
Abstract
Systemic lupus erythematosus (SLE) is a complex disease characterized by numerous autoantibodies and clinical involvement in multiple organ systems. The immunological events triggering the onset and progression of clinical manifestations are also complex and multi-step, including breach of tolerance in the adaptive immune system, amplification of autoimmunity through innate and adaptive immune system dysregulation, and end-organ damage. Studies of murine genetic manipulations and human risk variants have provided important clues to the cellular and molecular pathogenesis of SLE, operating at multiple of these steps. The breakdown of B-cell tolerance is probably a defining and early event in the disease process and may occur by multiple pathways, including alterations in factors that affect B-cell activation thresholds, B-cell longevity, and apoptotic cell processing. Examples of amplification of autoimmunity on the adaptive immune system side include disturbances in B-cell/T-cell collaboration. B cells can also amplify innate immune cell activation via antibody-dependent and antibody-independent mechanisms. Indeed, one of the key amplification loops in SLE is the activation of plasmacytoid dendritic cells via autoantibodies and RNA-containing and DNA-containing immune complexes, which act as Toll-like receptor ligands, stimulating the secretion of large quantities of IFNα. A more recent link between the innate and adaptive immune system in SLE includes the neutrophil, which can be primed by interferon and autoantibodies to release neutrophil extracellular traps as an additional source of immunogenic DNA, histones, and neutrophil proteins. The innate immune system activation then feeds back, driving autoreactive B-cell and T-cell survival and maturation. This self-perpetuating disease cycle creates the opportunity for targeted treatment inventions at multiple steps.
Similar content being viewed by others


Introduction
Systemic lupus erythematosus (SLE) is a complex autoimmune disease with heterogeneity in clinical manifestations and disease course, characterized by pathogenic autoantibody formation, immune complex deposition, and end-organ damage. Despite the fact that the mortality and morbidity of patients with SLE has improved significantly during the last few decades, mortality rates remain approximately three times those of the age-matched and sex-matched population in most studies [1]. The need for more effective therapies with less toxic side effects has propelled interest in targeted biologic therapies based on an expanding understanding about SLE disease pathogenesis. Until recently, this effort has been hampered by the challenges of clinical trial design given the low prevalence of disease, clinical heterogeneity, relapsing-remitting course, and lack of well-established endpoints [2–4]. Despite these difficulties, there have been great strides towards improving lupus clinical trial methodology [4], leading to recent successful outcomes in clinical trials of B-cell-targeted biologics in SLE. Moreover, our understanding about the pathogenesis of SLE has grown substantially in the past decade, leading to an explosion of promising biologic therapies. In this review, we will discuss our current understanding of SLE disease pathogenesis with a focus on B-cell biology and novel interactions between the adaptive and innate immune systems and how this has revealed new treatment targets.
Lessons about SLE disease pathogenesis from genetics
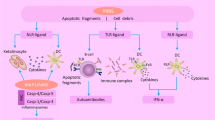
Nearly two decades ago, Wakeland and colleagues proposed a three-checkpoint model for the development of SLE based on their studies dissecting lupus genetic susceptibility using congenic mouse strains [5]. Although like all elegant models this is an oversimplification, their model does provide a very useful framework for understanding the genetics and pathogenesis of SLE. The three stages or events in disease development include: breach of tolerance in the adaptive immune system (loss of tolerance in B cells and T cells), amplification of autoimmunity through innate and adaptive immune system dysregulation, and end-organ damage [6] (Figure 1). These steps are highlighted below with correlations between genetics [7] and disease pathogenesis.

Pathogenesis-driven biologic targets in systemic lupus erythematosus. The three stages in disease development include breach of tolerance in the adaptive immune system (loss of tolerance in B cells and T cells), amplification of autoimmunity through innate and adaptive immune system dysregulation, and end-organ damage. Examples of genetic predispositions (exclusively mouse; blue) at each stage are shown. Disturbances in innate and adaptive immunity in the middle panel contribute to both emergence and progression of disease. NETosis of neutrophils and production of autoantibodies by plasma cells (PC) leads to interferon (IFN) production by plasmacytoid dendritic cells (pDCs). Potential treatment targets are shown. Antibodies against B-cell surface antigens such as CD20 and CD19 can cause death of B cells. Antibodies against other B-cell surface molecules, such as the inhibitory receptor CD22, are alternative targets for both inhibition of B cells and induction of cell death. Inhibition of co-stimulation is another approach; for example, monoclonal antibodies against CD40 ligand or CTLA4-Ig block co-stimulatory signals, thus inhibiting B-cell activation, but also T-cell activation. B-cell survival can be inhibited via monoclonal antibodies against the cytokine B-cell activating factor (BAFF; belimumab) or other approaches to disrupting the BAFF receptor family. The possibility of interfering with other pathways such as Toll-like receptors (TLRs), IFN, and other cytokines with inhibitors and causing direct PC and pDC effects with proteasome inhibitors is also suggested. CD40L, CD40 ligand; mDC, myeloid dendritic cell; NET, neutrophil extracellular trap.
Loss of immunologic tolerance
Because SLE is characterized by the generation of large amounts of autoantibodies directed against chromatin and a variety of other self-antigens, the loss of B-cell tolerance is believed to play a key role in the disease. Evidence that the breakdown of B-cell tolerance occurs very early in SLE and may precede or trigger other immune abnormalities is provided by the demonstration that SLE patients express anti-nuclear antibodies several years before the onset of clinical disease [8]. Although the precise nature of the immune abnormalities leading to loss of tolerance in human SLE remains to be defined, mouse models again provide a useful framework for understanding probable mechanisms. Three broad categories of defects that can lead to a lupus-like phenotype have thus been defined in the mouse and are instructive for thinking about B-cell abnormalities in human SLE. These defects may affect B-cell activation thresholds, B-cell longevity, or apoptotic cell/autoantigen processing.
As an example of altered B-cell activation, Mohan and colleagues further elucidated the mechanism by which the Sle1 susceptibility locus derived from Wakeland's lupus-prone New Zealand Mixed (NZM2410) mice contributes to the development of autoimmunity. A gene within this locus encoding a member of the signaling lymphocyte activation molecule family was found to be highly expressed in immature B cells and altered in these lupus-prone mice in such a way as to impair signaling and impede antigen-driven negative selection [9]. In contrast, other B-cell signaling defects may cause up-regulated signaling, as exemplified by loss of CD22 or inhibitory Fc receptor function. The latter is notable because decreased expression of FcγRIIb has been demonstrated in human lupus memory B cells [10], and genetic polymorphisms in this gene have been associated with human lupus. The observation that genetic variants in other B-cell signaling molecules such as BANK1 and BLK confer an increased risk for lupus in humans provides additional support for the critical role of B-cell signaling abnormalities in disease pathogenesis [7].
A prominent example of alterations in B-cell longevity leading to lupus is provided by B-cell activating factor (BAFF) transgenic mice. This is of great clinical relevance given the recent approval of BAFF blockade in the treatment of human disease [11], as further expanded in another review in this supplement [12].
Finally, the impaired clearance of apoptotic debris may also lead to SLE and does so in part by providing large amounts of self-antigen and immune complexes that deliver stimulatory signals to other cells, including B cells, as detailed later. Examples of genetic manipulations in mice that involve alterations in the clearance or response to apoptotic debris include C1q, Mer receptor tyrosine kinase, and serum amyloid P. In humans, homozygous C1q deficiency is the most highly penetrant complement deficiency leading to SLE. In another parallel with the mouse, genetic variants in human C-reactive protein - a protein that functions in the clearance of apoptotic debris in an analogous way to serum amyloid P - are also associated with an increased risk of lupus [13].
Amplification of autoimmunity
Role of adaptive immunity defects
B-cell/T-cell cross-talk
Disturbances in B-cell/T-cell collaboration are probably critical for amplification of the autoimmune process and clinical expression of disease [14]. For example, Sle3 congenic mice have accumulation of activated T cells - which in combination with the Sle1 interval augments the production of pathogenic autoantibodies, probably through epitope spreading. These mice also have increased activation of dendritic cells, an intrinsic defect that appears to drive the T-cell phenotype. Other data in human SLE indicate that T cells have abnormal intra-cellular signaling, and in murine lupus small molecule kinase inhibitors have clinical benefit. Inhibitors of kinases such as Syk and CaMK4 that are abnormally expressed in T cells of SLE patients may thus present a new therapeutic opportunity [15]. Of note, such an approach may have benefit regardless of whether T-lymphocyte signaling defects are direct or related to disturbances in B-cell/T-cell collaboration or other immune cell abnormalities.
The development of high-affinity plasma cells and memory B cells occurs in germinal center reactions, and disturbances in these follicular as well as extrafollicular immune responses are thought to be a key feature of SLE. For example, Sanz and colleagues have shown that censoring of autoreactive (9G4) B cells is defective in germinal center reactions in SLE [16]. There are a number of essential signals regulating the B-cell/T-cell interaction that may play a role in SLE and provide interesting treatment targets, including soluble factors such as IL-21 and cell-cell interactions via CD40-CD40 ligand and CD28-B7. The latter are co-stimulatory molecule pairs that have already been targeted as treatment approaches for SLE, with variable success [17]. IL-21 is a multi-faceted cytokine that promotes germinal center formation, somatic hypermutation, and plasma cell differentiation, the latter in synergy with BAFF [18, 19]. The role for germinal centers and IL-21 in SLE is further supported by data showing that polymorphisms of both IL-21 and the IL-21 receptor are associated with SLE [20]. IL-21 is produced by a subset of T cells termed T-follicular helper cells that provide critical assistance for follicular and germinal center B cells, inducing activation, differentiation, and antibody production. Excessive activity of T-follicular helper cells in mice induces hyperactive germinal center, breakdown of B-cell tolerance, autoantibody production, and a lupus-like phenotype [21].
Another interesting example of the importance of coordination of B-cell/T-cell interactions for the prevention of autoimmunity was recently provided by an elegant study demonstrating that deficiency of two regulatory molecules, DEF6 and SWAP70 (homologous intracellular signaling proteins in T cells and B cells regulated by IRF4), leads to the development of lupus in mice with simultaneous dysregulation of CD4+ T cell IL-21 production and increased IL-21 B-cell responsiveness [22]. These mice also demonstrate aberrant T-cell production of IL-17, which is interesting given that T-helper type (Th) 17 cells have been implicated in a number of autoimmune diseases, recently including SLE [23]. Th17 cells may have a role in some but not all lupus patients, raising the idea that disease heterogeneity may be defined by genetic or disease manifestation subsets of patients who would respond to anti-IL-17 therapy [24]. Finally, further support for the key role of T-cell abnormalities and B-cell/T-cell collaboration in the pathogenesis of SLE is provided by a number of genetic variants associated with human lupus that appear to affect this interaction, including PTPN22.
Contribution of dysregulated B cells to disease expression
The B-cell compartment is an active treatment target in SLE given the participation of B cells in the immune process at multiple levels. B cells may thus serve as the precursors of antibody-secreting cells, present auto-antigens to T cells, and regulate inflammatory responses and other immune cells through cytokine and chemokine secretion (such as IL-10, IL-6, IFNγ, and lymphotoxin-α). The importance of these latter autoantibody-independent functions has been demonstrated in murine SLE, where B cells have been found to be critical to the development of disease even when they are unable to secrete auto-antibodies [25]. The concept that B cells can play a pathogenic role in lupus independent of serum auto-antibody has been recently supported by our observations that B-cell depletion with anti-CD20 has robust effects in the treatment of murine lupus without changes in autoantibodies [26]. This notion has also been corroborated in humans by our own and others findings that clinical improvement in SLE patients treated with rituximab correlates with B-cell depletion and precedes by several months any decline in serum levels of relevant autoantibodies [27].
One of the key autoantibody-independent functions of B cells is their ability to produce cytokines in a polarized fashion and thus actively modulate both humoral and cellular immune responses [28]. So-called effector B cells (Be1 and Be2), by analogy with Th1/Th2 cells, can participate in feedback regulation of Th cells [29]. The notion that different B-cell subsets may induce separate T-cell subsets has recently been expanded by the demonstration that B1 cells may promote Th1 and Th17 differentiation [30]. B cells also play a key role in the recruitment of CXCR5+ follicular T-helper cells to the germinal center as well as regulation of their function via ICOSL and OX40L [31, 32].
The pathogenic role of B cells in SLE has been highlighted by mouse studies using either B-cell-deficient mice or B-cell depletion [25, 26]. On the contrary, B cells may also prevent or suppress autoimmunity through anti-inflammatory cytokines such as IL-10 and transforming growth factor beta and the expansion of regulatory T cells and/or inhibition of effector T cells [33–36]. At a population level, separate B-cell subsets may exert different functions [37]. Evidence is thus accumulating for regulatory B cells capable of suppressing autoimmunity in different mouse models including SLE [38, 39].
The concept of regulatory B cells is also probably of relevance to human SLE. A number of groups have thus identified a human regulatory B-cell counterpart, although there is still controversy in the field regarding the precise nature of this population [40, 41]. Further, it is important to realize that current B-cell-targeted therapies in SLE, such as rituximab (anti-CD20) and BAFF blockade, do not distinguish between pathogenic and protective B-cell subsets. Along these lines, our group has provided some of the original observations regarding how the timing and quality of B-cell depletion and repopulation after treatment of SLE with rituximab impacts response [42, 43]. We have proposed that a repopulation with transitional B cells, a putative regulatory population, is associated with sustained clinical remission while a quick resurgence of memory cells portends a poor outcome. These observations have recently been confirmed by Emery and colleagues [44]. Understanding the imbalance between opposing B-cell functions in disease and how this imbalance may be restored after targeted biologic therapy is an important area of ongoing research.
The expanding role of innate immunity in SLE: interferon, neutrophils, and more
It is important not to underestimate the role of autoantibodies in SLE disease pathogenesis. Indeed, one explanation for the failure of rituximab in the EXPLORER (anti-CD20 in general lupus) and LUNAR (anti-CD20 in lupus nephritis) trials to meet primary endpoints is the lack of effect on plasma cells and pathogenic auto-antibodies [45, 46]. This observation has raised interest in the possibility of targeting the plasma cell compartment, for example with proteasome inhibitors [47].
In addition to conventional roles of autoantibodies in SLE via type II (antibody-dependent cytotoxicity) and type III (immune complex) mechanisms, intriguing evidence has emerged that autoantibodies can play an active role in propagating the autoimmune process in SLE through B-cell and other immune cell activation involving RNA-containing or DNA-containing autoantigens and Toll-like receptors (TLRs) [48–51]. B-cell stimulation by self-antigen may be magnified by increased sensitivity to TLR activation, as exemplified by the TLR7 duplication that is mediated by the Y-linked autoimmune accelerator lupus susceptibility locus in mice [52]. Notably, plasmacytoid dendritic cells may also be activated by co-stimulation of TLRs and Fc receptors via immune complex binding, stimulating the secretion of large quantities of IFNα, a cytokine with important immune-modulatory functions that include activation and maturation of dendritic cells and stimulation of both T cells and B cells [53]. In combination with TLR7 and TLR9 activation of myeloid dendritic cells to produce BAFF, a feedback loop is generated that triggers further B-cell activation. Notably, one of the mechanisms by which anti-malarials probably exert their effect in SLE is via inhibition of TLR signaling, and novel TLR inhibitors are in active pharmaceutical development.
Additional evidence implicating a prominent role for IFNα activation in SLE and making it an attractive treatment target includes: the demonstration of serum elevations among patients with active SLE [54]; the more recent demonstration that high interferon levels are a heritable risk factor for SLE [55]; induction of auto-immunity with IFNα treatment of malignancy and hepatitis C [56]; and the presence of an IFNα gene expression signature in human SLE [57]. IFNα acts at the center of an amplification loop in the disease, contributing to B-cell abnormalities, promoting the differentiation of activated B cells into plasmablasts [58], and in conjunction with TLR stimulation triggering B-cell expansion [59] and a lowered activation threshold for autoreactive B cells [60]. We have also recently reported a novel role for IFNα activation in the bone marrow of SLE patients by decreasing B-cell lymphopoiesis, contributing to B-cell lymphopenia and theoretically decreasing the stringency of B-cell-negative selection (unpublished data). The importance of TLR and IFN signaling to SLE disease pathogenesis is highlighted by the identification of multiple risk variants in human lupus involving genes in these pathways, including IRAK1, IRF5, and STAT4 [55, 61–63].
When the interferon signature was first demonstrated in SLE, little attention was paid in the field to the immature neutrophil signature that was also described. Recent studies have elucidated how these cells may be important in SLE pathogenesis as a primary source of autoantigen modification and exposure to the immune system [64]. Neutrophil extracellular traps (NETs) are web-like structures released by neutrophils as a means of immobilizing and killing invading microbes that contain DNA, histones, and neutrophil proteins. Sera from SLE patients have recently been described to contain immune complexes of NET-derived self-DNA and the antimicrobial peptides LL37 and human neutrophil peptide that are immunogenic. In particular, FcγRII-mediated endocytosis of self-DNA through TLR9 in the endosomes of activated plasmacytoid dendritic cells is more effective when in complex with these antimicrobial peptides, leading to increased type I interferon release and additional neutrophil priming. In SLE, NETosis appears to be dysregulated due to an expansion of an abnormal neutrophil subset, the fact that type I interferons prime neutrophils for NETosis, and the presence of autoantibodies against LL37 and human neutrophil peptide that enhance NET formation and facilitate further plasmacytoid dendritic cells activation [65, 66]. Interestingly, other studies have demonstrated that active lupus patients have impaired degradation of NETs due to the presence of DNase I inhibitors and anti-NET antibodies [67]. Moreover, NETs can be potent activators of complement, further exacerbating disease [68].
Interestingly, netting neutrophils in SLE can also induce endothelial damage and infiltrate affected skin and kidneys, suggesting a link to both cardiovascular and end-organ damage [69]. Whether NETosis may serve as a biomarker or predictor of tissue damage in SLE and how this pathway can be targeted for treatment of disease remain to be shown.
Local autoimmunity and end-organ damage
Steps 1 and 2 of SLE disease pathogenesis in the adaptive and innate immune system generate pathogenic mediators including autoantibodies, immune complexes, effector B cells, T cells, and myeloid cells. However, additional events and genetic predispositions presumably have the potential to modulate the type and severity of end-organ disease. Key factors that have been shown to affect the severity of kidney disease in mouse models include the complement receptors [70], cytokines such as macrophage colony-stimulating factor and macrophage migration inhibitory factor [71], and chemokines such as monocyte chemoattractant protein-1 [72]. Identified genetic variants likely to affect end-organ damage in human SLE include FcγRIIa and FcγRIIIa [73], ITGAM (functioning in immune complex clearance and cell adhesion/migration) [62], and kallikrein polymorphisms [74].
Conclusion and perspectives
SLE is complex from both the pathogenesis perspective and in terms of the tremendous heterogeneity in clinical manifestations. Although multiple immunologic abnormalities can contribute to the emergence and amplification of disease, the predominant abnormalities will probably vary in different patients.
A recent paper took an interesting approach to categorizing subsets of SLE based on the association of 22 known genetic susceptibility loci and disease characteristics [75]. The authors found that certain subphenotypes such as the presence of anti-dsDNA autoantibodies, immunological abnormalities, young age at onset, hematological disorder, and absence of oral ulcers were associated with cumulative genetic risk scores, whereas others were associated with single genes (for example, HLADR3 and lupus nephritis; ITGAM alleles protecting against arthritis) or no genetic associations (malar rash, serositis, neurologic). This observation highlights the concept that distinct phenotypes of disease may be defined by genetics, although probably in combination with expanded genetic markers and other immunologic biomarkers. For example, a particular cytokine signature or peripheral blood flow cytometry signature may denote a patient with a B-cell-predominant versus Th17-predominant versus interferon-predominant disease.
Given the recent disappointing clinical trial results with targeted B-cell depletion and co-stimulatory blockade, targeting approaches based on biomarker-defined subsets of disease is an attractive future direction. Furthermore, combination therapy directed at multiple steps in disease pathogenesis needs to be considered.
Abbreviations
- BAFF:
-
B-cell activating factor
- FcγR:
-
Fcγ receptor
- IFN:
-
interferon
- IL:
-
interleukin
- NET:
-
neutrophil extracellular trap
- SLE:
-
systemic lupus erythematosus
- Th:
-
T-helper type
- TLR:
-
Toll-like receptor.
References
Bongu A, Chang E, Ramsey-Goldman R: Can morbidity and mortality of SLE be improved?. Best Practice Res Clin Rheumatol. 2002, 16: 313-332. 10.1053/berh.2001.0228.
Gordon C, Bertsias G, Ioannidis JP, Boletis J, Bombardieri S, Cervera R, Dostal C, Font J, Gilboe IM, Houssiau F, Huizinga TW, Isenberg D, Kallenberg CG, Khamashta MA, Piette JC, Schneider M, Smolen JS, Sturfelt G, Tincani A, Van Vollenhoven R, Boumpas DT: EULAR recommendations for points to consider in conducting clinical trials in systemic lupus erythematosus (SLE). Ann Rheum Dis. 2009, 68: 470-476. 10.1136/ard.2007.083022.
Dall'Era M, Wofsy D: Clinical trial design in systemic lupus erythematosus. Curr Opin Rheumatol. 2006, 18: 476-480. 10.1097/01.bor.0000240357.22680.63.
Bertsias G, Gordon C, Boumpas DT: Clinical trials in systemic lupus erythematosus (SLE): lessons from the past as we proceed to the future - the EULAR recommendations for the management of SLE and the use of end-points in clinical trials. Lupus. 2008, 17: 437-442. 10.1177/0961203308090031.
Morel L, Rudofsky UH, Longmate JA, Schiffenbauer J, Wakeland EK: Polygenic control of susceptibility to murine systemic lupus erythematosus. Immunity. 1994, 1: 219-229. 10.1016/1074-7613(94)90100-7.
Kanta H, Mohan C: Three checkpoints in lupus development: central tolerance in adaptive immunity, peripheral amplification by innate immunity and end-organ inflammation. Genes Immun. 2009, 10: 390-396. 10.1038/gene.2009.6.
Cheung YH, Loh C, Pau E, Kim J, Wither J: Insights into the genetic basis and immunopathogenesis of systemic lupus erythematosus from the study of mouse models. Semin Immunol. 2009, 21: 372-382. 10.1016/j.smim.2009.10.005.
Arbuckle M, McClain M, Rubertone M, Scofield R, Dennis G, James J, Harley J: Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003, 349: 1526-1533. 10.1056/NEJMoa021933.
Kumar KR, Li L, Yan M, Bhaskarabhatla M, Mobley AB, Nguyen C, Mooney JM, Schatzle JD, Wakeland EK, Mohan C: Regulation of B cell tolerance by the lupus susceptibility gene Ly108. Science. 2006, 312: 1665-1669. 10.1126/science.1125893.
Mackay M, Stanevsky A, Wang T, Aranow C, Li M, Koenig S, Ravetch JV, Diamond B: Selective dysregulation of the FcγIIB receptor on memory B cells in SLE. J Exp Med. 2006, 203: 2157-2164. 10.1084/jem.20051503.
Navarra SV, Guzman RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, Li EK, Thomas M, Kim HY, Leon MG, Tanasescu C, Nasonov E, Lan JL, Pineda L, Zhong ZJ, Freimuth W, Petri MA: Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011, 377: 721-731. 10.1016/S0140-6736(10)61354-2.
Boneparth A, Davidson A: B-cell activating factor targeted therapy and lupus. Ar thritis Res Therapy. 2012, 14 (Suppl 4): S2-10.1186/ar3920.
Edberg JC, Wu J, Langefeld CD, Brown EE, Marion MC, McGwin G, Petri M, Ramsey-Goldman R, Reveille JD, Frank SG, Kaufman KM, Harley JB, Alarcon GS, Kimberly RP: Genetic variation in the CRP promoter: association with systemic lupus erythematosus. Hum Mol Genet. 2008, 17: 1147-1155. 10.1093/hmg/ddn004.
Chan OT, Madaio MP, Shlomchik NJ: The central and multiple roles of B cells in lupus pathogenesis. Immunol Rev. 1999, 169: 107-121. 10.1111/j.1600-065X.1999.tb01310.x.
Tsokos GC: Systemic lupus erythematosus. N Engl J Med. 2011, 365: 2110-2121. 10.1056/NEJMra1100359.
Cappione A, Anolik JH, Pugh-Bernard A, Barnard J, Dutcher P, Silverman G, Sanz I: Germinal center exclusion of autoreactive B cells is defective in human systemic lupus erythematosus. J Clin Invest. 2005, 115: 3205-3216. 10.1172/JCI24179.
Merrill JT, Burgos-Vargas R, Westhovens R, Chalmers A, D'Cruz D, Wallace DJ, Bae SC, Sigal L, Becker JC, Kelly S, Raghupathi K, Li T, Peng Y, Kinaszczuk M, Nash P: The efficacy and safety of abatacept in patients with non-life-threatening manifestations of systemic lupus erythematosus: results of a twelve-month, multicenter, exploratory, phase IIb, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2010, 62: 3077-3087. 10.1002/art.27601.
Kuchen S, Robbins R, Sims GP, Sheng C, Phillips TM, Lipsky PE, Ettinger R: Essential role of IL-21 in B cell activation, expansion, and plasma cell generation during CD4+ T cell-B cell collaboration. J Immunol. 2007, 179: 5886-5896.
Ettinger R, Sims GP, Robbins R, Withers D, Fischer RT, Grammer AC, Kuchen S, Lipsky PE: IL-21 and BAFF/BLyS synergize in stimulating plasma cell differentiation from a unique population of human splenic memory B cells. J Immunol. 2007, 178: 2872-2882.
Webb R, Merrill JT, Kelly JA, Sestak A, Kaufman KM, Langefeld CD, Ziegler J, Kimberly RP, Edberg JC, Ramsey-Goldman R, Petri M, Reveille JD, Alarcón GS, Vilá LM, Alarcón-Riquelme ME, James JA, Gilkeson GS, Jacob CO, Moser KL, Gaffney PM, Vyse TJ, Nath SK, Lipsky P, Harley JB, Sawalha AH: A polymorphism within IL21R confers risk for systemic lupus erythematosus. Arthritis Rheum. 2009, 60: 2402-2407. 10.1002/art.24658.
Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, Yu D, Domaschenz H, Whittle B, Lambe T, Roberts IS, Copley RR, Bell JI, Cornall RJ, Goodnow CC: A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature. 2005, 435: 452-458. 10.1038/nature03555.
Biswas PS, Gupta S, Stirzaker RA, Kumar V, Jessberger R, Lu TT, Bhagat G, Pernis AB: Dual regulation of IRF4 function in T and B cells is required for the coordination of T-B cell interactions and the prevention of autoimmunity. J Exp Med. 2012, 209: 581-596. 10.1084/jem.20111195.
Doreau A, Belot A, Bastid J, Riche B, Trescol-Biemont MC, Ranchin B, Fabien N, Cochat P, Pouteil-Noble C, Trolliet P, Durieu I, Tebib J, Kassai B, Ansieau S, Puisieux A, Eliaou JF, Bonnefoy-Berard N: Interleukin 17 acts in synergy with B cell-activating factor to influence B cell biology and the pathophysiology of systemic lupus erythematosus. Nat Immunol. 2009, 10: 778-785.
Pathak S, Mohan C: Cellular and molecular pathogenesis of systemic lupus erythematosus: lessons from animal models. Arthritis Res Ther. 2011, 13: 241-10.1186/ar3465.
Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ: A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999, 189: 1639-1648. 10.1084/jem.189.10.1639.
Bekar KW, Owen T, Dunn R, Ichikawa T, Wang W, Wang R, Barnard J, Brady S, Nevarez S, Goldman BI, Kehry M, Anolik JH: Prolonged effects of short-term anti-CD20 B cell depletion therapy in murine systemic lupus erythematosus. Arthritis Rheum. 2010, 62: 2443-2457. 10.1002/art.27515.
Looney RJ, Anolik JH, Campbell D, Felgar RE, Young F, Arend LJ, Sloand J, Rosenblatt J, Sanz I: B cell depletion as a novel treatment for systemic lupus erythematosus: A phase I/II dose-escalation trial of rituximab. Arthritis Rheum. 2004, 50: 2580-2589. 10.1002/art.20430.
Lund FE: Cytokine-producing B lymphocytes - key regulators of immunity. Curr Opin Immunol. 2008, 20: 332-338. 10.1016/j.coi.2008.03.003.
Harris DP, Haynes L, Sayles PC, Duso DK, Eaton SM, Lepak NM, Johnson LL, Swain SL, Lund FE: Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat Immunol. 2000, 1: 475-482. 10.1038/82717.
Zhong X, Gao W, Degauque N, Bai C, Lu Y, Kenny J, Oukka M, Strom TB, Rothstein TL: Reciprocal generation of Th1/Th17 and T(reg) cells by B1 and B2 B cells. Eur J Immunol. 2007, 37: 2400-2404. 10.1002/eji.200737296.
Ebert LM, Horn MP, Lang AB, Moser B: B cells alter the phenotype and function of follicular-homing CXCR5+ T cells. Eur J Immunol. 2004, 34: 3562-3571. 10.1002/eji.200425478.
Akiba H, Takeda K, Kojima Y, Usui Y, Harada N, Yamazaki T, Ma J, Tezuka K, Yagita H, Okumura K: The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J Immunol. 2005, 175: 2340-2348.
Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM: B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002, 3: 944-950.
Mauri C, Gray D, Mushtaq N, Londei M: Prevention of arthritis by interleukin 10-producing B cells. J Exp Med. 2003, 197: 489-501. 10.1084/jem.20021293.
Reichardt P, Dornbach B, Rong S, Beissert S, Gueler F, Loser K, Gunzer M: Naive B cells generate regulatory T cells in the presence of a mature immunologic synapse. Blood. 2007, 110: 1519-1529. 10.1182/blood-2006-10-053793.
Wei B, Velazquez P, Turovskaya O, Spricher K, Aranda R, Kronenberg M, Birnbaumer L, Braun J: Mesenteric B cells centrally inhibit CD4+ T cell colitis through interaction with regulatory T cell subsets. Proc Natl Acad Sci. 2005, 102: 2010-2015. 10.1073/pnas.0409449102.
Anolik JH, Looney RJ, Lund FE, Randall TD, Sanz I: Insights into the heterogeneity of human B cells: diverse functions, roles in autoimmunity, and use as therapeutic targets. Immunol Res. 2009, 45: 144-158. 10.1007/s12026-009-8096-7.
Fillatreau S, Gray D, Anderton AM: Not always the bad guys: B cells as regulators of autoimmune pathology. Nat Rev Immunol. 2008, 8: 391-397. 10.1038/nri2315.
Watanabe R, Ishiura N, Nakashima H, Kuwano Y, Okochi H, Tamaki K, Sato S, Tedder TF, Fujimoto M: Regulatory B cells (B10 cells) have a suppressive role in murine lupus: CD19 and B10 cell deficiency exacerbates systemic autoimmunity. J Immunol. 2010, 184: 4801-4809. 10.4049/jimmunol.0902385.
Blair PA, Norena LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, Mauri C: CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity. 2010, 32: 129-140. 10.1016/j.immuni.2009.11.009.
Iwata Y, Matsushita T, Horikawa M, Dilillo DJ, Yanaba K, Venturi GM, Szabolcs PM, Bernstein SH, Magro CM, Williams AD, Hall RP, St Clair EW, Tedder TF: Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood. 2011, 117: 530-541. 10.1182/blood-2010-07-294249.
Anolik J, Barnard J, Owen T, Zheng B, Kemshett S, Looney J, Sanz I: Delayed memory B cell recovery in peripheral blood and lymphoid tissue in systemic lupus erythematosus after B cell depletion therapy. Arthritis Rheum. 2007, 56: 3044-3056. 10.1002/art.22810.
Palanichamy A, Barnard J, Zheng B, Owen T, Quach T, Wei C, Looney RJ, Sanz I, Anolik JH: Novel human transitional B cell populations revealed by B cell depletion therapy. J Immunol. 2009, 182: 5982-5993. 10.4049/jimmunol.0801859.
Vital EM, Dass S, Buch MH, Henshaw K, Pease CT, Martin MF, Ponchel F, Rawstron AC, Emery P: B cell biomarkers of rituximab responses in systemic lupus erythematosus. Arthritis Rheum. 2011, 63: 3038-3047. 10.1002/art.30466.
Rovin BH, Furie R, Latinis K, Looney RJ, Fervenza FC, Sanchez-Guerrero J, Maciuca R, Zhang D, Garg JP, Brunetta P, Appel G: Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012, 64: 1215-1226. 10.1002/art.34359.
Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, Utset TO, Gordon C, Isenberg DA, Hsieh HJ, Zhang D, Brunetta PG: Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010, 62: 222-233. 10.1002/art.27233.
Ichikawa HT, Conley T, Muchamuel T, Jiang J, Lee S, Owen T, Barnard J, Nevarez S, Goldman BI, Kirk CJ, Looney RJ, Anolik JH: Beneficial effect of novel proteasome inhibitors in murine lupus via dual inhibition of type I interferon and autoantibody-secreting cells. Arthritis Rheum. 2012, 64: 493-503. 10.1002/art.33333.
Bave U, Alm GV, Ronnblom L: The combination of apoptotic U937 cells and lupus IgG is a potent IFN-alpha inducer. J Immunol. 2000, 165: 3519-3526.
Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A: Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors [see comment]. Nature. 2002, 416: 603-607. 10.1038/416603a.
Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD: Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest. 2005, 115: 407-417.
Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A: Activation of autoreactive B cells by CpG dsDNA. Immunity. 2003, 19: 837-847. 10.1016/S1074-7613(03)00323-6.
Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S: Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006, 312: 1669-1672. 10.1126/science.1124978.
Ronnblom L, Alm GV: A pivotal role for the natural interferon alphaproducing cells (plasmacytoid dendritic cells) in the pathogenesis of lupus [comment]. J Exp Med. 2001, 194: F59-F63. 10.1084/jem.194.12.f59.
Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL: Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med. 1979, 301: 5-8. 10.1056/NEJM197907053010102.
Kariuki SN, Franek BS, Kumar AA, Arrington J, Mikolaitis RA, Utset TO, Jolly M, Crow MK, Skol AD, Niewold TB: Trait-stratified genome-wide association study identifies novel and diverse genetic associations with serologic and cytokine phenotypes in systemic lupus erythematosus. Arthritis Res Ther. 2010, 12: R151-10.1186/ar3101.
Kalkner KM, Ronnblom L, Karlsson Parra AK, Bengtsson M, Olsson Y, Oberg K: Antibodies against double-stranded DNA and development of polymyositis during treatment with interferon. QJM. 1998, 91: 393-399. 10.1093/qjmed/91.6.393.
Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V: Interferon and granulopoiesis signatures in systemic lupus erythematosus blood [see comment]. J Exp Med. 2003, 197: 711-723. 10.1084/jem.20021553.
Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J: Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003, 19: 225-234. 10.1016/S1074-7613(03)00208-5.
Bekeredjian-Ding IB, Wagner M, Hornung V, Giese T, Schnurr M, Endres S, Hartmann G: Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN. J Immunol. 2005, 174: 4043-4050.
Uccellini MB, Busconi L, Green NM, Busto P, Christensen SR, Shlomchik MJ, Marshak-Rothstein A, Viglianti GA: Autoreactive B cells discriminate CpG-rich and CpG-poor DNA and this response is modulated by IFN-alpha. J Immunol. 2008, 181: 5875-5884.
Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, de Bakker PI, Le JM, Lee HS, Batliwalla F: STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007, 357: 977-986. 10.1056/NEJMoa073003.
Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ, Langefeld CD, Nath SK, Guthridge JM, Cobb BL, Mirel DB, Marion MC, Williams AH, Divers J, Wang W, Frank SG, Namjou B, Gabriel SB, Lee AT, Gregersen PK, Behrens TW, Taylor KE, Fernando M, Zidovetzki R, Gaffney PM, Edberg JC, Rioux JD, Ojwang JO: Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008, 40: 204-210. 10.1038/ng.81.
Jacob CO, Zhu J, Armstrong DL, Yan M, Han J, Zhou XJ, Thomas JA, Reiff A, Myones BL, Ojwang JO, Kaufman KM, Klein-Gitelman M, McCurdy D, Wagner-Weiner L, Silverman E, Ziegler J, Kelly JA, Merrill JT, Harley JB, Ramsey-Goldman R, Vila LM, Bae SC, Vyse TJ, Gilkeson GS, Gaffney PM, Moser KL, Langefeld CD, Zidovetzki R, Mohan C: Identification of IRAK1 as a risk gene with critical role in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci USA. 2009, 106: 6256-6261. 10.1073/pnas.0901181106.
Dorner T: SLE in 2011: deciphering the role of NETs and networks in SLE. Nat Rev Rheumatol. 2012, 8: 68-70.
Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, Barrat FJ, Banchereau J, Pascual V: Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011, 3: 73ra20-10.1126/scitranslmed.3001201.
Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V, Bassett R, Amuro H, Fukuhara S, Ito T, Liu YJ, Gilliet M: Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011, 3: 73ra19-10.1126/scitranslmed.3001180.
Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A: Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. 2010, 107: 9813-9818. 10.1073/pnas.0909927107.
Leffler J, Martin M, Gullstrand B, Tyden H, Lood C, Truedsson L, Bengtsson AA, Blom AM: Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol. 2012, 188: 3522-3531. 10.4049/jimmunol.1102404.
Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, Rubin CJ, Zhao W, Olsen SH, Klinker M, Shealy D, Denny MF, Plumas J, Chaperot L, Kretzler M, Bruce AT, Kaplan MJ: Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011, 187: 538-552. 10.4049/jimmunol.1100450.
Wenderfer SE, Wang H, Ke B, Wetsel RA, Braun MC: C3a receptor deficiency accelerates the onset of renal injury in the MRL/lpr mouse. Mol Immunol. 2009, 46: 1397-1404. 10.1016/j.molimm.2008.12.004.
Hoi AY, Hickey MJ, Hall P, Yamana J, O'Sullivan KM, Santos LL, James WG, Kitching AR, Morand EF: Macrophage migration inhibitory factor deficiency attenuates macrophage recruitment, glomerulonephritis, and lethality in MRL/lpr mice. J Immunol. 2006, 177: 5687-5696.
Tesch GH, Maifert S, Schwarting A, Rollins BJ, Kelley VR: Monocyte chemoattractant protein 1-dependent leukocytic infiltrates are responsible for autoimmune disease in MRL-Fas(lpr) mice. J Exp Med. 1999, 190: 1813-1824. 10.1084/jem.190.12.1813.
Magnusson V, Johanneson B, Lima G, Odeberg J, Alarcon-Segovia D, Alarcon-Riquelme ME: Both risk alleles for FcγRIIA and FcγRIIIA are susceptibility factors for SLE: a unifying hypothesis. Genes Immun. 2004, 5: 130-137. 10.1038/sj.gene.6364052.
Liu K, Li QZ, Delgado-Vega AM, Abelson AK, Sanchez E, Kelly JA, Li L, Liu Y, Zhou J, Yan M, Ye Q, Liu S, Xie C, Zhou XJ, Chung SA, Pons-Estel B, Witte T, de Ramon E, Bae SC, Barizzone N, Sebastiani GD, Gregersen PK, Gilkeson GG, Kimberly RP, Vyse TJ, Kim I, D'Alfonso S, Martin J, Harley JB, Criswell LA, Wakeland EK, Alarcon-Riquelme ME, Mohan C: Kallikrein genes are associated with lupus and glomerular basement membrane-specific antibody-induced nephritis in mice and humans. J Clin Invest. 2009, 119: 911-923. 10.1172/JCI36728.
Taylor KE, Chung SA, Graham RR, Ortmann WA, Lee AT, Langefeld CD, Jacob CO, Kamboh MI, Alarcon-Riquelme ME, Tsao BP, Moser KL, Gaff ney PM, Harley JB, Petri M, Manzi S, Gregersen PK, Behrens TW, Criswell LA: Risk alleles for systemic lupus erythematosus in a large case-control collection and associations with clinical subphenotypes. PLoS Genet. 2011, 7: e1001311-10.1371/journal.pgen.1001311.
Acknowledgements
JHA has been supported by several grants including R01 AI077674-01, P01 AI1078907-01, U19 Autoimmunity Center of Excellence AI563262-06, the Lupus Foundation of American, and the Lupus Research Institute. The collaborations and thoughtful discussions with colleagues, especially Inaki Sanz and John Looney, are gratefully noted.
Declarations
This article has been published as part of Arthritis Research & Therapy Volume 14 Suppl 4, 2012: New therapeutic targets in systemic lupus erythematosus. The supplement was proposed and developed by the journal. Articles were commissioned by the journal, were independently prepared by the authors and have undergone the journal's standard peer review process. Publication of the supplement has been supported by an unrestricted educational grant from UCB. Completed articles underwent a data quality check by Darwin Healthcare Communications, funded by UCB
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
JHA has received grants from Amgen Pharmaceuticals, Genentech, Karyopharm, Proteolix, and Vaccinex. She has served as a consultant for Amgen, Boehringer Ingelheim, Genentech, MedImmune, Roche, and UCB.
Rights and permissions
About this article
Cite this article
Marian, V., Anolik, J.H. Treatment targets in systemic lupus erythematosus: biology and clinical perspective. Arthritis Res Ther 14 (Suppl 4), S3 (2012). https://doi.org/10.1186/ar3917
Published:
DOI: https://doi.org/10.1186/ar3917