the subchondral bone of the knee joint at an early age [33, 34]. Mice with Col9a1 gene inactivation (the type IX collagen gene) were also reported to prematurely develop OA, with fibrillation or cartilage erosion extending to subchondral bone [35]. However, the impact of genetic predisposition on the integrity of subchondral bone in humans has not been extensively investigated.
Gender
Men are reported to have a higher prevalence of OA than women before the age of 50 years, but after this age the prevalence is higher in women, which coincides with menopause [36–38]. Accordingly, estrogen deficiency occurring with menopause has been associated with an increased incidence and severity of OA in women [39]. Postmenopausal women taking estrogen replacement therapy exhibit a reduced risk of hip and knee OA, compared with those not taking it [40, 41].
In animal experiments, estrogen depletion by ovariectomy has been related to high bone remodeling, structure deterioration and weakened biomechanical properties in subchondral trabecular bone of joints [42, 43]. Estrogen depletion could also enhance subchondral bone plate thinning, especially when an additional OA trigger is applied [44]. In a cynomolgus macaque model, estrogen replacement therapy was reported to decrease bone remodeling and preserve bone mass in subchondral bone [45]. A cross-sectional study has also shown that elderly women receiving estrogen had a significantly decreased prevalence of knee OA- related subchondral bone attrition and bone marrow edema-like abnormalities [46].
Aging
Aging is considered as a primary risk factor for OA, due to loss of normal bone structure and accumulation of bone microdamage [47]. The microarchitecture of subchondral trabecular bone in joints has a significant dependence on aging, which is characterized by a decrease in trabecular thickness and bone volume fraction, loss of connectivity, increased trabecular separation and bone marrow space volume, transformation of trabeculae from plate-like into rod-like, and an increase in anisotropy degree [48].
However, opinions differ and Crane and colleagues [49] suggested that OA changed the close relationship between age and trabecular bone structure. To illustrate, bone volume fraction of trabecular bone in the proximal femur in those with OA did not depend on age. Further, Perilli and colleagues [50] recently demonstrated that neither structural parameters nor mechanical properties of trabecular bone in the osteoarthritic femoral head depend on age. This phenomenon may be due to altered mechanical factors in the joints with OA, which inhibit age-related bone loss when mechanical stresses are relatively higher [51].
Obesity
Obesity is a significant risk factor for OA, especially in the knee [52]. However, the relationship between obesity and OA in the hand and hip remains controversial [53, 54]. Traditionally, increased compressive stresses on weight-bearing joints due to increased body weight and fat mass is thought to influence obesity-associated OA progression [55]. Recently, attention has shifted to the potential influence of metabolic dysfunction on the progression of obesity-associated OA [56]. It has been suggested that the early stage of obesity-associated OA is more strongly influenced by body weight and fat mass, while metabolic dysfunction is more important in the late stage of OA in which cartilage degeneration becomes more severe [57].
A matched case-control cohort study evaluated the relationship between obesity and bone strength and microarchitecture in postmenopausal women. The increase of absolute values of all bone parameters was not in proportion to excess weight, leading to relative bone fragility [58]. The negative effect of obesity on bone health may be attributed to inflammatory cytokines, increased abnormal fat acid and adipokines [59]. Specifically, Dequeker and colleagues [60] speculated that subchondral bone stiffness was greater due to increased biomechanical loads in obese individuals and this rendered bone less able to cope with higher impact loads. However, the precise impact of obesity on the microarchitecture and pathology of subchondral bone requires further investigation.
Physical activity
Numerous studies have investigated the relationship between physical activity and OA. However, whether physical exercise alone induces OA remains highly controversial. In general, moderate bearing loading exercise protects against OA [61], while non-physiological loading or joint abuse is detrimental and increases the risk of developing OA [62].
In one cross-sectional study of runners, tibia diaphyseal volumetric bone mineral content, cortical area and polar moment of resistance were highest in sprinters, followed in descending order by middle and long distance runners, race-walkers and controls. This was considered to be due to an adaption to ‘exercise specific peak forces’ experienced during running [63]. In a canine experiment, running enhanced bone remodeling in subchondral bone regions, resulting in thicker subchondral bone plate and higher trabecular volume [64]. Subchondral bone responds to the stress of exercise by increasing bone formation and density in an attempt to increase strength [65]. Exercise during the critical years of growth and development may be instrumental for subchondral bone modeling, and might assist in reducing the risk of OA later in life [66].
However, exercise may have varying effects on joints, depending on the mode, rate, intensity and duration of the activity [14]. Abnormal impulsive loading was reported to induce high bone remodeling, microdamage and vascular invasion in subchondral bone, which eventually reduced its elasticity and led to subchondral bone sclerosis [9, 67, 68]. Racehorses running long distances at high speeds exhibited tremendous changes within the subchondral bone in the carpal and metacarpophalangeal joints, including osteochondral fragmentation and fracture, subchondral bone necrosis and sclerosis [69, 70].
Joint injury
Joint injuries, including intra-articular fractures, meniscal tears, ligamentous damages, and traumatic cartilage injuries, all increase the risk of progressive joint degeneration that causes post-traumatic OA [71].
Joint injuries not only lead to cartilage degeneration, but also have negative effects on subchondral bone. In a mouse model, intra-articular fractures caused subchondral bone thickening and sclerosis in the knee [72]. In a canine model, transarticular impact below the joint fracture threshold led to subchondral bone bruises [73]. Based on clinical experience and epidemiologic studies, many OA animal models have been developed through cruciate ligament transection and meniscus destabilization, in which abnormal changes of subchondral bone are detected [74, 75].
In humans, meniscal damages may lead to subchondral bone pathological changes, including increased bone mineral density, bone cysts and bone marrow lesions [76, 77]. Traumatic ligament injuries also jeopardized subchondral bone integrity, causing microdamage [78]. Radiological evidence also supports that traumatic cartilage injuries have a close relationship with subchondral bone damage (for example, subchondral edema) [79].
Joint malalignment
OA is a quantitative or qualitative abnormality in intra-articular stress that may be caused by a host of factors, such as joint malalignment. The mechanical abnormality overwhelms innate physiologic mechanisms for repairing damaged joint tissues [24].
In a population study of 2,644 knees, both valgus and varus alignment, resulting from genetic, developmental, and traumatic factors, were found to be highly associated with the development of OA [80]. It is not the malalignment per se that is detrimental to the joint, but the excessive concentration of stress on the articular cartilage and underlying bone. Malalignment seems to be associated with subchondral bone attrition [81]. Increased mechanical stress by malalignment would give rise to microcracks of subchondral bone, leading to abnormal bone remodeling [82]. In a longitudinal study of the knee, intra-osseous lesions were widely detected in subchondral bone adjacent to abnormal, malaligned joints [83].
Abnormal joint shape
Joint shape has been considered as a key predictor of OA for five decades, and joint shape is in turn altered by the disease, leading to an intricate interplay between joint shape and OA [84]. Joint shape is under tight genetic control, including genes such as those encoding bone morphogenetic proteins or members of the Wnt signaling family [85]. A cross-sectional survey across a wide age range (22 to 93 years), reported that hip dysplasia-associated malformations (for example, DDH, acetabular dysplasia, pistol-grip deformity) constituted significant risk factors for the progression of hip OA [86].
Subchondral bone is widely believed to be able to adapt its structure to acting loads as well as to joint shape [87]. During the development of hip dysplasia observed in a canine model, abnormal weight bearing forces caused subcondral bone fractures and subsequent sclerosis in both femoral head and acetabulum [31]. In a study of human femoral heads with late-phase DDH, synchrotron radiation micro-computed tomography identified many large and small cysts in sclerotic subchondral bone [88].
Microarchitectural changes of subchondral bone in osteoarthritis
Despite the focus on the contribution of subchondral bone to the pathogenesis of OA for over four decades, there remains a controversy over its role: is it a trigger factor or a secondary consequence of cartilage degeneration [3]? Irrespective of the precise mechanism, subchondral bone is believed to play a vital role in OA pathogenesis. Subchondral sclerosis is commonly considered an indisputable sign of OA [77]. However, some studies suggest that different microarchitectural alterations of subchondral bone occur during different stages of OA; subchondral sclerosis may be observed only during more advanced stages of OA [89].
In early stages of OA in humans, elevated bone remodeling and subchondral bone loss was observed, and was considered as a determinant of OA progression [6]. In a rabbit model with preceding osteoporosis, subchondral bone microstructural damage by increased remodeling aggravated experimental OA [23]. Temporal subchondral bone loss in early OA was also documented in a number of different animal models [78, 89, 90]. Specifically, thinning and increased porosity of the subchondral bone plate were detected, which are testified to be strongly associated with cartilage damage [89]. However, the subchondral bone plate remained thin in the feline OA knee joint [78], while other animals showed a subsequent thickening of the cortical plate with OA progression [77, 90]. In the underlying subchondral trabecular bone, increased trabecular separation, decreased bone volume fraction and trabecular thickness in subchondral trabecular bone were detected in these animal models, in contrast with previous concepts [77, 78, 89, 90]. A high incidence of microdamage (highly associated with subchondral bone stiffening and cartilage degeneration) was also reported in the subchondral bone of patients with early OA [91].
Although the underlying mechanism for the increased bone turnover and structural deterioration in the early phase of OA is not fully understood, several factors have been implicated, including microdamage repair [92], increased vascularity stimulated by angiogenic factors [93] and enhanced bone-cartilage crosstalk via increased subchondral plate pores [17, 18]. Further, it has been suggested that elevated bone remodeling and its associated stimulated vascularity are indispensable for the progression of OA, rather than subchondal sclerosis [4].
In the late stage of OA, microarchitectural characteristics of subchondral bone are elevated apparent density, increased bone volume, thickening of subchondral bone plate, increased trabecular thickness, decrease of trabecular separation and bone marrow spacing, and transformation of trabeculae from rod-like into plate-like [94]. The overlying calcified cartilage is also thickened, with advancement and duplication of the tidemark (Figure 2), which contributes to articular cartilage thinning and deterioration [20].

Reduplicated tidemarks in a human joint with osteoarthritis. (A) Histological and (B) backscattered scanning electron microscope manifestations of reduplicated tidemarks in a human joint with osteoarthritis. CC, calcified cartilage; NCC, non-calcified cartilage; SB, subchondral bone. Arrows denote reduplicated tidemarks.
Despite increased bone volume density in the so-called ‘sclerotic’ subchondral bone, its mineralization is reduced and lower than that in normal or even osteoporotic joints [95]. Although collagen synthesis is elevated in subchondral bone, the deposited collagen is hypomineralized and has a markedly reduced calcium-to-collagen ratio [96]. When tested mechanically, subchondral bone stiffness is also low, based on its higher volume fraction [95]. One theory is that bone volume density increases as a mechanoregulatory bone adaptation/compensation in response to decreased mineralization and reduced bone stiffness [97]. Decreased mineralization and reduced bone stiffness result from accelerated bone turnover in OA joints. The lowered subchondral bone modulus may contribute to cartilage degeneration [98], challenging the previous concept that increased subchondral stiffness drives the process of cartilage damage in OA [91].
Bone marrow edema-like lesions of subchondral bone in osteoarthritis
Bone marrow edema-like lesions (BMELs), which are strongly associated with pain among patients with OA, are frequently identified by magnetic resonance imaging (MRI) in patients with progressive OA [99]. BMELs are also observed in the healthy, asymptomatic population, and predict an increased risk of OA [100]. The exact pathogenesis of BMELs is unclear. However, damaged cartilage, an inflammatory reaction to cartilage breakdown products or other factors in intruded synovial fluid, and microtraumatic changes associated with altered biomechanics, may contribute to the formation of BMELs [101].
The term ‘bone marrow edema’ was introduced in 1988 by Wilson and colleagues [102] and was used to describe bone marrow hyperintensive signal lesions using T2-weighted MRI. It is increasingly common for radiologists to describe the abnormal signal lesion as bone marrow edema [103]. However, edema is not a major constituent of the so-called ‘bone marrow edema’ [8]. There are insufficient histologic data available on the MRI-detected BMELs in osteoarthritic joints (Table 1). BMELs are normally present in the sclerotic subchondral bone area, with increased bone volume fraction and increased trabecular thickness [104].
BMELs are consistently and fundamentally involved in development of OA, and are considered as an important risk factor for structural deterioration [8, 107]. Focal cartilage lesions are preferentially located in proximity to BMELs, and the level of cartilage degradation is proportional to BMEL signal intensity [111]. BMELs also have a profound relationship with subchondral bone cysts (SBCs), which could develop in pre-existing regions of BMELs [112].
Subchondral bone cysts in osteoarthritis
The cavitary lesions in subchondral bone, which are normally referred to as ‘subchondral bone cysts’, are commonly reported in patients with OA [113] (Figure 3A, B). Recent evidence suggests that patients with SBCs had greater disease severity and pain, and a higher risk of joint replacement [113].

Radiological and histological characterization of subchondral bone cysts in osteoarthritic joint. (A) Two-dimensional image of a subchondral bone cyst (SBC; dotted line) in a bone cylinder taken from the primary compressive region of osteoarthritic femoral head, from micro-computed tomography scan. (B) Three-dimensional reconstruction of the SBC (blue), from micro-computed tomography scan. (C) Osteoclastic bone resorption (arrowheads) in the trabeculae surrounding a SBC, from backscattered scanning electron microscope scan. (D) Histological features of new bone formation (arrows) on the surface of the trabeculae surrounding a SBC.
Plewes first identified SBCs in subchondral bone adjacent to osteoarthritic joint surfaces in 1940 [114]. Despite the continuous focus on SBCs in OA over seven decades, the exact etiology and pathogenesis of these lesions are still unclear. There are two main conflicting hypotheses that explain the origin of SBCs in OA. The ‘synovial fluid intrusion’ theory proposes that synovial fluid intrudes into subchondral bone and leads to formation of SBCs, which is due to the breach of the osteochondral junction [115]. The ‘bone contusion’ theory suggests that SBCs originate from the necrotic lesions in subchondral bone, which are induced by abnormal mechanical stress and subsequent microcracks, edema and focal bone resorption [116].
However, the term ‘subchondral bone cysts’ is not accurate, as the cavitary lesions in subchondral bone do not have an epithelial lining and are not uniformly fluid filled [117]. As such, they are also described as ‘intra-osseous lesions’, ‘pseudo-cysts’ or ‘geodes’ [117, 118]. SBCs are composed of fibroconnective tissue that may initially contain fluid but ossify in later stages [7]. SBCs appear as well-defined areas of fluid signal on MRI, which correspond to well-defined lucent areas with sclerotic rims on radiographic images [112]. They appear at sites of greatest cartilage loss, in both human [113] and animal models [7]. SBCs are also shown to be associated with high bone mineralization and bone turnover [88]. Osteoclastic bone resorption, activated osteoblasts and new bone formation were detected to be present surrounding SBCs in OA [119, 120] (Figure 3C, D).
Microdamage of calcified cartilage and subchondral bone in osteoarthritis
Microdamage is an important determinant of bone quality. It is widely detected in peri-articular mineralized tissues in osteoarthritic joints, including calcified cartilage, subchondral bone plate and trabecular bone [67]. It is normally induced by overloading [121] and appears in two different forms: linear microcracks and diffuse microdamage [122] (Figure 4).

Linear microcracks (arrows) and diffuse microdamage (arrowheads) in human trabecular bone bulk-stained with basic fuchsin, under confocal microscope with fluorescent light. Images kindly provided by Dr Julia S Kuliwaba, Discipline of Anatomy and Pathology, School of Medical Sciences, The University of Adelaide, Adelaide, Australia.
Linear microcracks, defined as short interstitial cracks, play a vital role in the initiation and progression of OA [9]. In general, microcracks have two main functions: first, they act as a nidus for the initiation of bone remodeling [92]; and second, they provide a conduit for the physiological communication between cartilage and subchondral bone, with more catabolic agents crossing the osteochondral junction, which may be accelerated by accompanying vascular invasion and cutting cones into cartilage [4]. The mechanism for the promotion of bone remodeling by microcracks may involve damage to osteocyte canalicular processes and subsequent osteocyte apoptosis, which may induce osteoclastic resorption and microcrack repair [92]. The high bone turnover induced by microcracks leads to the thickening of subchodral plates and calcified cartilage with tidemark advancement, which eventually causes thinning of cartilage [123].
Diffuse microdamage, present in the form of a range of submicron-sized cracks, is also detected in subchondral trabecular bone in osteoarthritic joints [124]. It is primarily located in tensile regions and contributes to the deterioration of bone mechanical properties [125]. However, diffuse damage has a different biomechanical response, compared with linear microcracks. Diffuse microdamage does not lead to osteocyte apoptosis, nor does it activate focal bone remodeling activities to remove and replace the damaged area [122].
Conclusion
Subchondral bone is an intricate structure consisting of a dome-like subchondral plate and underlying trabeculae, which enjoys a close biomechanical and biochemical relationship with overlying cartilage. Subchondral bone plays a vital role in the pathogenesis of OA. Strong evidence associates subchondral bone alterations with cartilage damage and loss in OA. At the microstructure level, subchondral bone changes are not consistent between the initiation and progression periods of OA. Subchondral sclerosis is widely considered as a main feature of late-stage OA, while early-stage OA is characterized by a thinning subchondral plate with increased porosity and deteriorated subchondral trabeculae with decreased bone density. Histopathological alterations in the subchondral bone, including BMELs, SBCs and microdamage, are also highly associated with the progression of OA.
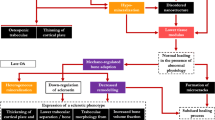
Despite the numerous pathophysiological alterations detected in subchondral bone with OA, we still lack a clear understanding of the mechanisms underpinning these phenomena and how these different aspects are interrelated to each other. Based on the current state of knowledge, one hypothesis for the pathogenesis of OA emerges (Figure 5). Subchondral bone plays an important role in the pathogenesis of OA, manifesting both microarchitectural and histopathological changes (BMELs, SBCs and microdamage). These histopathological changes not only have intimate interactions with each other, but also have a close relationship with bone remodeling that would subsequently lead to microarchitectural changes in the subchondral bone and the overlying cartilage.

Hypothetical model of osteoarthritis (OA) pathogenesis. Normal subchondral bone suffering from a non-physiological strain (induced by risk factors) starts a pathological cascade reaction, leading to osteoarthritic changes in different tissues. In early-stage OA, subchondral plate becomes thinner and more porous, together with initial cartilage degeneration. Subchondral trabecular bone also deteriorates, with increased separation and thinner trabeculae. At the same time, microdamage begins to appear in both calcified cartilage and subchondral bone, which will persist throughout the whole pathological process. In late-stage OA, calcified cartilage and subchondral plate become thicker, with reduplicated tidemarks and progressive non-calcified cartilage damage. Subchondral trabecular bone becomes sclerotic. The sclerosis of periarticular mineralized tissues may be a biomechanical compensational adaptation to the widespread cysts and microdamage in subchondral bone, which render subchondral bone structure more fragile.
As an important pathological lesion, SBCs have been underestimated in the pathogenesis of OA and should be closely considered more seriously in future research. SBCs may expand upwards, and damage subchondral plate and calcified cartilage, facilitating more biochemical catabolic molecule transportation between subchondral bone and cartilage. This would contribute to structural change across the whole joint. In addition, the inhomogeneity of the subchondral plate and calcified cartilage, which is caused by intruding SBCs, microdamage and penetrating pores, could lead to high tensile and shear stresses at the bone-cartilage interface. The abnormal stress distribution of the bone-cartilage interface will degrade the overlying cartilage and cartilage damage could, in turn, influence subchondral bone deterioration. This vicious circle leads to the progression of OA. Collectively, the optimal strategy may therefore be to promote the maintenance of subchondral bone integrity.
Given that the importance of subchondral bone in OA has been recognized for over four decades, the progression in this area is far from satisfying. The fundamental science and pathophysiology of subchondral bone in OA warrants further investigation. It is only through a better understanding and appreciation of the role of this often overlooked structure in the development and progression of OA that we will be able to effectively prevent, diagnose and treat this disease.
Abbreviations
- BMEL:
-
Bone marrow edema-like lesion
- DDH:
-
Developmental dysplasia of the hip
- MRI:
-
Magnetic resonance imaging
- OA:
-
Osteoarthritis
- SBC:
-
Subchondral bone cyst.
References
Grynpas MD, Alpert B, Katz I, Lieberman I, Pritzker KP: Subchondral bone in osteoarthritis. Calcif Tissue Int. 1991, 49: 20-26. 10.1007/BF02555898.
Suri S, Walsh DA: Osteochondral alterations in osteoarthritis. Bone. 2012, 51: 204-211. 10.1016/j.bone.2011.10.010.
Madry H, van Dijk CN, Mueller-Gerbl M: The basic science of the subchondral bone. Knee Surg Sports Traumatol Arthrosc. 2010, 18: 419-433. 10.1007/s00167-010-1054-z.
Burr DB, Gallant MA: Bone remodelling in osteoarthritis. Nat Rev Rheumatol. 2012, 8: 665-673. 10.1038/nrrheum.2012.130.
Henrotin Y, Pesesse L, Sanchez C: Subchondral bone and osteoarthritis: biological and cellular aspects. Osteoporos Int. 2012, 23: S847-S851. 10.1007/s00198-012-2162-z.
Bettica P, Cline G, Hart DJ, Meyer J, Spector TD: Evidence for increased bone resorption in patients with progressive knee osteoarthritis: longitudinal results from the Chingford study. Arthritis Rheum. 2002, 46: 3178-3184. 10.1002/art.10630.
McErlain DD, Ulici V, Darling M, Gati JS, Pitelka V, Beier F, Holdsworth DW: An in vivo investigation of the initiation and progression of subchondral cysts in a rodent model of secondary osteoarthritis. Arthritis Res Ther. 2012, 14: R26-10.1186/ar3727.
Felson DT, McLaughlin S, Goggins J, LaValley MP, Gale ME, Totterman S, Li W, Hill C, Gale D: Bone marrow edema and its relation to progression of knee osteoarthritis. Ann Intern Med. 2003, 139: 330-336. 10.7326/0003-4819-139-5_Part_1-200309020-00008.
Burr DB, Radin EL: Microfractures and microcracks in subchondral bone: are they relevant to osteoarthrosis?. Rheum Dis Clin North Am. 2003, 29: 675-685. 10.1016/S0889-857X(03)00061-9.
Castaneda S, Roman-Blas JA, Largo R, Herrero-Beaumont G: Subchondral bone as a key target for osteoarthritis treatment. Biochem Pharmacol. 2012, 83: 315-323. 10.1016/j.bcp.2011.09.018.
Goldring M, Goldring S: Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann NY Acad Sci. 2010, 1192: 230-237. 10.1111/j.1749-6632.2009.05240.x.
Milz S, Putz R: Quantitative morphology of the subchondral plate of the tibial plateau. J Anat. 1994, 185: 103-110.
Holmdahl DE, Ingelmark BE: The contact between the articular cartilage and the medullary cavities of the bone. Acta Orthop Scand. 1950, 20: 156-165. 10.3109/17453675009043414.
Holopainen JT, Brama PA, Halmesmaki E, Harjula T, Tuukkanen J, van Weeren PR, Helminen HJ, Hyttinen MM: Changes in subchondral bone mineral density and collagen matrix organization in growing horses. Bone. 2008, 43: 1108-1114. 10.1016/j.bone.2008.07.254.
Walker WT, Kawcak CE, Hill AE: Medial femoral condyle morphometrics and subchondral bone density patterns in thoroughbred racehorses. Am J Vet Res. 2013, 74: 691-699. 10.2460/ajvr.74.5.691.
Goldring SR: Alterations in periarticular bone and cross talk between subchondral bone and articular cartilage in osteoarthritis. Ther Adv Musculoskelet Dis. 2012, 4: 249-258. 10.1177/1759720X12437353.
Pan J, Zhou X, Li W, Novotny JE, Doty SB, Wang L: In situ measurement of transport between subchondral bone and articular cartilage. J Orthop Res. 2009, 27: 1347-1352. 10.1002/jor.20883.
Lyons TJ, McClure SF, Stoddart RW, McClure J: The normal human chondro-osseous junctional region: evidence for contact of uncalcified cartilage with subchondral bone and marrow spaces. BMC Musculoskelet Disord. 2006, 7: 52-10.1186/1471-2474-7-52.
Imhof H, Breitenseher M, Kainberger F, Rand T, Trattnig S: Importance of subchondral bone to articular cartilage in health and disease. Top Magn Reson Imaging. 1999, 10: 180-192. 10.1097/00002142-199906000-00002.
Goldring SR: Role of bone in osteoarthritis pathogenesis. Med Clin North Am. 2009, 93: 25-35. 10.1016/j.mcna.2008.09.006. xv
Neogi T, Nevitt M, Niu J, Sharma L, Roemer F, Guermazi A, Lewis CE, Torner J, Javaid K, Felson D: Subchondral bone attrition may be a reflection of compartment-specific mechanical load: the MOST Study. Ann Rheum Dis. 2010, 69: 841-844. 10.1136/ard.2009.110114.
Lajeunesse D, Reboul P: Subchondral bone in osteoarthritis: a biologic link with articular cartilage leading to abnormal remodeling. Curr Opin Rheumatol. 2003, 15: 628-633. 10.1097/00002281-200309000-00018.
Bellido M, Lugo L, Roman-Blas JA, Castaneda S, Caeiro JR, Dapia S, Calvo E, Largo R, Herrero-Beaumont G: Subchondral bone microstructural damage by increased remodelling aggravates experimental osteoarthritis preceded by osteoporosis. Arthritis Res Ther. 2010, 12: R152-10.1186/ar3103.
Brandt KD, Dieppe P, Radin E: Etiopathogenesis of osteoarthritis. Med Clin North Am. 2009, 93: 1-24. 10.1016/j.mcna.2008.08.009. xv
Chapman K, Valdes AM: Genetic factors in OA pathogenesis. Bone. 2012, 51: 258-264. 10.1016/j.bone.2011.11.026.
Spector TD, Cicuttini F, Baker J, Loughlin J, Hart D: Genetic influences on osteoarthritis in women: a twin study. BMJ. 1996, 312: 940-943. 10.1136/bmj.312.7036.940.
Bijkerk C, Houwing-Duistermaat JJ, Valkenburg HA, Meulenbelt I, Hofman A, Breedveld FC, Pols HA, van Duijn CM, Slagboom PE: Heritabilities of radiologic osteoarthritis in peripheral joints and of disc degeneration of the spine. Arthritis Rheum. 1999, 42: 1729-1735. 10.1002/1529-0131(199908)42:8<1729::AID-ANR23>3.0.CO;2-H.
Jordan JM, Kraus VB, Hochberg MC: Genetics of osteoarthritis. Curr Rheumatol Rep. 2004, 6: 7-13. 10.1007/s11926-004-0078-0.
Doherty M: How important are genetic factors in osteoarthritis?. J Rheumatol Suppl. 2004, 70: 22-27.
Sandell LJ: Etiology of osteoarthritis: genetics and synovial joint development. Nat Rev Rheumatol. 2012, 8: 77-89.
Fries CL, Remedios AM: The pathogenesis and diagnosis of canine hip dysplasia: a review. Can Vet J. 1995, 36: 494-502.
Kannu P, Bateman JF, Belluoccio D, Fosang AJ, Savarirayan R: Employing molecular genetics of chondrodysplasias to inform the study of osteoarthritis. Arthritis Rheum. 2009, 60: 325-334. 10.1002/art.24251.
Saamanen AK, Salminen HJ, Dean PB, De Crombrugghe B, Vuorio EI, Metsaranta MP: Osteoarthritis-like lesions in transgenic mice harboring a small deletion mutation in type II collagen gene. Osteoarthritis Cartilage. 2000, 8: 248-257. 10.1053/joca.2000.0298.
Salminen H, Perala M, Lorenzo P, Saxne T, Heinegard D, Saamanen AM, Vuorio E: Up-regulation of cartilage oligomeric matrix protein at the onset of articular cartilage degeneration in a transgenic mouse model of osteoarthritis. Arthritis Rheum. 2000, 43: 1742-1748. 10.1002/1529-0131(200008)43:8<1742::AID-ANR10>3.0.CO;2-U.
Allen KD, Griffin TM, Rodriguiz RM, Wetsel WC, Kraus VB, Huebner JL, Boyd LM, Setton LA: Decreased physical function and increased pain sensitivity in mice deficient for type IX collagen. Arthritis Rheum. 2009, 60: 2684-2693. 10.1002/art.24783.
Wilson MG, Michet CJ, Ilstrup DM, Melton LJ: Idiopathic symptomatic osteoarthritis of the hip and knee: a population-based incidence study. Mayo Clin Proc. 1990, 65: 1214-1221. 10.1016/S0025-6196(12)62745-1.
McAlindon TE, Snow S, Cooper C, Dieppe PA: Radiographic patterns of osteoarthritis of the knee joint in the community: the importance of the patellofemoral joint. Ann Rheum Dis. 1992, 51: 844-849. 10.1136/ard.51.7.844.
Oliveria SA, Felson DT, Reed JI, Cirillo PA, Walker AM: Incidence of symptomatic hand, hip, and knee osteoarthritis among patients in a health maintenance organization. Arthritis Rheum. 1995, 38: 1134-1141. 10.1002/art.1780380817.
Wluka AE, Cicuttini FM, Spector TD: Menopause, oestrogens and arthritis. Maturitas. 2000, 35: 183-199. 10.1016/S0378-5122(00)00118-3.
Nevitt MC, Felson DT: Sex hormones and the risk of osteoarthritis in women: epidemiological evidence. Ann Rheum Dis. 1996, 55: 673-676. 10.1136/ard.55.9.673.
Zhang Y, McAlindon TE, Hannan MT, Chaisson CE, Klein R, Wilson PW, Felson DT: Estrogen replacement therapy and worsening of radiographic knee osteoarthritis: the Framingham Study. Arthritis Rheum. 1998, 41: 1867-1873. 10.1002/1529-0131(199810)41:10<1867::AID-ART20>3.0.CO;2-W.
Sniekers YH, Weinans H, Bierma-Zeinstra SM, van Leeuwen JP, van Osch GJ: Animal models for osteoarthritis: the effect of ovariectomy and estrogen treatment - a systematic approach. Osteoarthritis Cartilage. 2008, 16: 533-541. 10.1016/j.joca.2008.01.002.
Yang JH, Woo DG, Tyagi V, Oh KJ: Architectural changes in subchondral bone and its compliance in response to estrogen and progesterone: a micro-computed tomography study. Tissue Eng Regener Med. 2011, 8: 380-389.
Sniekers YH, Weinans H, van Osch GJ, van Leeuwen JP: Oestrogen is important for maintenance of cartilage and subchondral bone in a murine model of knee osteoarthritis. Arthritis Res Ther. 2010, 12: R182-10.1186/ar3148.
Ham KD, Carlson CS: Effects of estrogen replacement therapy on bone turnover in subchondral bone and epiphyseal metaphyseal cancellous bone of ovariectomized cynomolgus monkeys. J Bone Miner Res. 2004, 19: 823-829. 10.1359/jbmr.040309.
Carbone LD, Nevitt MC, Wildy K, Barrow KD, Harris F, Felson D, Peterfy C, Visser M, Harris TB, Wang BW, Kritchevsky SB, Health, Aging and Body Composition Study: The relationship of antiresorptive drug use to structural findings and symptoms of knee osteoarthritis. Arthritis Rheum. 2004, 50: 3516-3525. 10.1002/art.20627.
Shane Anderson A, Loeser RF: Why is osteoarthritis an age-related disease?. Best Pract Res Clin Rheumatol. 2010, 24: 15-26. 10.1016/j.berh.2009.08.006.
Ding M, Odgaard A, Linde F, Hvid I: Age-related variations in the microstructure of human tibial cancellous bone. J Orthop Res. 2002, 20: 615-621. 10.1016/S0736-0266(01)00132-2.
Crane GJ, Fazzalari NL, Parkinson IH, Vernon-Roberts B: Age-related changes in femoral trabecular bone in arthrosis. Acta Orthop Scand. 1990, 61: 421-426. 10.3109/17453679008993554.
Perilli E, Baleani M, Ohman C, Baruffaldi F, Viceconti M: Structural parameters and mechanical strength of cancellous bone in the femoral head in osteoarthritis do not depend on age. Bone. 2007, 41: 760-768. 10.1016/j.bone.2007.07.014.
Martin B: Aging and strength of bone as a structural material. Calcif Tissue Int. 1993, 53: S34-S39. 10.1007/BF01673400. discussion S39-S40
Lohmander LS: Gerhardsson de Verdier M, Rollof J, Nilsson PM, Engstrom G: Incidence of severe knee and hip osteoarthritis in relation to different measures of body mass: a population-based prospective cohort study. Ann Rheum Dis. 2009, 68: 490-496. 10.1136/ard.2008.089748.
Lievense AM, Bierma-Zeinstra SM, Verhagen AP, van Baar ME, Verhaar JA, Koes BW: Influence of obesity on the development of osteoarthritis of the hip: a systematic review. Rheumatology (Oxford). 2002, 41: 1155-1162. 10.1093/rheumatology/41.10.1155.
Grotle M, Hagen KB, Natvig B, Dahl FA, Kvien TK: Obesity and osteoarthritis in knee, hip and/or hand: an epidemiological study in the general population with 10 years follow-up. BMC Musculoskelet Disord. 2008, 9: 132-10.1186/1471-2474-9-132.
Guilak F, Fermor B, Keefe FJ, Kraus VB, Olson SA, Pisetsky DS, Setton LA, Weinberg JB: The role of biomechanics and inflammation in cartilage injury and repair. Clin Orthop Relat Res. 2004, 423: 17-26.
Killock D: Osteoarthritis: the influence of obesity on OA - does size matter or is metabolic dysfunction more important?. Nat Rev Rheumatol. 2012, 8: 61-
Mooney RA, Sampson ER, Lerea J, Rosier RN, Zuscik MJ: High-fat diet accelerates progression of osteoarthritis after meniscal/ligamentous injury. Arthritis Res Ther. 2011, 13: R198-10.1186/ar3529.
Sornay-Rendu E, Boutroy S, Vilayphiou N, Claustrat B, Chapurlat RD: In obese postmenopausal women, bone microarchitecture and strength are not commensurate to greater body weight: the Os des Femmes de Lyon (OFELY) study. J Bone Miner Res. 2013, 28: 1679-1687. 10.1002/jbmr.1880.
Cao JJ: Effects of obesity on bone metabolism. J Orthop Surg Res. 2011, 6: 30-10.1186/1749-799X-6-30.
Dequeker J, Goris P, Uytterhoeven R: Osteoporosis and osteoarthritis (osteoarthrosis). Anthropometric distinctions. JAMA. 1983, 249: 1448-1451. 10.1001/jama.1983.03330350024020.
Wijayaratne SP, Teichtahl AJ, Wluka AE, Hanna F, Bell R, Davis SR, Adams J, Cicuttini FM: The determinants of change in patella cartilage volume - a cohort study of healthy middle-aged women. Rheumatology (Oxford). 2008, 47: 1426-1429. 10.1093/rheumatology/ken244.
Sun HB: Mechanical loading, cartilage degradation, and arthritis. Ann N Y Acad Sci. 2010, 1211: 37-50. 10.1111/j.1749-6632.2010.05808.x.
Wilks DC, Winwood K, Gilliver SF, Kwiet A, Chatfield M, Michaelis I, Sun LW, Ferretti JL, Sargeant AJ, Felsenberg D, Rittweger J: Bone mass and geometry of the tibia and the radius of master sprinters, middle and long distance runners, race-walkers and sedentary control participants: a pQCT study. Bone. 2009, 45: 91-97. 10.1016/j.bone.2009.03.660.
Oettmeier R, Arokoski J, Roth AJ, Helminen HJ, Tammi M, Abendroth K: Quantitative study of articular cartilage and subchondral bone remodeling in the knee joint of dogs after strenuous running training. J Bone Miner Res. 1992, 7: S419-S424. 10.1002/jbmr.5650071410.
Young DR, Richardson DW, Markel MD, Nunamaker DM: Mechanical and morphometric analysis of the third carpal bone of thoroughbreds. Am J Vet Res. 1991, 52: 402-409.
Helminen HJ, Hyttinen MM, Lammi MJ, Arokoski JP, Lapvetelainen T, Jurvelin J, Kiviranta I, Tammi MI: Regular joint loading in youth assists in the establishment and strengthening of the collagen network of articular cartilage and contributes to the prevention of osteoarthrosis later in life: a hypothesis. J Bone Miner Metab. 2000, 18: 245-257. 10.1007/PL00010638.
Burr DB, Schaffler MB: The involvement of subchondral mineralized tissues in osteoarthrosis: quantitative microscopic evidence. Microsc Res Tech. 1997, 37: 343-357. 10.1002/(SICI)1097-0029(19970515)37:4<343::AID-JEMT9>3.0.CO;2-L.
Radin EL, Burr DB, Fyhrie D, Brown TD, Boyd RD: Characteristics of joint loading as it applies to osteoarthrosis. Biomechanics of Diarthrodial Joints, Volume 1. Edited by: Ratcliffe A, Woo SLY, Mow VC. 1990, New York: Springer New York, 437-451.
Colon JL, Bramlage LR, Hance SR, Embertson RM: Qualitative and quantitative documentation of the racing performance of 461 thoroughbred racehorses after arthroscopic removal of dorsoproximal first phalanx osteochondral fractures (1986-1995). Equine Vet J. 2000, 32: 475-481.
Parente EJ, Richardson DW, Spencer P: Basal sesamoidean fractures in horses: 57 cases (1980-1991). J Am Vet Med Assoc. 1993, 202: 1293-1297.
Gelber AC, Hochberg MC, Mead LA, Wang NY, Wigley FM, Klag MJ: Joint injury in young adults and risk for subsequent knee and hip osteoarthritis. Ann Intern Med. 2000, 133: 321-328. 10.7326/0003-4819-133-5-200009050-00007.
Furman BD, Strand J, Hembree WC, Ward BD, Guilak F, Olson SA: Joint degeneration following closed intraarticular fracture in the mouse knee: a model of posttraumatic arthritis. J Orthop Res. 2007, 25: 578-592. 10.1002/jor.20331.
Mrosek EH, Lahm A, Erggelet C, Uhl M, Kurz H, Eissner B, Schagemann JC: Subchondral bone trauma causes cartilage matrix degeneration: an immunohistochemical analysis in a canine model. Osteoarthritis Cartilage. 2006, 14: 171-178. 10.1016/j.joca.2005.08.004.
Hayami T, Pickarski M, Zhuo Y: Characterization of articular cartilage and subchondral bone changes in the rat anterior cruciate ligament transection and meniscectomized models of osteoarthritis. Bone. 2006, 38: 234-243. 10.1016/j.bone.2005.08.007.
Boyd SK, Muller R, Leonard T, Herzog W: Long-term periarticular bone adaptation in a feline knee injury model for post-traumatic experimental osteoarthritis. Osteoarthritis Cartilage. 2005, 13: 235-242. 10.1016/j.joca.2004.11.004.
Lo GH, Niu J, McLennan CE, Kiel DP, McLean RR, Guermazi A, Genant HK, McAlindon TE, Hunter DJ: Meniscal damage associated with increased local subchondral bone mineral density: a Framingham study. Osteoarthritis Cartilage. 2008, 16: 261-267. 10.1016/j.joca.2007.07.007.
Wang Y, Wluka AE, Pelletier JP, Martel-Pelletier J, Abram F, Ding C, Cicuttini FM: Meniscal extrusion predicts increases in subchondral bone marrow lesions and bone cysts and expansion of subchondral bone in osteoarthritic knees. Rheumatology (Oxford). 2010, 49: 997-1004. 10.1093/rheumatology/keq034.
Meyer EG, Baumer TG, Slade JM, Smith WE, Haut RC: Tibiofemoral contact pressures and osteochondral microtrauma during anterior cruciate ligament rupture due to excessive compressive loading and internal torque of the human knee. Am J Sports Med. 2008, 36: 1966-1977. 10.1177/0363546508318046.
Rubin DA, Harner CD, Costello JM: Treatable chondral injuries in the knee: frequency of associated focal subchondral edema. AJR Am J Roentgenol. 2000, 174: 1099-1106. 10.2214/ajr.174.4.1741099.
Brouwer GM, van Tol AW, Bergink AP, Belo JN, Bernsen RM, Reijman M, Pols HA, Bierma-Zeinstra SM: Association between valgus and varus alignment and the development and progression of radiographic osteoarthritis of the knee. Arthritis Rheum. 2007, 56: 1204-1211. 10.1002/art.22515.
Heijink A, Gomoll AH, Madry H, Drobnic M, Filardo G, Espregueira-Mendes J, Van Dijk CN: Biomechanical considerations in the pathogenesis of osteoarthritis of the knee. Knee Surg Sports Traumatol Arthrosc. 2012, 20: 423-435. 10.1007/s00167-011-1818-0.
Kraus VB, McDaniel G, Worrell TW, Feng S, Vail TP, Varju G, Coleman RE: Association of bone scintigraphic abnormalities with knee malalignment and pain. Ann Rheum Dis. 2009, 68: 1673-1679. 10.1136/ard.2008.094722.
Hunter DJ, Zhang Y, Niu J, Goggins J, Amin S, LaValley MP, Guermazi A, Genant H, Gale D, Felson DT: Increase in bone marrow lesions associated with cartilage loss: a longitudinal magnetic resonance imaging study of knee osteoarthritis. Arthritis Rheum. 2006, 54: 1529-1535. 10.1002/art.21789.
Baker-LePain JC, Lane NE: Relationship between joint shape and the development of osteoarthritis. Curr Opin Rheumatol. 2010, 22: 538-543. 10.1097/BOR.0b013e32833d20ae.
Herrero-Beaumont G, Roman-Blas JA, Castaneda S, Jimenez SA: Primary osteoarthritis no longer primary: three subsets with distinct etiological, clinical, and therapeutic characteristics. Semin Arthritis Rheum. 2009, 39: 71-80. 10.1016/j.semarthrit.2009.03.006.
Jacobsen S, Sonne-Holm S: Hip dysplasia: a significant risk factor for the development of hip osteoarthritis. A cross-sectional survey. Rheumatology (Oxford). 2005, 44: 211-218. 10.1093/rheumtology/keh436.
Swieszkowski W, Tuan BH, Kurzydlowski KJ, Hutmacher DW: Repair and regeneration of osteochondral defects in the articular joints. Biomol Eng. 2007, 24: 489-495. 10.1016/j.bioeng.2007.07.014.
Chiba K, Nango N, Kubota S, Okazaki N, Taguchi K, Osaki M, Ito M: Relationship between microstructure and degree of mineralization in subchondral bone of osteoarthritis: a synchrotron radiation microCT study. J Bone Miner Res. 2012, 27: 1511-1517. 10.1002/jbmr.1609.
Intema F, Hazewinkel HA, Gouwens D, Bijlsma JW, Weinans H, Lafeber FP, Mastbergen SC: In early OA, thinning of the subchondral plate is directly related to cartilage damage: results from a canine ACLT-meniscectomy model. Osteoarthritis Cartilage. 2010, 18: 691-698. 10.1016/j.joca.2010.01.004.
Batiste DL, Kirkley A, Laverty S, Thain LM, Spouge AR, Holdsworth DW: Ex vivo characterization of articular cartilage and bone lesions in a rabbit ACL transection model of osteoarthritis using MRI and micro-CT. Osteoarthritis Cartilage. 2004, 12: 986-996. 10.1016/j.joca.2004.08.010.
Radin EL, Paul IL, Tolkoff MJ: Subchondral bone changes in patients with early degenerative joint disease. Arthritis Rheum. 1970, 13: 400-405. 10.1002/art.1780130406.
Verborgt O, Gibson GJ, Schaffler MB: Loss of osteocyte integrity in association with microdamage and bone remodeling after fatigue in vivo. J Bone Miner Res. 2000, 15: 60-67. 10.1359/jbmr.2000.15.1.60.
Luyten FP, Lories RJ, Verschueren P, de Vlam K, Westhovens R: Contemporary concepts of inflammation, damage and repair in rheumatic diseases. Best Pract Res Clin Rheumatol. 2006, 20: 829-848. 10.1016/j.berh.2006.06.009.
Ding M: Microarchitectural adaptations in aging and osteoarthrotic subchondral bone issues. Acta Orthop Suppl. 2010, 81: 1-53.
Li B, Aspden RM: Composition and mechanical properties of cancellous bone from the femoral head of patients with osteoporosis or osteoarthritis. J Bone Miner Res. 1997, 12: 641-651. 10.1359/jbmr.1997.12.4.641.
Mansell JP, Bailey AJ: Abnormal cancellous bone collagen metabolism in osteoarthritis. J Clin Invest. 1998, 101: 1596-1603. 10.1172/JCI867.
Day J, Ding M, van der Linden J: A decreased subchondral trabecular bone tissue elastic modulus is associated with pre-arthritic cartilage damage. J Orthop Res. 2001, 19: 914-918. 10.1016/S0736-0266(01)00012-2.
Karsdal MA, Leeming DJ, Dam EB, Henriksen K, Alexandersen P, Pastoureau P, Altman RD, Christiansen C: Should subchondral bone turnover be targeted when treating osteoarthritis?. Osteoarthritis Cartilage. 2008, 16: 638-646. 10.1016/j.joca.2008.01.014.
Link TM, Steinbach LS, Ghosh S, Ries M, Lu Y, Lane N, Majumdar S: Osteoarthritis: MR imaging findings in different stages of disease and correlation with clinical findings. Radiology. 2003, 226: 373-381. 10.1148/radiol.2262012190.
Wluka AE, Hanna F, Davies-Tuck M, Wang Y, Bell RJ, Davis SR, Adams J, Cicuttini FM: Bone marrow lesions predict increase in knee cartilage defects and loss of cartilage volume in middle-aged women without knee pain over 2 years. Ann Rheum Dis. 2009, 68: 850-855. 10.1136/ard.2008.092221.
Garnero P, Peterfy C, Zaim S, Schoenharting M: Bone marrow abnormalities on magnetic resonance imaging are associated with type II collagen degradation in knee osteoarthritis: a three-month longitudinal study. Arthritis Rheum. 2005, 52: 2822-2829. 10.1002/art.21366.
Wilson AJ, Murphy WA, Hardy DC, Totty WG: Transient osteoporosis: transient bone marrow edema?. Radiology. 1988, 167: 757-760.
Lecouvet FE, van de Berg BC, Maldague BE, Lebon CJ, Jamart J, Saleh M, Noel H, Malghem J: Early irreversible osteonecrosis versus transient lesions of the femoral condyles: prognostic value of subchondral bone and marrow changes on MR imaging. AJR Am J Roentgenol. 1998, 170: 71-77. 10.2214/ajr.170.1.9423603.
Hunter DJ, Gerstenfeld L, Bishop G, Davis AD, Mason ZD, Einhorn TA, Maciewicz RA, Newham P, Foster M, Jackson S, Morgan EF: Bone marrow lesions from osteoarthritis knees are characterized by sclerotic bone that is less well mineralized. Arthritis Res Ther. 2009, 11: R11-10.1186/ar2601.
Bergman AG, Willen HK, Lindstrand AL, Pettersson HT: Osteoarthritis of the knee: correlation of subchondral MR signal abnormalities with histopathologic and radiographic features. Skeletal Radiol. 1994, 23: 445-448.
Zanetti M, Bruder E, Romero J, Hodler J: Bone marrow edema pattern in osteoarthritic knees: correlation between MR imaging and histologic findings. Radiology. 2000, 215: 835-840. 10.1148/radiology.215.3.r00jn05835.
Kazakia GJ, Kuo D, Schooler J, Siddiqui S, Shanbhag S, Bernstein G, Horvai A, Majumdar S, Ries M, Li X: Bone and cartilage demonstrate changes localized to bone marrow edema-like lesions within osteoarthritic knees. Osteoarthritis Cartilage. 2013, 21: 94-101. 10.1016/j.joca.2012.09.008.
Nolte-Ernsting CC, Adam G, Buhne M, Prescher A, Gunther RW: MRI of degenerative bone marrow lesions in experimental osteoarthritis of canine knee joints. Skeletal Radiol. 1996, 25: 413-420. 10.1007/s002560050108.
Baird DK, Hathcock JT, Kincaid SA, Rumph PF, Kammermann J, Widmer WR, Visco D, Sweet D: Low-field magnetic resonance imaging of early subchondral cyst-like lesions in induced cranial cruciate ligament deficient dogs. Vet Radiol Ultrasound. 1998, 39: 167-173. 10.1111/j.1740-8261.1998.tb00334.x.
Martig S, Boisclair J, Konar M, Spreng D, Lang J: MRI characteristics and histology of bone marrow lesions in dogs with experimentally induced osteoarthritis. Vet Radiol Ultrasound. 2007, 48: 105-112. 10.1111/j.1740-8261.2007.00213.x.
Zhao J, Li X, Bolbos RI, Link TM, Majumdar S: Longitudinal assessment of bone marrow edema-like lesions and cartilage degeneration in osteoarthritis using 3 T MR T1rho quantification. Skeletal Radiol. 2010, 39: 523-531. 10.1007/s00256-010-0892-6.
Carrino JA, Blum J, Parellada JA, Schweitzer ME, Morrison WB: MRI of bone marrow edema-like signal in the pathogenesis of subchondral cysts. Osteoarthritis Cartilage. 2006, 14: 1081-1085. 10.1016/j.joca.2006.05.011.
Tanamas SK, Wluka AE, Pelletier JP, Martel-Pelletier J, Abram F, Wang Y, Cicuttini FM: The association between subchondral bone cysts and tibial cartilage volume and risk of joint replacement in people with knee osteoarthritis: a longitudinal study. Arthritis Res Ther. 2010, 12: R58-10.1186/ar2971.
Plewes LW: Osteo-arthritis of the hip. Br J Surg. 1940, 27: 682-695. 10.1002/bjs.18002710807.
Durr HD, Martin H, Pellengahr C, Schlemmer M, Maier M, Jansson V: The cause of subchondral bone cysts in osteoarthrosis: a finite element analysis. Acta Orthop Scand. 2004, 75: 554-558. 10.1080/00016470410001411.
Ondrouch AS: Cyst formation in osteoarthritis. J Bone Joint Surg Br. 1963, 45: 755-760.
Bancroft LW, Peterson JJ, Kransdorf MJ: Cysts, geodes, and erosions. Radiol Clin North Am. 2004, 42: 73-87. 10.1016/S0033-8389(03)00165-9.
Resnick D, Niwayama G, Coutts RD: Subchondral cysts (geodes) in arthritic disorders: pathologic and radiographic appearance of the hip joint. AJR Am J Roentgenol. 1977, 128: 799-806. 10.2214/ajr.128.5.799.
Havdrup T, Hulth A, Telhag H: The subchondral bone in osteoarthritis and rheumatoid arthritis of the knee. A histological and microradiographical study. Acta Orthop Scand. 1976, 47: 345-350. 10.3109/17453677608992003.
Sabokbar A, Crawford R, Murray DW, Athanasou NA: Macrophage-osteoclast differentiation and bone resorption in osteoarthrotic subchondral acetabular cysts. Acta Orthop Scand. 2000, 71: 255-261. 10.1080/000164700317411843.
Vener MJ, Thompson RC, Lewis JL, Oegema TR: Subchondral damage after acute transarticular loading: an in vitro model of joint injury. J Orthop Res. 1992, 10: 759-765. 10.1002/jor.1100100603.
Herman BC, Cardoso L, Majeska RJ, Jepsen KJ, Schaffler MB: Activation of bone remodeling after fatigue: differential response to linear microcracks and diffuse damage. Bone. 2010, 47: 766-772. 10.1016/j.bone.2010.07.006.
Kawcak CE, McIlwraith CW, Norrdin RW, Park RD, James SP: The role of subchondral bone in joint disease: a review. Equine Vet J. 2001, 33: 120-126.
Fazzalari NL, Kuliwaba JS, Forwood MR: Cancellous bone microdamage in the proximal femur: influence of age and osteoarthritis on damage morphology and regional distribution. Bone. 2002, 31: 697-702. 10.1016/S8756-3282(02)00906-7.
Vashishth D, Koontz J, Qiu SJ, Lundin-Cannon D, Yeni YN, Schaffler MB, Fyhrie DP: In vivo diffuse damage in human vertebral trabecular bone. Bone. 2000, 26: 147-152. 10.1016/S8756-3282(99)00253-7.
Acknowledgments
The authors thank Prof. Heather A McKay from the Department of Orthopedics, Centre for Hip Health and Mobility, University of British Columbia for critical comments of important intellectual content and Dr Julia S Kuliwaba from Discipline of Anatomy and Pathology, School of Medical Sciences, The University of Adelaide for provision of microdamage images.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Li, G., Yin, J., Gao, J. et al. Subchondral bone in osteoarthritis: insight into risk factors and microstructural changes. Arthritis Res Ther 15, 223 (2013). https://doi.org/10.1186/ar4405
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar4405