
Abstract
Systemic lupus erythematosus is a prototypic autoimmune disease characterized by autoantibody production and immune complex formation/deposition in target organs such as the kidney. Resultant local inflammation then leads to organ damage. Nephritis, a major cause of morbidity and mortality in patients with lupus, occurs in approximately 50% of lupus patients. In the present review, we provide an overview of the current research and knowledge concerning mechanisms of renal injury in both lupus-prone mouse models and human lupus patients.
Similar content being viewed by others


Introduction
Nephritis is a major cause of morbidity and mortality in patients with lupus. Nephritis occurs in approximately 50% of lupus patients, but rates vary significantly between genders (men more than women) and ethnicities (more common in people of color). Men and minorities with lupus nephritis are also more likely to progress to end-stage renal disease than women or people of European ancestry. The multiple factors underlying these demographic differences are unclear at this time [1].
The International Society of Nephrology revised the World Health Organization classification of lupus nephritis recently, although maintaining six classes [2]. The pathologic classes vary from mild mesangial involvement (Class I) to diffuse proliferative disease (Class IV) to membranous disease (Class V) to end-stage fibrosis (Class VI). Although most attention in lupus nephritis is focused on glomerular disease, there is also significant tubular disease that impacts prognosis and renal function [3]. For the purposes of the present review, we will primarily focus on the proliferative forms of lupus nephritis (focal proliferative, Class III disease; and diffuse proliferative, Class IV disease), highlighting several contributors to tissue injury.
Much of what is known about pathogenic factors in tissue damage in lupus nephritis was derived from studies of murine models of lupus, with confirmation as possible in humans. These studies utilize multigenic models of lupus (that is, MRL/lpr, NZB/NZW, and NZM congenic strains) as well as single gene mutants (that is, DNAse 1, Nrf2, or Fcγ receptor (FCγR) knockouts) [4, 5]. These models share common features of human disease such as anti-double-stranded DNA (anti-dsDNA) antibodies and proliferative nephritis, but differ in their renal cytokine/chemokine profile, cellular infiltration and acuity/chronicity of disease [5]. Thus, as in human disease, there is heterogeneity of pathogenic mechanisms in murine lupus nephritis.
Autoantibodies and renal immune complex deposition
The presence of autoantibodies is a requirement for development of lupus nephritis [6]. Antibodies to dsDNA/nucleosomes are most closely linked with development of nephritis [7], although what separates pathogenic from nonpathogenic anti-dsDNA antibodies is not clear [8]. Pathogenic anti-dsDNA antibodies deposit as immune complexes (IC) [6]. When anti-C1q antibodies are present along with anti-dsDNA antibodies, development of renal disease is accelerated [9, 10].
There are three postulated mechanisms for formation of glomerular ICs, all of which probably contribute to disease in some patients, given the heterogeneity of disease [11]. The first mechanism is deposition of preformed serum ICs [12]. This mechanism is hard to confirm, as ICs are difficult to isolate or quantify in lupus patient sera and thus are not felt to play a major role in the pathogenesis of lupus nephritis. Binding of autoantibodies to in situ glomerular antigens such as laminin, annexin II or heparin is a second mechanism postulated for IC deposition. This crossreactivity is demonstrated via the elution of antibodies from glomeruli that bind these antigens in addition to dsDNA/chromatin [13, 14].
A recent series of investigations implicates a third mechanism, anti-dsDNA/chromatin antibodies binding to nucleosomes/DNA present in the glomerular matrix, as the most compelling [13]. Due to charge/charge inter-actions, circulating DNA/nucleosomes can deposit in the glomerular basement membrane and serve as antigen for autoantibodies. Another source of glomerular DNA/nucleosomes is retention of nucleosomes from necrotic intrinsic glomerular cells [7]. Indeed, recent electron microscopic co-localization experiments in human and mouse lupus kidneys indicated that antibodies present in the glomerulus are bound to electron-dense deposits that were identified to be nucleosomal material [15]. Following the formation of these ICs, there is downregulation of DNAse I in the kidney, which allows for enhanced amounts of nucleosomal material in the glomerulus [16]. These complexes can then lead to further activation of immune pathways by co-stimulation of FcγRs and endosomal Toll-like receptors (TLRs) and/or by activating the complement cascade [7]. Although the latter mechanism of antibodies binding nucleosomal material from necrotic glomerular cells provides a compelling story, it is likely that any of these mechanisms may be present in a given patient [11].
Complement and tissue injury in lupus nephritis
Complement has a dual role in lupus. Deposition of complement proteins in glomeruli is a key feature of lupus nephritis. There is strong evidence that complement activation is deleterious in lupus nephritis [17]. This is in contrast to the known association of early complement component deficiency with lupus. Individuals deficient in C1 components, C2 and C4, have a high prevalence of lupus due to impaired clearance of ICs/apoptotic bodies leading to breaking of tolerance. Activation of the classical pathway of complement activation thus appears protective against lupus due to enhanced clearance of ICs and cellular debris [18].
Recent findings implicate the alternative complement pathway as a key component of complement-mediated damage in lupus nephritis [19, 20]. Activation of the alternative complement pathway triggers an amplification loop that accelerates cleavage of C3 to C3b, covalent binding to cellular surfaces, with release of the anaphylotoxin C3a and C5a, and formation of the complement membrane attack complex. It is currently unclear which of the outcomes of complement activation are most important in lupus nephritis: generation of C3a and C5a or formation of the membrane attack complex.
Blocking the alternative complement pathway either genetically or pharmacologically leads to significantly decreased severity of renal disease in murine lupus models [20–22]. Eliminating the natural inhibitor of the alternative pathway, Factor H, leads to acceleration of lupus-like renal disease [23]. Pharmacologic inhibition of the alternative pathway is effective in both MRL/lpr mice and NZM congenic mice [19, 21]. These results suggest that the alternative complement pathway is a key mechanism for tissue injury in lupus nephritis. Genetic deletion of C3 has minimal effect on murine lupus nephritis, probably due to diminished clearance of ICs enhancing immune activation by noncomplement-mediated mechanisms [24]. Blocking the C3a receptor has minimal impact on disease [25], while blocking complement activation further downstream is effective, as studies of C5aR-deficient mice or using a C5aR blocking antibody also led to decreased severity of renal disease in murine models of lupus [26, 27].
Complement may also play a role in tubular damage in lupus. Development of proteinuria leads to spilling of complement components into the urine. Complement C3 is activated in urine via pH and urea, resulting in formation of membrane attack complexes on the epithelial side of tubular cells [28]. There are no complement protective mechanisms present on the epithelial side of renal tubular epithelial cells, resulting in unchecked complement activation and tubular damage. These experiments were performed under adriamycin-induced proteinuria in mice, not in lupus, but similar mechanisms may explain some of the tubular damage that occurs in lupus.
Fcγ receptors and Toll-like receptors in lupus nephritis
Another mechanism by which ICs may lead to tissue damage is via activation of activating FcγRs, upon binding of immunoglobulin Fc regions by FcγR-expressing cells [29]. Although FcγRs are clearly implicated in the development of lupus in genetic studies of gain-of-function and loss-of-function mutations and copy numbers of FcγR genes, their role in predisposition to lupus nephritis and/or tissue injury is not as clear [29–32]. In mice, knockout of specific FcγR can lead to accentuation or diminution of disease; most of the effect, however, is on development of lupus rather than specific tissue injury [33, 34]. Any impact of FcγR on disease is highly dependent on background strain [35]. Similarly, reports of associations of FcγR genetic changes with nephritis appear linked to specific ethnicities [29].
FcγR may be important in association with TLRs in mediating IC-induced inflammation in the kidney [36]. As noted above, dsDNA-containing ICs may activate kidney resident cells via a co-signaling mechanism of FcγR activation via the autoantibody and TLR9 activation via dsDNA. This type of two-step activation is known to activate B cells by ICs containing either TLR9 or TLR7 activators such as dsDNA or single-stranded RNA [37]. Inhibition of TLR7/9 is effective in treating murine lupus, although whether primarily at the level of systemic autoimmunity or via blocking specifically renal tissue damage is not clear [38].
Immune cells in lupus nephritis
Following the formation and/or deposition of ICs in the kidney, interactions between resident renal cells and infiltrating inflammatory cells promote tissue injury. Local cytokine, chemokine and adhesion molecule production leads to further influx of inflammatory cells and production of proinflammatory cytokines, ultimately resulting in renal inflammation, tissue injury and fibrosis. T cells are important mediators in both mouse models and human patients in the progression of lupus nephritis. Lupus T cells express increased levels of molecules necessary for homing and/or demonstrate increased homing to the kidney [39–42]. Mechanisms by which T cells contribute to tissue injury include activating and providing help to nephritogenic antibody-producing B cells, recruiting macrophages and dendritic cells (DCs), and producing cytokines. Indeed, kidney-infiltrating T cells - including CD4+, CD8+ and IL17-producing CD4-CD8- double-negative T cells - are activated and express a wide array of proinflammatory cytokines [43–46]. Depleting T cells or blocking T-cell activation reduces progression of nephritis in lupus mouse models [47, 48].
Pathogenic B cells have a variety of functions that contribute to lupus nephritis. Namely they produce autoantibodies that can cause renal damage via disruption of cellular functions, cytotoxicity mediated by interactions with complement and release of inflammatory mediators. Studies in lupus mouse models demonstrated that infiltrating B cells in the kidney secrete antibodies with various Ag specificities, contributing to increased in situ ICs [49–51]. Similarly, germinal center-like structures and T-cell-B-cell aggregates present in the kidney suggest in situ secretion of pathogenic antibodies, including nephritogenic antibodies, and ICs in human lupus patients [52–54]. Depleting B cells either prior to or after disease onset prevented and/or delayed the onset of nephritis in several different lupus mouse models [55–58] and resulted in complete or partial clinical remission in patients [59]. MRL/lpr lupus-prone mice that have B cells unable to secrete antibodies still develop nephritis, however, although less severely [60] - indicating that additional B-cell functions, such as antigen presentation and activation of pathogenic T cells and proinflammatory cytokine production (IL-6 and TNFα), contribute significantly to kidney injury.
Neutrophils, macrophages and DCs, present in nephritic kidneys, also are contributors to injury. Neutrophils are a source of neutrophil extracellular traps that contain self-antigens such as histones and DNA, and are present in ICs deposited in the kidney of systemic lupus erythematosus (SLE) patients [61–64]. The response to neutrophil extracellular traps contributes to kidney injury through the activation of plasmacytoid DCs and production of type I interferon [63, 64]. DCs and macrophages produce T-helper type 1 proinflammatory cytokines (IL-12 and IFNγ), express chemokine receptors and interact with autoreactive T cells to recruit additional inflammatory cells. Reduction of CD11c+ DCs in the MRL/lpr lupus-prone model resulted in improved kidney disease [65], and the presence of plasmacytoid DCs was correlated with high IL-18 expression in the glomeruli of patients with active nephritis [66]. An activated macrophage population with a type II phenotype (M2b) that expresses high amounts of proinflammatory cytokines and exhibits tissue degradation is associated with the onset of proteinuria in NZB/NZW F1 mice [67–69]. Similarly, in lupus patients with nephritis, macrophage infiltration in the kidney correlates with disease [70] (reviewed in [71]).
Cytokines and chemokines
Production of cytokines and chemokines in glomeruli early during lupus nephritis precedes inflammatory cell infiltration and proteinuria [72, 73]. T-helper type 1 cytokines are predominantly present in nephritic kidneys in SLE patients [74, 75]. T-helper type 1 proinflammatory cytokines that contribute to tissue damage include IL-12, IL-18 and IFNγ. High IL-18 and/or IL-12 production is observed in glomeruli of human and mouse lupus nephritis. IL-18 overexpression in kidneys of predisease MRL/lpr lupus-prone mice resulted in accumulation of leukocytes in the kidney and increased renal pathology and proteinuria [76]. Similarly, MRL/lpr mice in which IL-12 was overexpressed presented increased T-cell infil-tration, specifically IFNγ-producing T cells, and accelerated nephritis [77], while MRL/lpr IL12-/- mice showed reduced IFNγ levels and delayed nephritis [78]. Higher IL-18, IL-12 and IFNγ levels were demonstrated in SLE patients compared with healthy controls, and specifically in SLE patients with nephritis compared with patients without nephritis. Urinary IL-12 levels correlated with onset and severity of nephritis in these patients [66, 79]. The major mechanism of renal injury by IL-18 and IL-12 is probably through their upregulation of IFNγ. The levels of IFNγ in nephritic MRL/lpr mice are increased compared with controls, and kidney pathology in mice overexpressing IL-12 requires IFNγ [77]. Importantly, IFNγ signaling was demonstrated to directly induce cell death of tubular epithelial cells in MRL/lpr kidneys [80].
Chemokines contribute to renal damage by recruiting inflammatory cells to the kidney. Proinflammatory chemokines/growth factors including monocyte chemoattractant protein-1 (MCP-1, CCL2), macrophage inflammatory protein-1β (CCL4), RANTES (CCL5), macrophage colony-stimulating factor and IFNγ-induced protein-10 (CXCL10) were demonstrated to be upregulated in the kidney of lupus-prone mice prior to proteinuria and renal damage [81]. Their expression was followed by mononuclear infiltration and increased expression of their respective receptors (CCR1, CCR2 and CCR5). Increased levels of macrophage inflammatory protein-1α (CCL3), MCP-1, RANTES and IFNγ-induced protein-10 were also observed in the serum of lupus patients [82, 83]. Of these chemokines, MCP-1 was demonstrated to be associated with kidney damage in lupus. MCP-1 levels increase in the kidney as nephritis progresses in the MRL/lpr lupus model [84]. A knockout of MCP-1 on the MRL/lpr background resulted in reduced macrophage and T-cell infiltration in the kidney, reduced proteinuria and renal pathology and prolonged survival [84]. Blockade of MCP-1 after disease onset improved renal disease and prolonged survival, characterized by decreased renal infiltration by macrophages and T cells [85, 86]. In lupus nephritis patients, tubulointerstitial expression of MCP-1 was demonstrated to be associated with chronic renal damage [75] and urinary MCP-1 levels were associated with renal disease activity [87, 88].
Transcription factors
The signal transducers and activators of transcription (STAT) factor family is part of the Jak/STAT signaling pathway activated by cytokines and contains several members identified as playing roles in lupus nephritis, including STAT1 and STAT4.
STAT1, when activated, binds to IFNγ-activated sequences in the promoters of IFNγ-inducible genes, and IFNγ induced the activation of STAT1 in mesangial cells of MRL/lpr mice [89]. Elevated STAT1 expression, both total and activated forms, is present in kidneys of nephritic lupus mice with predominant expression in glomeruli [89]. In SLE patients, STAT1 expression is present in renal biopsies of lupus nephritis patients and expression levels correlated with disease activity [90].
STAT4 was identified as a lupus risk gene. A polymorphism identified in STAT4 is associated with dsDNA antibodies and severe nephritis in human SLE [91]. In NZM2410 and NZM2328 lupus-prone mouse strains, loss of STAT4 results in lower levels of IgG anti-dsDNA antibodies, but development of more severe renal disease [92, 93].
Transcription factors such as STAT factors influence the expression of an array of genes that play a role in the cellular function of immune cells and/or the response of cells in target tissues to inflammation, influencing the extent of tissue injury. Dysregulation of transcription in lupus nephritis is further indicated by the profound effect of alterations in Ets factor/Fli-1 expression and the impact of histone deacetylase inhibitors, which diminish gene transcription, on development and severity of renal disease.
Reactive intermediates in tissue injury
Several studies utilizing competitive inhibitors of inducible nitric oxide synthase (iNOS) suggest that iNOS activity is pathogenic in murine lupus [94, 95]. Inhibiting iNOS activity in MRL/lpr mice, before disease onset, with the nonspecific arginine analog L-NG-monomethyl-L-arginine reduced 3-nitrotyrosine formation in the kidney, partially restored renal catalase activity, and inhibited cellular proliferation and necrosis within the glomerulus [94, 95]. The partially selective iNOS inhibitor L-N6-(1-iminoethyl)lysine had a similar effect when used to treat these mice prior to disease onset [96]. L-NG-monomethyl-L-arginine therapy in NZB/W mice that were already suffering from nephritis had a similar but less profound effect on proteinuria and renal histopathology than did preventative therapy [97]. However, L-NG-monomethyl-L-arginine as monotherapy for the treatment of active disease was less effective in the rapidly progressive MRL/lpr model [97]. These findings suggest that overproduction of nitric oxide is deleterious and mediates tissue damage in lupus nephritis.
The mechanisms through which iNOS activity may be pathogenic in SLE were studied in animal models and in vitro. Peroxynitrite (ONOO-), a byproduct of iNOS activity, can nitrate amino acids and change the catalytic activity of enzymes [97]. One such enzyme, catalase, serves to protect host tissues from free radical attack [98]. In vascular tissue, prostacyclin synthase and endothelial nitric oxide synthase are inactivated by peroxynitrite, leading to vasoconstriction [99]. These observations suggest that one mechanism through which iNOS activity is pathogenic is via deactivation of tissue protective enzymes. Nitrosylation is also being increasingly recognized as a mechanism for impacting gene regulation similar to methylation and acetylation. Nitrosylation of NF-κB modulates its function, altering resultant inflammatory gene transcription. Such nitrosylation does not appear to impact the nuclear migration of NF-κB, but rather modulates its transcriptional activity once inside the nucleus [100]. Such nitrosylation can be achieved in vivo by administering S-nitrosoglutathione, providing a potential therapeutic pathway via modulation of reactive intermediates [100].
Markers of systemic nitric oxide production are elevated in patients with SLE in a manner that parallels disease activity [101]. Those patients with lupus nephritis had the most elevated markers of systemic nitric oxide production among SLE subjects [102]. This observation spawned the hypothesis that glomerular proliferative lesions were a source of increased nitric oxide production, as well as a potential result of inappropriate nitric oxide production. Several reports supported this hypothesis, with renal biopsy studies showing increased iNOS expression in the glomeruli of lupus nephritis subjects [101, 103] - particularly in mesangial cells, glomerular epithelial cells, and infiltrating inflammatory cells [101]. When 3-nitrotyrosine was used as a surrogate for iNOS activity, the association with disease activity was greater in African Americans [104], suggesting a possible difference between Caucasians and African Americans in reactive oxygen intermediate production versus reactive nitrogen intermediate production that may impact outcome.
To assess whether genes involved in reactive oxygen intermediate production are associated with lupus nephritis, polymorphisms in the gene for myeloperoxidase were assessed. There was a significant correlation between the low expressing myeloperoxidase 463A allele and the risk for developing nephritis in African Americans [105]. This association was subsequently confirmed in two other cohorts. This finding may seem paradoxical until one considers that reactive oxygen intermediates can sequester reactive nitrogen intermediates and that low myeloperoxidase activity can lead to increased OH radical stress. Polymorphisms of iNOS and endothelial nitric oxide synthase are also reported to be associated with genetic risk of developing lupus, although associations with renal disease are less clear [106, 107]. A recent study demonstrated that inhibiting reactive intermediate production in diabetics improved renal function, suggesting that a similar strategy may also be effective in lupus [108].
Renal regeneration/fibrosis
End-stage renal disease in lupus is secondary to loss of glomerular and tubular function due to renal cell death and resultant fibrosis. The factors important in the inflammatory process are more clearly defined than the factors resulting in progressive glomerular/tubular loss and fibrosis. As in other fibrotic processes, transforming growth factor beta expression is associated with renal fibrosis [109]. Co-factors such as hypertension, production of vasoactive substances such as kallikrein, ongoing proteinuria and nephrotoxic drugs play an important role in progression of renal disease in lupus. Genetic factors are probably also a major determinant of progression to end-stage renal disease. Factors involved in renal regeneration post injury are even less well defined. Recruitment of stromal cells to the kidney via chemokine receptors and C3a may result in repair of some tissue damage, but further research is needed in this area to define therapeutic strategies [110].
Conclusion
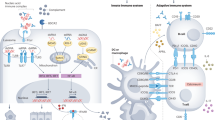
In summary, the pathogenesis of lupus nephritis and mechanisms of resultant renal injury remains an active field of investigation, with much knowledge gained but many questions still left to answer. The complexity and number of factors involved in disease make it difficult to derive a clear step-by-step pathogenic pathway. A summary of proposed pathogenic mechanisms is illustrated in Figure 1. Autoantibodies and ICs are important first mediators that are required for disease expression in human disease. Deposition of ICs, however, is not sufficient for disease expression, as numerous studies report lack of proliferative disease despite significant IgG/IC deposition in glomeruli. Downstream mediators are blocked in these pharmacologic/genetic studies, inhibiting disease activity without impacting IC deposition. Complement, TLRs and FcγRs play an amplification role in the initiation and propagation of disease. IC deposition with complement, TLR and/or FcγR activation stimulates intrinsic immune active glomerular cells to release inflammatory cytokines and chemoattractant chemokines, resulting in the influx of the spectrum of inflammatory cells. The end mediators of disease appear to be the reactive intermediates produced by both inflammatory cells and intrinsic glomerular cells. Although tissue repair post inflammatory injury is also probably a key prognostic process, very little is known regarding factors involved in tissue repair. These multiple mediators provide a host of targets for therapeutic intervention. Only 50% of patients respond to current standards of therapy. Clearly there is room for improvement, but no one therapy will probably be effective in most patients. Determining which pathway is key to a given patient is the challenge for the immediate future, as well as developing safe mechanisms for blocking these pathways.

Summary of proposed pathogenic mechanisms in tissue injury in lupus nephritis. Top right: The first step leading to nephritis involves the production of autoantibodies to self-antigens followed by formation of immune complexes (ICs) in glomeruli. IC deposition leads to complement activation and intrinsic renal cell activation (mesangial cells (MC) and endothelial cells (EC)), both leading to local chemokine and cytokine inflammation. Chemokine expression leads to an influx of inflammatory cells such as lymphocytes and macrophages. Left: Influx of immune cells leads to interstitial as well as further glomeruli inflammation and EC activation. Activated renal cells (MC and podocytes) and infiltrating immune cells (macrophages and dendritic cells (DCs)) produce reactive nitrogen (nitric oxide (NO)) and reactive oxygen species (ROS). The combined expression of cytokines and ROS results in further renal inflammation and fibrosis, resulting in cumulative tissue destruction both at the glomerular level (top right) and the tubular level (left). Bottom right: Lymphocyte interactions and functions such as cytokine expression and antibody production contribute to inflammation and damage in the tubules and glomeruli. BCR, B-cell receptor; FcR, Fc receptor; MCP-1, monocyte chemoattractant protein-1; STAT, signal transducers and activators of transcription; TCR, T-cell receptor.
Note
Autoimmune Basis of Rheumatic Diseases
This article is part of a series on Systemic lupus erythematosus, edited by David Pisetsky, which can be found online at http://arthritis-research.com/series/lupus
This series forms part of a special collection of reviews covering major autoimmune rheumatic diseases, available at: http://arthritis-research.com/series/abrd
Abbreviations
- DC:
-
dendritic cell
- dsDNA:
-
double-stranded DNA
- FCγR:
-
Fcγ receptor
- IC:
-
immune complex
- IFN:
-
interferon
- IL:
-
interleukin
- iNOS:
-
inducible nitric oxide synthase
- MCP-1:
-
monocyte chemoattractant protein-1
- NF:
-
nuclear factor
- RANTES:
-
regulated upon activation, normal T-cell expressed and secreted
- SLE:
-
systemic lupus erythematosus
- STAT:
-
signal transducers and activators of transcription
- TLR:
-
Toll-like receptor
- TNF:
-
tumor necrosis factor.
References
Austin HA, Boumpas DT, Vaughan EM, Balow JE: High-risk features of lupus nephritis: importance of race and clinical and histological factors in 166 patients. Nephrol Dial Transplant. 1995, 10: 1620-1628.
Markowitz GS, D'Agati VD: The ISN/RPS 2003 classification of lupus nephritis: an assessment at 3 years. Kidney Int. 2007, 71: 491-495. 10.1038/sj.ki.5002118.
Yu F, Wu LH, Tan Y, Li LH, Wang CL, Wang WK, Qu Z, Chen MH, Gao JJ, Li ZY, Zheng X, Ao J, Zhu SN, Wang SX, Zhao MH, Zou WZ, Liu G: Tubulointerstitial lesions of patients with lupus nephritis classified by the 2003 International Society of Nephrology and Renal Pathology Society system. Kidney Int. 2010, 77: 820-829. 10.1038/ki.2010.13.
Ma Q, Battelli L, Hubbs AF: Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am J Pathol. 2006, 168: 1960-1974. 10.2353/ajpath.2006.051113.
Davidson A, Diamond B: Activated basophils give lupus a booster shot. Nat Med. 2010, 16: 635-636. 10.1038/nm0610-635.
Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, Harley JB: Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N Engl J Med. 2003, 349: 1526-1533. 10.1056/NEJMoa021933.
Fenton KA, Rekvig OP: A central role of nucleosomes in lupus nephritis. Ann N Y Acad Sci. 2007, 1108: 104-113. 10.1196/annals.1422.012.
Zykova SN, Seredkina NE, Rekvig OP: Glomerular targets for autoantibodies in lupus nephritis - an apoptotic origin. Ann N Y Acad Sci. 2007, 1108: 1-10. 10.1196/annals.1422.001.
Trouw LA, Groeneveld TW, Seelen MA, Duijs JM, Bajema IM, Prins FA, Kishore U, Salant DJ, Verbeek JS, van Kooten C, Daha MR: Anti-C1q autoantibodies deposit in glomeruli but are only pathogenic in combination with glomerular C1q-containing immune complexes. J Clin Invest. 2004, 114: 679-688.
Sinico RA, Rimoldi L, Radice A, Bianchi L, Gallelli B, Moroni G: Anti-C1q autoantibodies in lupus nephritis. Ann N Y Acad Sci. 2009, 1173: 47-51. 10.1111/j.1749-6632.2009.04746.x.
Lefkowith JB, Gilkeson GS: Nephritogenic autoantibodies in lupus: current concepts and continuing controversies. Arthritis Rheum. 1996, 39: 894-903. 10.1002/art.1780390605.
Alexander JJ, Hack BK, Jacob A, Chang A, Haas M, Finberg RW, Quigg RJ: Abnormal immune complex processing and spontaneous glomerulonephritis in complement factor H-deficient mice with human complement receptor 1 on erythrocytes. J Immunol. 2010, 185: 3759-3767. 10.4049/jimmunol.1000683.
Kalaaji M, Mortensen E, Jorgensen L, Olsen R, Rekvig OP: Nephritogenic lupus antibodies recognize glomerular basement membrane-associated chromatin fragments released from apoptotic intraglomerular cells. Am J Pathol. 2006, 168: 1779-1792. 10.2353/ajpath.2006.051329.
Mjelle JE, Rekvig OP, Van Der Vlag J, Fenton KA: Nephritogenic antibodies bind in glomeruli through interaction with exposed chromatin fragments and not with renal cross-reactive antigens. Autoimmunity. 2011, 44: 373-383. 10.3109/08916934.2010.541170.
Fenton KA, Tommeras B, Marion TN, Rekvig OP: Pure anti-dsDNA mAbs need chromatin structures to promote glomerular mesangial deposits in BALB/c mice. Autoimmunity. 2010, 43: 179-188. 10.3109/08916930903305633.
Fenton K, Fismen S, Hedberg A, Seredkina N, Fenton C, Mortensen ES, Rekvig OP: Anti-dsDNA antibodies promote initiation, and acquired loss of renal Dnase1 promotes progression of lupus nephritis in autoimmune (NZBxNZW)F1 mice. PLoS One. 2009, 4: e8474-10.1371/journal.pone.0008474.
Boackle SA, Holers VM: Role of complement in the development of autoimmunity. Curr Dir Autoimmun. 2003, 6: 154-168.
Truedsson L, Bengtsson AA, Sturfelt G: Complement deficiencies and systemic lupus erythematosus. Autoimmunity. 2007, 40: 560-566. 10.1080/08916930701510673.
Watanabe H, Garnier G, Circolo A, Wetsel RA, Ruiz P, Holers VM, Boackle SA, Colten HR, Gilkeson GS: Modulation of renal disease in MRL/lpr mice genetically deficient in the alternative complement pathway factor B. J Immunol. 2000, 164: 786-794.
Elliott MK, Jarmi T, Ruiz P, Xu Y, Holers VM, Gilkeson GS: Effects of complement factor D deficiency on the renal disease of MRL/lpr mice. Kidney Int. 2004, 65: 129-138. 10.1111/j.1523-1755.2004.00371.x.
Sekine H, Kinser TT, Qiao F, Martinez E, Paulling E, Ruiz P, Gilkeson GS, Tomlinson S: The benefit of targeted and selective inhibition of the alternative complement pathway for modulating autoimmunity and renal disease in MRL/lpr mice. Arthritis Rheum. 2011, 63: 1076-1085. 10.1002/art.30222.
Sekine H, Ruiz P, Gilkeson GS, Tomlinson S: The dual role of complement in the progression of renal disease in NZB/W F(1) mice and alternative pathway inhibition. Molecular Immunol. 2011, 49: 317-323. 10.1016/j.molimm.2011.09.015.
Bao L, Haas M, Quigg RJ: Complement factor H deficiency accelerates development of lupus nephritis. J Am Soc Nephrol. 2011, 22: 285-295. 10.1681/ASN.2010060647.
Sekine H, Reilly CM, Molano ID, Garnier G, Circolo A, Ruiz P, Holers VM, Boackle SA, Gilkeson GS: Complement component C3 is not required for full expression of immune complex glomerulonephritis in MRL/lpr mice. J Immunol. 2001, 166: 6444-6451.
Wenderfer SE, Wang H, Ke B, Wetsel RA, Braun MC: C3a receptor deficiency accelerates the onset of renal injury in the MRL/lpr mouse. Mol Immunol. 2009, 46: 1397-1404. 10.1016/j.molimm.2008.12.004.
Wenderfer SE, Ke B, Hollmann TJ, Wetsel RA, Lan HY, Braun MC: C5a receptor deficiency attenuates T cell function and renal disease in MRLlpr mice. J Am Soc Nephrol. 2005, 16: 3572-3582. 10.1681/ASN.2005040373.
Bao L, Osawe I, Puri T, Lambris JD, Haas M, Quigg RJ: C5a promotes development of experimental lupus nephritis which can be blocked with a specific receptor antagonist. Eur J Immunol. 2005, 35: 2496-2506. 10.1002/eji.200526327.
Lenderink AM, Liegel K, Ljubanovic D, Coleman KE, Gilkeson GS, Holers VM, Thurman JM: The alternative pathway of complement is activated in the glomeruli and tubulointerstitium of mice with adriamycin nephropathy. Am J Physiol Renal Physiol. 2007, 293: F555-F564. 10.1152/ajprenal.00403.2006.
Niederer HA, Clatworthy MR, Willcocks LC, Smith KG: FcγRIIB, FcγRIIIB, and systemic lupus erythematosus. Ann N Y Acad Sci. 2010, 1183: 69-88. 10.1111/j.1749-6632.2009.05132.x.
Grossman JM, Tsao BP: Genetics and systemic lupus erythematosus. Curr Rheumatol Rep. 2000, 2: 13-18. 10.1007/s11926-996-0063-x.
Zhou XJ, Lv JC, Bu DF, Yu L, Yang YR, Zhao J, Cui Z, Yang R, Zhao MH, Zhang H: Copy number variation of FCGR3A rather than FCGR3B and FCGR2B is associated with susceptibility to anti-GBM disease. Int Immunol. 2010, 22: 45-51. 10.1093/intimm/dxp113.
Tarzi RM, Cook HT: Role of Fcγ receptors in glomerulonephritis. Nephron Exp Nephrol. 2003, 95: e7-e12. 10.1159/000073018.
Gergely P, Isaak A, Szekeres Z, Prechl J, Erdei A, Nagy ZB, Gergely J, Poor G: Altered expression of Fcγ and complement receptors on B cells in systemic lupus erythematosus. Ann N Y Acad Sci. 2007, 1108: 183-192. 10.1196/annals.1422.020.
Fukuyama H, Nimmerjahn F, Ravetch JV: The inhibitory Fcγ receptor modulates autoimmunity by limiting the accumulation of immunoglobulin G+ anti-DNA plasma cells. Nat Immunol. 2005, 6: 99-106.
Clynes R, Calvani N, Croker BP, Richards HB: Modulation of the immune response in pristane-induced lupus by expression of activation and inhibitory Fc receptors. Clin Exp Immunol. 2005, 141: 230-237. 10.1111/j.1365-2249.2005.02847.x.
Lee PY, Kumagai Y, Li Y, Takeuchi O, Yoshida H, Weinstein J, Kellner ES, Nacionales D, Barker T, Kelly-Scumpia K, van Rooijen N, Kumar H, Kawai T, Satoh M, Akira S, Reeves WH: TLR7-dependent and FcγR-independent production of type I interferon in experimental mouse lupus. J Exp Med. 2008, 205: 2995-3006. 10.1084/jem.20080462.
Rahman AH, Eisenberg RA: The role of toll-like receptors in systemic lupus erythematosus. Springer Semin Immunopathol. 2006, 28: 131-143. 10.1007/s00281-006-0034-3.
Sun S, Rao NL, Venable J, Thurmond R, Karlsson L: TLR7/9 antagonists as therapeutics for immune-mediated inflammatory disorders. Inflamm Allergy Drug Targets. 2007, 6: 223-235. 10.2174/187152807783334300.
Hase K, Tani K, Shimizu T, Ohmoto Y, Matsushima K, Sone S: Increased CCR4 expression in active systemic lupus erythematosus. J Leukoc Biol. 2001, 70: 749-755.
Yamada M, Yagita H, Inoue H, Takanashi T, Matsuda H, Munechika E, Kanamaru Y, Shirato I, Tomino Y, Matushima K, Okumura K, Hashimoto H: Selective accumulation of CCR4+ T lymphocytes into renal tissue of patients with lupus nephritis. Arthritis Rheum. 2002, 46: 735-740. 10.1002/art.10112.
Li Y, Harada T, Juang YT, Kyttaris VC, Wang Y, Zidanic M, Tung K, Tsokos GC: Phosphorylated ERM is responsible for increased T cell polarization, adhesion, and migration in patients with systemic lupus erythematosus. J Immunol. 2007, 178: 1938-1947.
Wang Y, Ito S, Chino Y, Goto D, Matsumoto I, Murata H, Tsutsumi A, Hayashi T, Uchida K, Usui J, Yamagata K, Sumida T: Laser microdissection-based analysis of cytokine balance in the kidneys of patients with lupus nephritis. Clin Exp Immunol. 2010, 159: 1-10. 10.1111/j.1365-2249.2009.04031.x.
Wang Y, Ito S, Chino Y, Iwanami K, Yasukochi T, Goto D, Matsumoto I, Hayashi T, Uchida K, Sumida T: Use of laser microdissection in the analysis of renalinfiltrating T cells in MRL/lpr mice. Mod Rheumatol. 2008, 18: 385-393. 10.1007/s10165-008-0074-8.
Crispin JC, Keenan BT, Finnell MD, Bermas BL, Schur P, Massarotti E, Karlson EW, Fitzgerald LM, Ergin S, Kyttaris VC, Tsokos GC, Costenbader KH: Expression of CD44 variant isoforms CD44v3 and CD44v6 is increased on T cells from patients with systemic lupus erythematosus and is correlated with disease activity. Arthritis Rheum. 2010, 62: 1431-1437.
Crispin JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, Kyttaris VC, Juang YT, Tsokos GC: Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. 2008, 181: 8761-8766.
Apostolidis SA, Crispin JC, Tsokos GC: IL-17-producing T cells in lupus nephritis. Lupus. 2011, 20: 120-124. 10.1177/0961203310389100.
Schiffer L, Sinha J, Wang X, Huang W, von Gersdorff G, Schiffer M, Madaio MP, Davidson A: Short term administration of costimulatory blockade and cyclophosphamide induces remission of systemic lupus erythematosus nephritis in NZB/W F1 mice by a mechanism downstream of renal immune complex deposition. J Immunol. 2003, 171: 489-497.
Wofsy D, Ledbetter JA, Hendler PL, Seaman WE: Treatment of murine lupus with monoclonal anti-T cell antibody. J Immunol. 1985, 134: 852-857.
Cassese G, Lindenau S, de Boer B, Arce S, Hauser A, Riemekasten G, Berek C, Hiepe F, Krenn V, Radbruch A, Manz RA: Inflamed kidneys of NZB/W mice are a major site for the homeostasis of plasma cells. Eur J Immunol. 2001, 31: 2726-2732. 10.1002/1521-4141(200109)31:9<2726::AID-IMMU2726>3.0.CO;2-H.
Sekine H, Watanabe H, Gilkeson GS: Enrichment of anti-glomerular antigen antibody-producing cells in the kidneys of MRL/MpJ-Fas(lpr) mice. J Immunol. 2004, 172: 3913-3921.
Espeli M, Bokers S, Giannico G, Dickinson HA, Bardsley V, Fogo AB, Smith KG: Local renal autoantibody production in lupus nephritis. J Am Soc Nephrol. 2011, 22: 296-305. 10.1681/ASN.2010050515.
Hutloff A, Buchner K, Reiter K, Baelde HJ, Odendahl M, Jacobi A, Dorner T, Kroczek RA: Involvement of inducible costimulator in the exaggerated memory B cell and plasma cell generation in systemic lupus erythematosus. Arthritis Rheum. 2004, 50: 3211-3220. 10.1002/art.20519.
Steinmetz OM, Velden J, Kneissler U, Marx M, Klein A, Helmchen U, Stahl RA, Panzer U: Analysis and classification of B-cell infiltrates in lupus and ANCA-associated nephritis. Kidney Int. 2008, 74: 448-457. 10.1038/ki.2008.191.
Chang A, Henderson SG, Brandt D, Liu N, Guttikonda R, Hsieh C, Kaverina N, Utset TO, Meehan SM, Quigg RJ, Meffre E, Clark MR: In situ B cell-mediated immune responses and tubulointerstitial inflammation in human lupus nephritis. J Immunol. 2011, 186: 1849-1860. 10.4049/jimmunol.1001983.
Ramanujam M, Bethunaickan R, Huang W, Tao H, Madaio MP, Davidson A: Selective blockade of BAFF for the prevention and treatment of systemic lupus erythematosus nephritis in NZM2410 mice. Arthritis Rheum. 2010, 62: 1457-1468.
Shlomchik MJ, Madaio MP, Ni D, Trounstein M, Huszar D: The role of B cells in lpr/lpr-induced autoimmunity. J Exp Med. 1994, 180: 1295-1306. 10.1084/jem.180.4.1295.
Chan OT, Madaio MP, Shlomchik MJ: B cells are required for lupus nephritis in the polygenic, Fas-intact MRL model of systemic autoimmunity. J Immunol. 1999, 163: 3592-3596.
Bekar KW, Owen T, Dunn R, Ichikawa T, Wang W, Wang R, Barnard J, Brady S, Nevarez S, Goldman BI, Kehry M, Anolik JH: Prolonged effects of short-term anti-CD20 B cell depletion therapy in murine systemic lupus erythematosus. Arthritis Rheum. 2010, 62: 2443-2457. 10.1002/art.27515.
Sfikakis PP, Boletis JN, Lionaki S, Vigklis V, Fragiadaki KG, Iniotaki A, Moutsopoulos HM: Remission of proliferative lupus nephritis following B cell depletion therapy is preceded by down-regulation of the T cell costimulatory molecule CD40 ligand: an open-label trial. Arthritis Rheum. 2005, 52: 501-513. 10.1002/art.20858.
Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ: A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999, 189: 1639-1648. 10.1084/jem.189.10.1639.
Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, Herrmann M, Voll RE, Zychlinsky A: Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. 2010, 107: 9813-9818. 10.1073/pnas.0909927107.
Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, Rubin CJ, Zhao W, Olsen SH, Klinker M, Shealy D, Denny MF, Plumas J, Chaperot L, Kretzler M, Bruce AT, Kaplan MJ: Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011, 187: 538-552. 10.4049/jimmunol.1100450.
Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL, Barrat FJ, Banchereau J, Pascual V: Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011, 3: 73ra20-10.1126/scitranslmed.3001201.
Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R, Riccieri V, Bassett R, Amuro H, Fukuhara S, Ito T, Liu YJ, Gilliet M: Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011, 3: 73ra19-10.1126/scitranslmed.3001180.
Iwata Y, Furuichi K, Sakai N, Yamauchi H, Shinozaki Y, Zhou H, Kurokawa Y, Toyama T, Kitajima S, Okumura T, Yamada S, Maruyama I, Matsushima K, Kaneko S, Wada T: Dendritic cells contribute to autoimmune kidney injury in MRL-Faslpr mice. J Rheumatol. 2009, 36: 306-314. 10.3899/jrheum.080318.
Tucci M, Quatraro C, Lombardi L, Pellegrino C, Dammacco F, Silvestris F: Glomerular accumulation of plasmacytoid dendritic cells in active lupus nephritis: role of interleukin-18. Arthritis Rheum. 2008, 58: 251-262. 10.1002/art.23186.
Schiffer L, Bethunaickan R, Ramanujam M, Huang W, Schiffer M, Tao H, Madaio MP, Bottinger EP, Davidson A: Activated renal macrophages are markers of disease onset and disease remission in lupus nephritis. J Immunol. 2008, 180: 1938-1947.
Triantafyllopoulou A, Franzke CW, Seshan SV, Perino G, Kalliolias GD, Ramanujam M, van Rooijen N, Davidson A, Ivashkiv LB: Proliferative lesions and metalloproteinase activity in murine lupus nephritis mediated by type I interferons and macrophages. Proc Natl Acad Sci USA. 2010, 107: 3012-3017. 10.1073/pnas.0914902107.
Bethunaickan R, Berthier CC, Ramanujam M, Sahu R, Zhang W, Sun Y, Bottinger EP, Ivashkiv L, Kretzler M, Davidson A: A unique hybrid renal mononuclear phagocyte activation phenotype in murine systemic lupus erythematosus nephritis. J Immunol. 2011, 186: 4994-5003. 10.4049/jimmunol.1003010.
Hill GS, Delahousse M, Nochy D, Remy P, Mignon F, Mery JP, Bariety J: Predictive power of the second renal biopsy in lupus nephritis: significance of macrophages. Kidney Int. 2001, 59: 304-316.
Katsiari CG, Liossis SN, Sfikakis PP: The pathophysiologic role of monocytes and macrophages in systemic lupus erythematosus: a reappraisal. Semin Arthritis Rheum. 2010, 39: 491-503. 10.1016/j.semarthrit.2008.11.002.
Fan X, Oertli B, Wuthrich RP: Up-regulation of tubular epithelial interleukin-12 in autoimmune MRL-Fas(lpr) mice with renal injury. Kidney Int. 1997, 51: 79-86. 10.1038/ki.1997.10.
Fan X, Wuthrich RP: Upregulation of lymphoid and renal interferon-gamma mRNA in autoimmune MRL-Fas(lpr) mice with lupus nephritis. Inflammation. 1997, 21: 105-112. 10.1023/A:1027399027170.
Chan RW, Lai FM, Li EK, Tam LS, Chow KM, Li PK, Szeto CC: Imbalance of Th1/Th2 transcription factors in patients with lupus nephritis. Rheumatology (Oxford). 2006, 45: 951-957. 10.1093/rheumatology/kel029.
Chan RW, Lai FM, Li EK, Tam LS, Chow KM, Lai KB, Li PK, Szeto CC: Intrarenal cytokine gene expression in lupus nephritis. Ann Rheum Dis. 2007, 66: 886-892. 10.1136/ard.2006.063123.
Menke J, Bork T, Kutska B, Byrne KT, Blanfeld M, Relle M, Kelley VR, Schwarting A: Targeting transcription factor Stat4 uncovers a role for interleukin-18 in the pathogenesis of severe lupus nephritis in mice. Kidney Int. 2011, 79: 452-463. 10.1038/ki.2010.438.
Schwarting A, Tesch G, Kinoshita K, Maron R, Weiner HL, Kelley VR: IL-12 drives IFN-gamma-dependent autoimmune kidney disease in MRLFas(lpr) mice. J Immunol. 1999, 163: 6884-6891.
Kikawada E, Lenda DM, Kelley VR: IL-12 deficiency in MRL-Fas(lpr) mice delays nephritis and intrarenal IFN-gamma expression, and diminishes systemic pathology. J Immunol. 2003, 170: 3915-3925.
Tucci M, Lombardi L, Richards HB, Dammacco F, Silvestris F: Overexpression of interleukin-12 and T helper 1 predominance in lupus nephritis. Clin Exp Immunol. 2008, 154: 247-254. 10.1111/j.1365-2249.2008.03758.x.
Schwarting A, Wada T, Kinoshita K, Tesch G, Kelley VR: IFN-gamma receptor signaling is essential for the initiation, acceleration, and destruction of autoimmune kidney disease in MRL-Fas(lpr) mice. J Immunol. 1998, 161: 494-503.
Perez de Lema G, Maier H, Nieto E, Vielhauer V, Luckow B, Mampaso F, Schlondorff D: Chemokine expression precedes inflammatory cell infiltration and chemokine receptor and cytokine expression during the initiation of murine lupus nephritis. J Am Soc Nephrol. 2001, 12: 1369-1382.
Eriksson C, Eneslatt K, Ivanoff J, Rantapaa-Dahlqvist S, Sundqvist KG: Abnormal expression of chemokine receptors on T-cells from patients with systemic lupus erythematosus. Lupus. 2003, 12: 766-774. 10.1191/0961203303lu467oa.
Narumi S, Takeuchi T, Kobayashi Y, Konishi K: Serum levels of IFN-inducible PROTEIN-10 relating to the activity of systemic lupus erythematosus. Cytokine. 2000, 12: 1561-1565. 10.1006/cyto.2000.0757.
Tesch GH, Maifert S, Schwarting A, Rollins BJ, Kelley VR: Monocyte chemoattractant protein 1-dependent leukocytic infiltrates are responsible for autoimmune disease in MRL-Fas(lpr) mice. J Exp Med. 1999, 190: 1813-1824. 10.1084/jem.190.12.1813.
Shimizu S, Nakashima H, Masutani K, Inoue Y, Miyake K, Akahoshi M, Tanaka Y, Egashira K, Hirakata H, Otsuka T, Harada M: Anti-monocyte chemoattractant protein-1 gene therapy attenuates nephritis in MRL/lpr mice. Rheumatology (Oxford). 2004, 43: 1121-1128. 10.1093/rheumatology/keh277.
Kulkarni O, Pawar RD, Purschke W, Eulberg D, Selve N, Buchner K, Ninichuk V, Segerer S, Vielhauer V, Klussmann S, Anders HJ: Spiegelmer inhibition of CCL2/MCP-1 ameliorates lupus nephritis in MRL-(Fas)lpr mice. J Am Soc Nephrol. 2007, 18: 2350-2358. 10.1681/ASN.2006121348.
Kiani AN, Johnson K, Chen C, Diehl E, Hu H, Vasudevan G, Singh S, Magder LS, Knechtle SJ, Petri M: Urine osteoprotegerin and monocyte chemoattractant protein-1 in lupus nephritis. J Rheumatol. 2009, 36: 2224-2230. 10.3899/jrheum.081112.
Marks SD, Shah V, Pilkington C, Tullus K: Urinary monocyte chemoattractant protein-1 correlates with disease activity in lupus nephritis. Pediatr Nephrol (Berlin). 2010, 25: 2283-2288. 10.1007/s00467-010-1605-z.
Dong J, Wang QX, Zhou CY, Ma XF, Zhang YC: Activation of the STAT1 signalling pathway in lupus nephritis in MRL/lpr mice. Lupus. 2007, 16: 101-109. 10.1177/0961203306075383.
Martinez-Lostao L, Ordi-Ros J, Balada E, Segarra-Medrano A, Majo-Masferrer J, Labrador-Horrillo M, Vilardell-Tarres M: Activation of the signal transducer and activator of transcription-1 in diffuse proliferative lupus nephritis. Lupus. 2007, 16: 483-488. 10.1177/0961203307079618.
Taylor KE, Remmers EF, Lee AT, Ortmann WA, Plenge RM, Tian C, Chung SA, Nititham J, Hom G, Kao AH, Demirci FY, Kamboh MI, Petri M, Manzi S, Kastner DL, Seldin MF, Gregersen PK, Behrens TW, Criswell LA: Specificity of the STAT4 genetic association for severe disease manifestations of systemic lupus erythematosus. PLoS Genet. 2008, 4: e1000084-10.1371/journal.pgen.1000084.
Singh RR, Saxena V, Zang S, Li L, Finkelman FD, Witte DP, Jacob CO: Differential contribution of IL-4 and STAT6 vs STAT4 to the development of lupus nephritis. J Immunol. 2003, 170: 4818-4825.
Jacob CO, Zang S, Li L, Ciobanu V, Quismorio F, Mizutani A, Satoh M, Koss M: Pivotal role of Stat4 and Stat6 in the pathogenesis of the lupus-like disease in the New Zealand mixed 2328 mice. J Immunol. 2003, 171: 1564-1571.
Weinberg JB, Granger DL, Pisetsky DS, Seldin MF, Misukonis MA, Mason SN, Pippen AM, Ruiz P, Wood ER, Gilkeson GS: The role of nitric oxide in the pathogenesis of spontaneous murine autoimmune disease: increased nitric oxide production and nitric oxide synthase expression in MRL-lpr/lpr mice, and reduction of spontaneous glomerulonephritis and arthritis by orally administered NG-monomethyl-l-arginine. J Exp Med. 1994, 179: 651-660. 10.1084/jem.179.2.651.
Weinberg JB, Gilkeson GS, Mason RP, Chamulitrat W: Nitrosylation of blood hemoglobin and renal nonheme proteins in autoimmune MRL-lpr/lpr mice. Free Radic Biol Med. 1998, 24: 191-196. 10.1016/S0891-5849(97)00217-7.
Reilly CM, Olgun S, Goodwin D, Gogal RM, Santo A, Romesburg JW, Ahmed SA, Gilkeson GS: Interferon regulatory factor-1 gene deletion decreases glomerulonephritis in MRL/lpr mice. Eur J Immunol. 2006, 36: 1296-1308. 10.1002/eji.200535245.
Oates JC, Ruiz P, Alexander A, Pippen AM, Gilkeson GS: Effect of late modulation of nitric oxide production on murine lupus. Clin Immunol Immunopathol. 1997, 83: 86-92. 10.1006/clin.1997.4332.
Keng T, Privalle CT, Gilkeson GS, Weinberg JB: Peroxynitrite formation and decreased catalase activity in autoimmune MRL-lpr/lpr mice. Mol Med. 2000, 6: 779-792.
Oates JC, Gilkeson GS: The biology of nitric oxide and other reactive intermediates in systemic lupus erythematosus. Clin Immunol. 2006, 121: 243-250. 10.1016/j.clim.2006.06.001.
Prasad R, Giri S, Nath N, Singh I, Singh AK: GSNO attenuates EAE disease by S-nitrosylation-mediated modulation of endothelial-monocyte interactions. Glia. 2007, 55: 65-77. 10.1002/glia.20436.
Oates JC, Christensen EF, Reilly CM, Self SE, Gilkeson GS: Prospective measure of serum 3-nitrotyrosine levels in systemic lupus erythematosus: correlation with disease activity. Proc Assoc Am Phys. 1999, 111: 611-621. 10.1046/j.1525-1381.1999.99110.x.
Oates JC, Shaftman SR, Self SE, Gilkeson GS: Association of serum nitrate and nitrite levels with longitudinal assessments of disease activity and damage in systemic lupus erythematosus and lupus nephritis. Arthritis Rheum. 2008, 58: 263-272. 10.1002/art.23153.
Wang JS, Tseng HH, Shih DF, Jou HS, Ger LP: Expression of inducible nitric oxide synthase and apoptosis in human lupus nephritis. Nephron. 1997, 77: 404-411. 10.1159/000190316.
Oates JC, Farrelly LW, Hofbauer AF, Wang W, Gilkeson GS: Association of reactive oxygen and nitrogen intermediate and complement levels with apoptosis of peripheral blood mononuclear cells in lupus patients. Arthritis Rheum. 2007, 56: 3738-3747. 10.1002/art.22955.
Bouali H, Nietert P, Nowling TM, Pandey J, Dooley MA, Cooper G, Harley J, Kamen DL, Oates J, Gilkeson G: Association of the G-463A myeloperoxidase gene polymorphism with renal disease in African Americans with systemic lupus erythematosus. J Rheumatol. 2007, 34: 2028-2034.
Oates JC, Levesque MC, Hobbs MR, Smith EG, Molano ID, Page GP, Hill BS, Weinberg JB, Cooper GS, Gilkeson GS: Nitric oxide synthase 2 promoter polymorphisms and systemic lupus erythematosus in African-Americans. J Rheumatol. 2003, 30: 60-67.
Douglas G, Reilly C, Dooley MA, Page G, Cooper G, Gilkeson G: Angiotensin-converting enzyme (insertion/deletion) and endothelial nitric oxide synthase polymorphisms in patients with systemic lupus erythematosus. J Rheumatol. 2004, 31: 1756-1762.
Thomas MC, Cooper ME: Diabetes: bardoxolone improves kidney function in type 2 diabetes. Nat Rev. 2011, 7: 552-553.
Yang W, Shen N, Ye DQ, Liu Q, Zhang Y, Qian XX, Hirankarn N, Ying D, Pan HF, Mok CC, Chan TM, Wong RW, Lee KW, Mok MY, Wong SN, Leung AM, Li XP, Avihingsanon Y, Wong CM, Lee TL, Ho MH, Lee PP, Chang YK, Li PH, Li RJ, Zhang L, Wong WH, Ng IO, Lau CS, Sham PC, et al: Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet. 2010, 6: e1000841-10.1371/journal.pgen.1000841.
Dussaule JC, Guerrot D, Huby AC, Chadjichristos C, Shweke N, Boffa JJ, Chatziantoniou C: The role of cell plasticity in progression and reversal of renal fibrosis. Int J Exp Pathol. 2011, 92: 151-157. 10.1111/j.1365-2613.2011.00760.x.
Acknowledgements
The authors thank Lisa Fennessy for help with production of Figure 1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
TKN and GSG contributed equally to the drafting and editing of the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Nowling, T.K., Gilkeson, G.S. Mechanisms of tissue injury in lupus nephritis . Arthritis Res Ther 13, 250 (2011). https://doi.org/10.1186/ar3528
Published:
DOI: https://doi.org/10.1186/ar3528