
Abstract
MicroRNAs (miRNAs) are endogenous, non-coding, single-stranded RNAs about 21 nucleotides in length. miRNAs have been shown to regulate gene expression and thus influence a wide range of physiological and pathological processes. Moreover, they are detected in a variety of sources, including tissues, serum, and other body fluids, such as saliva. The role of miRNAs is evident in various malignant and nonmalignant diseases, and there is accumulating evidence also for an important role of miRNAs in systemic rheumatic diseases. Abnormal expression of miRNAs has been reported in autoimmune diseases, mainly in systemic lupus erythematosus and rheumatoid arthritis. miRNAs can be aberrantly expressed even in the different stages of disease progression, allowing miRNAs to be important biomarkers, to help understand the pathogenesis of the disease, and to monitor disease activity and effects of treatment. Different groups have demonstrated a link between miRNA expression and disease activity, as in the case of renal flares in lupus patients. Moreover, miRNAs are emerging as potential targets for new therapeutic strategies of autoimmune disorders. Taken together, recent data demonstrate that miRNAs can influence mechanisms involved in the pathogenesis, relapse, and specific organ involvement of autoimmune diseases. The ultimate goal is the identification of a miRNA target or targets that could be manipulated through specific therapies, aiming at activation or inhibition of specific miRNAs responsible for the development of disease.
Similar content being viewed by others
Introduction
Since their initial discovery in 1993 [1], microRNAs (miRNAs) have been studied extensively due to their role in the regulation of almost every cellular process thus far investigated. miRNAs are noncoding RNAs about 21 nucleotides in length that function as post-transcriptional regulators of gene expression [2]. They can influence the activity of about 50% or more of all protein-coding genes in mammals [2], and their change in expression is associated with human diseases, including infectious diseases, cancer, and rheumatic diseases [3–5]. Over 800 human miRNAs have been identified so far [2], and they have been shown to negatively regulate protein expression through the inhibition of translation and/or decrease in mRNA stability [6–8]. It is now apparent that miRNAs can potentially regulate every aspect of cellular activity, from dierentiation and proliferation to apoptosis, and they can also modulate a large range of physiological and pathological processes [6].
Biogenesis and function of miRNAs
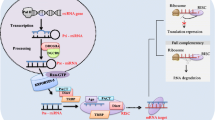
The first step in the biogenesis of mammalian miRNAs is the generation of primary miRNA transcripts (pri-miRNAs) in the nucleus [2]. The pri-miRNAs fold into hairpins and act as substrate for Drosha, which is one of the two members of the RNase III family involved in the miRNA maturation process. The product of Drosha cleavage, an approximately 70-nucleotide precursor miRNA (pre-miRNA), is exported to the cytoplasm where Dicer, the second RNase III family member, processes the pre-miRNA to a 20- to 23-nucleotide miRNA/miRNA* duplex [2]. The preferential loading of one miRNA strand (the guiding strand or mature miRNA) onto the RNA-induced silencing complex (RISC) over the other strand (passenger strand, miRNA*) apparently is based on the thermo-dynamic stability of the miRNA duplex. The mature miRNA with RISC will bind to and silence its target mRNA based on seed sequence complementarity, generally at the 3' UTR. The miRNA* may be discarded and eventually degraded [8, 9], but recent reports are showing that some miRNA* are stably expressed and they are implicated in important functions as well [9]. An interesting example is the report of Zhou and colleagues [10] demonstrating upregulation of miR-155 and miR-155* in human plasmacytoid dendritic cells to co-ordinate function in stimulating type I IFN production.
Since the first miRNAs discovered (lin-4 and let-7) were shown to bind the 3' UTR of target mRNAs, it has been widely believed that miRNAs exert their effects through a perfect or imperfect complementarity with sequences in the 3' UTR only. The imperfect complementarity still requires perfect target matching of the second through the seventh nucleotides ('seed sequence') starting from the 5' end of the miRNA [3]. However, it has been recently shown that miRNAs can also bind to the 5' UTR region and to protein coding sequences, albeit causing relatively weak repression [3]. Another very recent study challenged the traditional seed match principle by demonstrating a novel centered pairing between miRNA and mRNA, the 'centered sites', which consist of a class of miRNA target sites that lack both perfect seed pairing and 3' compensatory pairing and have 11 to 12 contiguous Watson-Crick pairs of miRNA nucleotides 4 to 15 [11]. This leads to more versatility in miRNA regulation of specific targets and, more importantly, may fail to be predicted by the most common algorithms designed to detect miRNA binding sites in the 3' UTR [11]. Other recent studies have also shown the ability of certain miRNAs in translational activation [12], which suggests that our knowledge of overall biological function for miRNAs remains some-what incomplete.
Key macromolecules of RISC are targets of human autoantibodies
The two best characterized protein families in the RISC complex, the Argonaute family and GW182 (glycine-tryptophan dipeptide-rich protein of 182 kDa), are known to play a central role in silencing mRNA translation as well as triggering mRNA degradation. They are essential components of the GW bodies (also known as mammalian processing bodies, or P bodies). Interestingly, both are known autoantigens recognized by autoantibodies in various disease states [13–15]. The Argonaute family comprises four Argonaute (Ago) proteins (Ago1 to 4) in mammals, and they have all been shown to interact with miRNAs [16, 17] and repress protein translation when artificially tethered to the 3' UTR of reporter mRNAs [18, 19]. However, Ago-mediated repression requires them to interact with another protein, GW182, which is the key silencer downstream of Ago2 [19]. GW182 (also known as TNRC6A) is a 182- kDa protein characterized by multiple glycine (G) and tryptophan (W) motifs and is a very important constituent of GW bodies [20, 21]. The GW182 family includes three paralogues of TNRC6 (GW182-related) proteins, GW182/TNGW1, TNRC6B (containing three isoforms), and TNRC6C, in mammals [22, 23]. A number of different models have been proposed for the GW182 silencing mechanism in the miRNA pathway, including its interference with translational initiation and 80S complex assembly as well as post-initiation steps, but the detailed molecular process remains to be explored [8, 9]. Recent studies also demonstrated that GW182 interacts with Poly-A binding protein (PABP) and further recruits deadenylase complex to promote miRNA-targeted mRNA decay [24, 25]. GW182 was originally identified and cloned in 2002 as a novel protein recognized by an autoimmune serum from a patient with motor and sensory neuropathy [15, 26]. In 2006, Jakymiw and colleagues [13] showed that the Ago2 protein corresponds to the 100-kDa component of the so-called 'anti-Su antibodies', and for this reason we now call these antibodies 'anti-Ago2/Su'. Since their identification [27, 28], anti-Ago2/Su antibodies have been detected in various diseases, including autoimmune and infectious disease [13, 15, 28–30]. However, the clinical significance of anti-Ago2/Su antibodies has not been established yet [15, 30].
MicroRNAs in rheumatic diseases
As miRNAs emerge to play important roles in many biological processes, they have been referred to as master regulators of gene expression, with a concept where a single miRNA may regulate an entire pathway or even multiple pathways [6]. Regulation of the immune system is vital to prevent many pathogenic disorders and mammals have developed a complex system of checks and balances for immune regulation in order to maintain self-tolerance while allowing immune responses to foreign pathogens [5]. Only in recent years has more evidence emerged to support a central role for miRNAs also in abnormal immune processes and in rheumatic diseases. In fact, the potential of miRNAs as biomarkers in rheumatic diseases is a new and growing area of research [3, 5]. The identification of candidate miRNAs that target genes implicated in rheumatic disorders and the evaluation of the consequences of mutations in their target sites coupled to phenotypic and gene expression studies should improve our understanding of the molecular mechanisms responsible for rheumatic diseases [31]. Increased knowledge of miRNAs has led to the development of mouse models for studying in vivo therapeutic approaches using specific miRNAs [32]. In particular, Nagata and colleagues [32] have performed the intraarticular injection of double-stranded miR-15a in the synovium of mice with autoantibody-mediated arthritis. Through this experiment, they have shown that this miRNA is capable of cell entry and induces cell apoptosis through targeting Bcl-2, which is known normally to suppress apoptotic processes [32].
miR-146a appears to be an interesting example of a master regulator in several aspects of immunity. Specifically, it contributes to controlling the overproduction of cytokines, such as TNF-α, and it functions as a negative feedback control of innate immunity in toll-like receptor (TLR) signaling during recurrent bacterial infection by establishing endotoxin tolerance [33] and cross-tolerance [34]. Lu and colleagues [35] recently demonstrated that miR-146a is critical for the suppressor functions of regulatory T (Treg) cells. In fact, a miR-146a knockout mouse showed some loss of immunological tolerance, responsible for fatal IFNγ-dependent immune-mediated lesions in different organs [35]. This is an example of how specific cellular aspects can also be controlled by a single miRNA, where the lack of function of miRNAs can be responsible for the onset of autoimmune disease. In another study, Curtale and colleagues [36] showed that miR-146a is involved in T-cell activation and is highly expressed in mature human memory T cells. miR-146a can modulate activation-induced cell death processes, thus acting as an anti-apoptotic factor in T cells, and it is also able to reduce the expression of cytokines, such as IL-2, induced by T-cell receptor engagement in the adaptive immune response [36].
Another miRNA widely studied for its key role in autoimmunity is miR-155. It functions in the hemato-poietic compartment to promote the development of inflammatory T cells, including the T helper (Th)17 and Th1 cell subset [37]. Divekar and colleagues have investigated the influence of miR-155 on Treg cells in a mouse model (MRL/lpr) of systemic lupus erythematosus (SLE) [38]. These investigators have shown an increase in CD4+CD25+Foxp3+ Treg cells that have an altered phenotype and reduced suppressive capacity. Searching for the reason for this alteration, they detected a significant reduction of Dicer expression and the over-expression of some miRNAs in MRL/lpr Treg cells, including miR-155, which is able to target CD62L in Treg cells. The results of this study show that elevated miR-155 expression together with a reduced level of Dicer can be responsible for the Treg cell phenotype in MRL/lpr mice [38]. This study also introduces a new concept that some miRNAs may be produced in this SLE model independently of Dicer, as described recently in mouse embryonic stem cells [39]. miR-155 also plays an important role in mouse models of collagen-induced arthritis and K/BxN serum transfer arthritis [40]. In fact, miR-155 knockout mice do not develop collagen-induced arthritis. In the K/BxN serum transfer arthritis model, the miR-155-/- mice show a reduction in pathogenic autoreactive B and T cells and cytokine production (IL-6, IL-17 and IL-22) and local bone destruction is reduced because of a decreased generation of osteoclasts [40]. These results support a possible therapeutic role for miRNAs in rheumatoid arthritis (RA).
Beside immune and autoimmune mechanisms, the study of miRNAs as biomarkers is most advanced in oncology [3]. Initial reports showed that cancer cells and tissues have different miRNA profiles from normal cells and tissues, suggesting that they could be used for diagnosis, prognosis and therapeutic outcome [3]. By the regulation of gene expression at the post-transcriptional level, they affect various signaling cascades during the progression of neoplastic diseases [41]. Sustained angiogenesis is one of the mechanisms leading to cancer progression. Recently, a role of the secreted protein epidermal growth factor-like domain 7 (EGFL7) in the control of vascular tubulogenesis has been suggested. Interestingly, the two biologically active miRNAs miR-126 and its complement miR-126*, which are encoded by intron 7 of the EGFL7 gene, have been shown to mediate vascular functions [41], promoting blood vessel growth and inflammation. Complex networks of reprogramming of miRNAs have been detected in cancer and leukemia and, given the critical role that miRNAs play in tumorigenesis processes and their disease-specific expression, they have the potential to become therapeutic targets and specific cancer biomarkers [42, 43].
In the present review, we will focus our attention on recent developments in understanding the role of miRNAs in autoimmune rheumatic diseases, such as SLE, RA, systemic sclerosis (SSc; scleroderma), Sjögren's syndrome (SS) and polymyositis/dermatomyositis (PM/DM).
Systemic lupus erythematosus
SLE is a systemic inflammatory autoimmune disease characterized by the presence of autoantibodies against a large number of self-antigens, including chromatin, ribonucleoproteins, and phospholipids. Clinical manifestations are heterogeneous and include malar rash, photo-sensitivity, arthritis, glomerulonephritis, and neurological disorders [5, 44, 45]. Since 2007, different groups have reported altered miRNA expression in tissues and peripheral blood mononuclear cells (PBMCs) from SLE patients [46, 47], but these first reports mainly identified groups of miRNAs that were aberrantly expressed through microarray chip analysis, without defining potential pathways they participate in. Table 1 summarizes studies that are more focused on the identification of specific aberrant miRNAs in SLE and other diseases. For example, Tang and colleagues [48] have studied the role of miR-146a, showing that it is down-regulated in SLE. They have evaluated the IFN score through the expression levels of the IFN signature genes OAS1, MX1, and LY6E [48]. Since it is known that miR-146a targets adaptors TRAF6 (TNF receptor-associated factor 6) and IRAK1 (IL-1 receptor-associated kinase 1) in the pathway to NF-κB activation (Figure 1a), they have postulated that the lower expression levels of miR-146a in lupus PBMCs is inversely correlated with the IFN score and may be responsible for IFN overproduction in SLE [48]. They have also demonstrated that low miR-146a and high IFN expression correlate with SLE disease activity, in particular with renal disease [48].

Contribution of aberrant miRNA expression in SLE PBMCs. (A) miR-146a is down-regulated in systemic lupus erythematosus (SLE) peripheral blood mononuclear cells (PBMCs) and this may amplify the activation of NF-kB through its direct regulation of NF-kB upstream regulators IRAK1 (IL-1 receptor-associated kinase 1) and TRAF6 (TNF receptor-associated factor 6). Activation of NF-kB leads to increased type I IFN production and thus increased expression of 'IFN signature genes', including LY6E, OAS1, and MX1 [48]. (B) miR-125a is down-regulated in PBMCs from SLE patients, which leads to elevated expression of its target transcriptional factor KLF13. KLF13 can trigger the expression of the pro-inflammatory chemokine RANTES (Regulated upon activation, normal T-cell expressed, and secreted), which is known to enhance inflammatory processes such as arthritis and nephritis [49]. (C) Up-regulation of miR-21, miR-148, and miR-126 can either directly or indirectly inhibit DNA methyltransferase 1 (DNMT1) levels. This inhibition in turn reduces the CpG methylation level and causes over-expression of autoimmune-associated genes in SLE, such as those encoding CD70, LFA-1 (CD11a) and EGFL7 (epidermal growth factor-like domain 7) [50, 51]. An, poly-A tail; CH3, methyl groups; RASGRP1, RAS guanyl-releasing protein 1.
The same group studied another miRNA, miR-125a, reporting that the miR-125a level is reduced in PBMCs from SLE patients, and the expression of the predicted target of miR-125a, KLF13, was increased [49]. The final result is the significant over-expression of the inflammatory chemokine RANTES (Regulated upon activation, normal T-cell expressed, and secreted; also called CCL5), which is known to be highly expressed and have detrimental effects in inflammatory processes, including arthritis and nephritis (Figure 1b). This study demonstrated that miR-125a negatively regulates RANTES expression by targeting KLF13, as shown by manipulation studies of activated T cells from lupus patients [49].
While miR-146a and miR-125a are down-regulated in SLE patients, other miRNAs can be up-regulated, as is the case for miR-21, miR-148, and miR-126 (Figure 1c) [50, 51]. In contrast, miR-21 and miR-148 are over-expressed in PBMCs of SLE patients, and it has been demonstrated that they can target the DNA-methylation pathway, causing DNA hypomethylation and over-expression of autoimmune-associated methylation-sensitive genes, such as CD70 and LFA-1 (CD11a) [50]. The same targets are also influenced by another miRNA, miR-126, which is also directed to the EGFL7 gene. In this case, the final result is the DNA hypomethylation and overexpression of autoimmune-associated genes, leading to the autoimmune response in SLE [51].
Rheumatoid arthritis
RA is a systemic autoimmune disorder characterized by chronic inflammation of synovial tissue that results in irreversible joint damage [52]. Inflammatory cytokines, especially TNF -α, IL-1β, and IL-6, are known to play an important role in the pathogenesis of RA, as the inhibition of these cytokines can ameliorate disease symptoms in patients [53]. In recent years, many studies have focused on the identification of altered miRNA expression in RA patients compared to healthy controls or patients affected by osteoarthritis [54–56]. Some studies mainly considered miRNA expression in plasma and serum, while others mainly focused on tissue analysis (Table 1) [57]. Two of these studies examined miRNA expression in RA synovial tissue and fibroblasts. Stanczyk and colleagues [56] reported an increase of miR-155 and miR-146a expression in both RA synovial fibroblasts (RASFs) and RA synovial tissue compared to osteoarthritis patients. These investigators concluded that the inflammatory milieu of RA may alter miRNA expression profiles in resident cells of the rheumatoid joints.
Considering that miR-155 had a repressive effect on the expression of two matrix metalloproteinases (MMP-3 and MMP-1) in RASFs, Stanczyk and colleagues [56] hypothesize that miR-155 may be involved in the modulation of joint destructive properties of RASFs, and in the control of the excessive tissue destruction due to inflammation. The same group has recently identified another miRNA, miR-203, highly expressed in RASFs and they demonstrated methylation-dependent regulation of miR-203 expression. Moreover, high expression of miR-203 led to increased secretion of MMP-1 and IL-6 via the NF-kB pathway, contributing to the activated phenotype of synovial fibroblasts in RA [58].
Two other miRNAs have been detected at high levels in RA. In particular, miR-223 is up-regulated in CD4+ naïve T lymphocytes of RA patients, and a possible role of this miRNA in the pathogenesis of the disease has been hypothesized [59]. Alsaleh and colleagues [60] studied the overexpression of miR-346 in RA fibroblast-like synoviocytes, showing that miR-346 indirectly regulates the release of the pro-inflammatory cytokine IL-18.
Nakasa and colleagues [54] have studied the expression pattern of miR-146a in synovial tissue from patients with RA. They reported increased expression of mature miR-146a and primary miR-146a/b in RA synovial tissue, which also expressed TNF-α [54]. Cells positive for miR-146a are primarily CD68+ macrophages, but also some CD3+ T cell subsets and CD79a+ B cells [54]. The expression of miR-146a/b is markedly up-regulated in RASFs after stimulation with TNF-α and IL-1β [54] (Figure 2a). Our group has implemented a different approach to examine miRNA expression in RA patients compared to healthy controls [55]. Pauley and colleagues [55] have shown increased expression of miR-146a, miR-155, miR-132 and miR-16 in RA PBMCs. In addition, two targets of miR-146a, TRAF6 and IRAK1, are similarly expressed between RA patients and control individuals, despite increased expression of miR-146a in patients with RA. Repression of TRAF6 and/or IRAK1 in THP-1 cells resulted in up to an 86% reduction in TNF-α production, implying that normal miR-146a function is critical for the regulation of TNF-α production. Our data thus demonstrate that miRNA expression in RA PBMCs mimics that of synovial tissue/fibroblasts, and our hypothesis is that miR-146a is upregulated but unable to properly regulate TRAF6/IRAK1, leading to prolonged TNF-α production in RA patients [55]. More recently, Nimoto and colleagues [61] confirmed the upregulation of miR-146 a/b in PBMCs of RA patients, which seems to be involved in the overexpression of the pro-inflammatory cytokine IL-17. Other groups have also demonstrated the overexpression of miR-146a in CD4+ T cells from RA patients, which is closely related to TNF-α expression and to regulation of T-cell apoptosis, thus maintaining the pro-inflammatory milieu typical of RA patients [62]. A recent report by Nakamachi and colleagues [63] has shown another miRNA, miR-124a, is involved in RA. They have found that miR-124a levels are significantly decreased in RA synoviocytes compared to osteoarthritis synoviocytes. Transfection of precursor miR-124a into RA synoviocytes led to the significant suppression of cell proliferation and arrest of the cell cycle at the G1 phase. They identified a putative consensus site for miR-124a binding in the 3' UTR of cyclin-dependent kinase 2 (CDK2) and monocyte chemo-attractant protein 1 (MCP-1) mRNA. In fact, induction of miR-124a in RA synoviocytes significantly suppressed the production of the CDK2 and MCP-1 proteins [63]. Thus, these investigators show that miR-124a is also a key miRNA in the post-transcriptional regulatory mechanism of RA synoviocytes (Figure 2b).

Aberrant expression of miRNAs in rheumatoid arthritis synoviocytes. (A) Contrary to peripheral blood mononuclear cells (PBMCs) from systemic lupus erythematosus patients, miR-146a is up-regulated in rheumatoid arthritis (RA) synoviocytes and PBMCs. miR-146a is a known regulator of IRAK1 (IL-1 receptor-associated kinase 1) and TRAF6 (TNF receptor-associated factor 6) mRNA and this may be responsible for the altered regulation of IRAK1 and TRAF6, both of which act through the NF-kB pathway to prolong the production of proinflammatory cytokines and chemokines, including TNF-α and IL-1β [3, 55]. (B) miR-124a is down-regulated in synoviocytes from RA patients. Its target proteins, CDK2 (cyclin-dependent kinase 2) and MCP-1 (monocyte chemoattractant protein 1), are up-regulated and this leads to increased synovial proliferation, angiogenesis and chemotaxis [63].
Other autoimmune diseases
Sjögren's syndrome
SS is an autoimmune inflammatory exocrinopathy affecting the lacrimal and salivary glands, leading to dry eyes and mouth [64]. It is often associated with positive anti-SSA/Ro and anti-SSB/La antibodies and with other systemic symptoms, such as arthritis, lymphadenopathy, interstitial pneumonia, and renal disease [64]. The role of miRNAs in SS has not been widely explored yet (Table 1). Alevizos and colleagues [65] identified miRNA signatures from the minor salivary glands of patients with SS and normal controls. This analysis allowed them to distinguish between these two populations, as well as between subsets of SS patients with low-grade or high-grade inflammation [65]. Michael and colleagues [66] explored the presence of miRNAs in saliva exosomes isolated from parotid and submandibular glands of patients with SS. They have shown that miRNAs can be identified in saliva, which suggests it may be possible to obtain information from these target organs without the need for invasive methods, such as biopsies. The same group also identified the miR-17-92 cluster as responsible for the accumulation of pro-B cells and the marked reduction of pre-B and more mature B cells in patients affected by SS [67] (Table 1). Alevizos and colleagues [68] also reported the importance of miRNAs as biomarkers in SS, as they identified a specific pattern of miRNA expression in inflamed salivary glands from SS patients with different degrees of inflammation. This opens the possibility to use predicted target pathways of differentially expressed miRNAs to identify either inflammation or exocrine gland dysfunction. Recently, Pauley and colleagues [69] reported the altered expression of miR-146a in PBMCs of SS patients and an established mouse model of SS. In this report, miR-146a was significantly overexpressed in SS patients compared with healthy controls, and functional experiments conducted on THP-1 cells have shown the influence of miR-146a on increased phagocytic activity and suppressed inflammation cytokine production. This is another example of how altered miRNAs can influence pathogenetic mechanisms in autoimmune diseases such as SS.
Scleroderma
Another autoimmune disease in which miRNAs have not been widely studied is SSc, a multisystemic fibrotic disorder with high morbidity and mortality rates [70]. The progressive replacement of normal tissue by collagen-rich extracellular matrix leads to impairment and, ultimately, to functional failure of affected organs. Fibroblasts are activated by profibrotic cytokines and growth factors, such as IL-4, transforming growth factor (TGF)-β, and platelet-derived growth factor (PDGF)-B [71]. Maurer and colleagues [71] identified miR-29 as one key regulator of collagen expression in SSc (Table 1). This miRNA is strongly downregulated in SSc fibroblasts and skin sections, and transfection experiments showed a possible direct regulation of collagen by miR-29a. Moreover, TGF-β, PDGF-B, and IL-4 reduce the levels of miR-29a in normal fibroblasts to those seen in SSc fibroblasts, while inhibition of PDGF-B and TGF-β pathways by treatment with imatinib restored the levels of miR-29a in vitro [71].
Polymyositis/dermatomyositis
PM/DM is a T-cell mediated inflammatory myopathy in which the cellular immune response is a key feature in promoting muscle damage [72–74]. As in other systemic autoimmune diseases, a strong association of autoantibodies with distinct clinical phenotypes is found in patients with PM/DM [75]. The study of miRNAs in this disease is mainly limited to work by Eisenberg and colleagues [73] showing the possible influence of miR-146b, miR-221, miR-155, miR-214, and miR-222 on the NF-kB pathway leading to muscle inflammation (Table 1).
Conclusion
miRNAs play important roles in fundamental cellular processes, and their dysregulated expression is observed in different pathological conditions, including rheumatic diseases, inflammation, and tumorigenesis [31]. The use of miRNAs or miRNA-mimic oligonucleotides has been tested in different cancer cell lines, in mice, and in nonhuman primates [31]. These previous investigations have shown that miRNA-based gene therapies targeting dysregulated miRNAs have the potential to become therapeutic tools. It will be interesting if these miRNA-based gene therapies will be developed to treat patients with rheumatic diseases, such as RA and SLE, in the future. However, further studies in multiple populations and conducted by independent investigators are needed to validate and elucidate these mechanisms and whether or not miRNAs could serve as useful disease markers or therapeutic targets.
Abbreviations
- Ago:
-
Argonaute
- CDK2:
-
cyclin-dependent kinase 2
- EGFL7:
-
epidermal growth factor-like domain 7
- IFN:
-
interferon
- IL:
-
interleukin
- IRAK1:
-
IL-1 receptor-associated kinase 1
- MCP-1:
-
monocyte chemoattractant protein 1
- miRNA:
-
microRNA
- MMP:
-
matrix metalloproteinase
- NF:
-
nuclear factor
- PBMC:
-
peripheral blood mononuclear cell
- PDGF:
-
platelet-derived growth factor
- PM/DM:
-
polymyositis/dermatomyositis
- pre-miRNA:
-
precursor miRNA
- pri-miRNA:
-
primary microRNA
- RA:
-
rheumatoid arthritis
- RANTES:
-
Regulated upon activation: normal T-cell expressed: and secreted
- RASF:
-
RA synovial fibroblast
- RISC:
-
RNA-induced silencing complex
- SLE:
-
systemic lupus erythematosus
- SS:
-
Sjögren's syndrome
- SSc:
-
Scleroderma: systemic sclerosis
- TGF:
-
transforming growth factor
- TNF:
-
tumor necrosis factor
- TRAF6:
-
TNF receptor-associated factor 6
- Treg:
-
regulatory T cells
- UTR:
-
untranslated region.
References
Lee RC, Feinbaum RL, Ambros V: The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993, 75: 843-854. 10.1016/0092-8674(93)90529-Y.
Krol J, Loedige I, Filipowicz W: The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010, 11: 597-610.
Alevizos I, Illei GG: MicroRNAs as biomarkers in rheumatic diseases. Nat Rev Rheumatol. 2010, 6: 391-398. 10.1038/nrrheum.2010.81.
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K, Rassenti L, Kipps T, Negrini M, Bullrich F, Croce CM: Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002, 99: 15524-15529. 10.1073/pnas.242606799.
Pauley KM, Cha S, Chan EK: MicroRNA in autoimmunity and autoimmune diseases. J Autoimmun. 2009, 32: 189-194. 10.1016/j.jaut.2009.02.012.
Ambros V: The functions of animal microRNAs. Nature. 2004, 431: 350-355. 10.1038/nature02871.
Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T: Identification of novel genes coding for small expressed RNAs. Science. 2001, 294: 853-858. 10.1126/science.1064921.
Filipowicz W, Bhattacharyya SN, Sonenberg N: Mechanisms of posttranscriptional regulation by microRNAs: are the answers in sight?. Nat Rev Genet. 2008, 9: 102-114.
Fabian MR, Sonenberg N, Filipowicz W: Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010, 79: 351-379. 10.1146/annurev-biochem-060308-103103.
Zhou H, Huang X, Cui H, Luo X, Tang Y, Chen S, Wu L, Shen N: miR-155 and its star-form partner miR-155* cooperatively regulate type I interferon production by human plasmacytoid dendritic cells. Blood. 2010, 116: 5885-5894. 10.1182/blood-2010-04-280156.
Shin C, Nam JW, Farh KK, Chiang HR, Shkumatava A, Bartel DP: Expanding the microRNA targeting code: functional sites with centered pairing. Mol Cell. 2010, 38: 789-802. 10.1016/j.molcel.2010.06.005.
Yao B, Li S, Jung HM, Lian SL, Abadal GX, Han F, Fritzler MJ, Chan EK: Divergent GW182 functional domains in the regulation of translational silencing. Nucleic Acids Res. 2010, 39: 2534-2547.
Jakymiw A, Ikeda K, Fritzler MJ, Reeves WH, Satoh M, Chan EK: Autoimmune targeting of key components of RNA interference. Arthritis Res Ther. 2006, 8: R87-10.1186/ar1959.
Eystathioy T, Chan EK, Takeuchi K, Mahler M, Luft LM, Zochodne DW, Fritzler MJ: Clinical and serological associations of autoantibodies to GW bodies and a novel cytoplasmic autoantigen GW182. J Mol Med. 2003, 81: 811-818. 10.1007/s00109-003-0495-y.
Bhanji RA, Eystathioy T, Chan EK, Bloch DB, Fritzler MJ: Clinical and serological features of patients with autoantibodies to GW/P bodies. Clin Immunol. 2007, 125: 247-256. 10.1016/j.clim.2007.07.016.
Liu J, Carmell MA, Rivas FV, Marsden CG, Thomson JM, Song JJ, Hammond SM, Joshua-Tor L, Hannon GJ: Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004, 305: 1437-1441. 10.1126/science.1102513.
Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T: Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell. 2004, 15: 185-197. 10.1016/j.molcel.2004.07.007.
Pillai RS, Artus CG, Filipowicz W: Tethering of human Ago proteins to mRNA mimics the miRNA-mediated repression of protein synthesis. RNA. 2004, 10: 1518-1525. 10.1261/rna.7131604.
Li S, Lian SL, Moser JJ, Fritzler ML, Fritzler MJ, Satoh M, Chan EK: Identification of GW182 and its novel isoform TNGW1 as translational repressors in Ago2-mediated silencing. J Cell Sci. 2008, 121: 4134-4144. 10.1242/jcs.036905.
Jakymiw A, Pauley KM, Li S, Ikeda K, Lian S, Eystathioy T, Satoh M, Fritzler MJ, Chan EK: The role of GW/P-bodies in RNA processing and silencing. J Cell Sci. 2007, 120: 1317-1323. 10.1242/jcs.03429.
Yang Z, Jakymiw A, Wood MR, Eystathioy T, Rubin RL, Fritzler MJ, Chan EK: GW182 is critical for the stability of GW bodies expressed during the cell cycle and cell proliferation. J Cell Sci. 2004, 117: 5567-5578. 10.1242/jcs.01477.
Eulalio A, Tritschler F, Izaurralde E: The GW182 protein family in animal cells: new insights into domains required for miRNA-mediated gene silencing. RNA. 2009, 15: 1433-1442. 10.1261/rna.1703809.
Tritschler F, Huntzinger E, Izaurralde E: Role of GW182 proteins and PABPC1 in the miRNA pathway: a sense of deja vu. Nat Rev Mol Cell Biol. 2010, 11: 379-384.
Fabian MR, Mathonnet G, Sundermeier T, Mathys H, Zipprich JT, Svitkin YV, Rivas F, Jinek M, Wohlschlegel J, Doudna JA, Chen CY, Shyu AB, Yates JR, Hannon GJ, Filipowicz W, Duchaine TF, Sonenberg N: Mammalian miRNA RISC recruits CAF1 and PABP to affect PABP-dependent deadenylation. Mol Cell. 2009, 35: 868-880. 10.1016/j.molcel.2009.08.004.
Zekri L, Huntzinger E, Heimstadt S, Izaurralde E: The silencing domain of GW182 interacts with PABPC1 to promote translational repression and degradation of microRNA targets and is required for target release. Mol Cell Biol. 2009, 29: 6220-6231. 10.1128/MCB.01081-09.
Eystathioy T, Chan EK, Tenenbaum SA, Keene JD, Griffith K, Fritzler MJ: A phosphorylated cytoplasmic autoantigen, GW182, associates with a unique population of human mRNAs within novel cytoplasmic speckles. Mol Biol Cell. 2002, 13: 1338-1351. 10.1091/mbc.01-11-0544.
Treadwell EL, Alspaugh MA, Sharp GC: Characterization of a new antigenantibody system (Su) in patients with systemic lupus erythematosus. Arthritis Rheum. 1984, 27: 1263-1271. 10.1002/art.1780271108.
Satoh M, Langdon JJ, Chou CH, McCauliffe DP, Treadwell EL, Ogasawara T, Hirakata M, Suwa A, Cohen PL, Eisenberg RA, et al: Characterization of the Su antigen, a macromolecular complex of 100/102 and 200-kDa proteins recognized by autoantibodies in systemic rheumatic diseases. Clin Immunol Immunopathol. 1994, 73: 132-141. 10.1006/clin.1994.1179.
Vázquez-Del Mercado M, Sánchez-Orozco LV, Pauley BA, Chan JY, Chan EK, Panduro A, Maldonado González M, Jiménez-Luévanos MA, Martin-Márquez BT, Palafox-Sánchez CA, Dávalos-Rodríguez IP, Salazar-Páramo M, González-López L, Gámez-Nava JI, Satoh M: Autoantibodies to a miRNA-binding protein Argonaute2 (Su antigen) in patients with hepatitis C virus infection. Clin Exp Rheumatol. 2010, 28: 842-848.
Ceribelli A, Tincani A, Cavazzana I, Franceschini F, Cattaneo R, Pauley BA, Chan JY, Chan EK, Satoh M: Anti-argonaute2 (Ago2/Su) and -Ro antibodies identified by immunoprecipitation in primary anti-phospholipid syndrome (PAPS). Autoimmunity. 2010, 44: 90-97.
Tili E, Michaille JJ, Costinean S, Croce CM: MicroRNAs, the immune system and rheumatic disease. Nat Clin Pract Rheumatol. 2008, 4: 534-541. 10.1038/ncprheum0885.
Nagata Y, Nakasa T, Mochizuki Y, Ishikawa M, Miyaki S, Shibuya H, Yamasaki K, Adachi N, Asahara H, Ochi M: Induction of apoptosis in the synovium of mice with autoantibody-mediated arthritis by the intraarticular injection of double-stranded MicroRNA-15a. Arthritis Rheum. 2009, 60: 2677-2683. 10.1002/art.24762.
Nahid MA, Pauley KM, Satoh M, Chan EK: miR-146a is critical for endotoxininduced tolerance: implication in innate immunity. J Biol Chem. 2009, 284: 34590-34599. 10.1074/jbc.M109.056317.
Nahid MA, Satoh M, Chan EK: Mechanistic role of microRNA-146a in endotoxin-induced differential cross-regulation of TLR signaling. J Immunol. 2011, 186 (3): 1723-1734.
Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, Yoshimura A, Baltimore D, Rudensky AY: Function of miR-146a in controlling Treg cellmediated regulation of Th1 responses. Cell. 2010, 142: 914-929. 10.1016/j.cell.2010.08.012.
Curtale G, Citarella F, Carissimi C, Goldoni M, Carucci N, Fulci V, Franceschini D, Meloni F, Barnaba V, Macino G: An emerging player in the adaptive immune response: microRNA-146a is a modulator of IL-2 expression and activation-induced cell death in T lymphocytes. Blood. 2010, 115: 265-273. 10.1182/blood-2009-06-225987.
O'Connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, Kahn ME, Rao DS, Baltimore D: MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity. 2010, 33: 607-619. 10.1016/j.immuni.2010.09.009.
Divekar AA, Dubey S, Gangalum PR, Singh RR: Dicer insufficiency and microRNA-155 overexpression in lupus regulatory T cells: an apparent paradox in the setting of an inflammatory milieu. J Immunol. 2011, 186: 924-930. 10.4049/jimmunol.1002218.
Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R: Mouse ES cells express endogenous shRNAs, siRNAs, and other microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008, 22: 2773-2785. 10.1101/gad.1705308.
Bluml S, Bonelli M, Niederreiter B, Puchner A, Mayr G, Hayer S, Koenders MI, van den Berg WB, Smolen J, Redlich K: Essential role for micro-RNA 155 in the pathogenesis of autoimmune arthritis. Arthritis Rheum. 2011, 63: 1281-1288. 10.1002/art.30281.
Meister J, Schmidt MH: miR-126 and miR-126*: new players in cancer. Scientific World Journal. 2010, 10: 2090-2100.
Volinia S, Galasso M, Costinean S, Tagliavini L, Gamberoni G, Drusco A, Marchesini J, Mascellani N, Sana ME, Abu Jarour R, Desponts C, Teitell M, Baffa R, Aqeilan R, Iorio MV, Taccioli C, Garzon R, Di Leva G, Fabbri M, Catozzi M, Previati M, Ambs S, Palumbo T, Garofalo M, Veronese A, Bottoni A, Gasparini P, Harris CC, Visone R, Pekarsky Y, et al: Reprogramming of miRNA networks in cancer and leukemia. Genome Res. 2010, 20: 589-599. 10.1101/gr.098046.109.
Farazi TA, Spitzer JI, Morozov P, Tuschl T: miRNAs in human cancer. J Pathol. 2010, 223: 102-115.
Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, Schaller JG, Talal N, Winchester RJ: The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982, 25: 1271-1277. 10.1002/art.1780251101.
Hochberg MC: Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997, 40: 1725-
Dai Y, Huang YS, Tang M, Lv TY, Hu CX, Tan YH, Xu ZM, Yin YB: Microarray analysis of microRNA expression in peripheral blood cells of systemic lupus erythematosus patients. Lupus. 2007, 16: 939-946. 10.1177/0961203307084158.
Dai Y, Sui W, Lan H, Yan Q, Huang H, Huang Y: Comprehensive analysis of microRNA expression patterns in renal biopsies of lupus nephritis patients. Rheumatol Int. 2009, 29: 749-754. 10.1007/s00296-008-0758-6.
Tang Y, Luo X, Cui H, Ni X, Yuan M, Guo Y, Huang X, Zhou H, de Vries N, Tak PP, Chen S, Shen N: MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009, 60: 1065-1075. 10.1002/art.24436.
Zhao X, Tang Y, Qu B, Cui H, Wang S, Wang L, Luo X, Huang X, Li J, Chen S, Shen N: MicroRNA-125a contributes to elevated inflammatory chemokine RANTES via targeting KLF13 in systemic lupus erythematosus. Arthritis Rheum. 2010, 62: 3425-3435. 10.1002/art.27632.
Pan W, Zhu S, Yuan M, Cui H, Wang L, Luo X, Li J, Zhou H, Tang Y, Shen N: MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol. 2010, 184: 6773-6781. 10.4049/jimmunol.0904060.
Zhao S, Wang Y, Liang Y, Zhao M, Long H, Ding S, Yin H, Lu Q: MicroRNA-126 regulates DNA methylation in CD4+ T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase I. Arthritis Rheum. 2011, 63: 1376-1386. 10.1002/art.30196.
Smolen JS, Aletaha D, Koeller M, Weisman MH, Emery P: New therapies for treatment of rheumatoid arthritis. Lancet. 2007, 370: 1861-1874. 10.1016/S0140-6736(07)60784-3.
Lipsky PE, van der Heijde DM, St Clair EW, Furst DE, Breedveld FC, Kalden JR, Smolen JS, Weisman M, Emery P, Feldmann M, Harriman GR, Maini RN, Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group: Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med. 2000, 343: 1594-1602. 10.1056/NEJM200011303432202.
Nakasa T, Miyaki S, Okubo A, Hashimoto M, Nishida K, Ochi M, Asahara H: Expression of microRNA-146 in rheumatoid arthritis synovial tissue. Arthritis Rheum. 2008, 58: 1284-1292. 10.1002/art.23429.
Pauley KM, Satoh M, Chan AL, Bubb MR, Reeves WH, Chan EK: Upregulated miR-146a expression in peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Res Ther. 2008, 10: R101-10.1186/ar2344.
Stanczyk J, Pedrioli DM, Brentano F, Sanchez-Pernaute O, Kolling C, Gay RE, Detmar M, Gay S, Kyburz D: Altered expression of MicroRNA in synovial fibroblasts and synovial tissue in rheumatoid arthritis. Arthritis Rheum. 2008, 58: 1001-1009. 10.1002/art.23386.
Murata K, Yoshitomi H, Tanida S, Ishikawa M, Nishitani K, Ito H, Nakamura T: Plasma and synovial fluid microRNAs as potential biomarkers of rheumatoid arthritis and osteoarthritis. Arthritis Res Ther. 2010, 12: R86-10.1186/ar3013.
Stanczyk J, Ospelt C, Karouzakis E, Filer A, Raza K, Kolling C, Gay R, Buckley CD, Tak PP, Gay S, et al: Altered expression of miR-203 in rheumatoid arthritis synovial fibroblasts and its role in fibroblast activation. Arthritis Rheum. 2011, 63: 373-381. 10.1002/art.30115.
Fulci V, Scappucci G, Sebastiani GD, Giannitti C, Franceschini D, Meloni F, Colombo T, Citarella F, Barnaba V, Minisola G, Galeazzi M, Macino G: miR-223 is overexpressed in T-lymphocytes of patients affected by rheumatoid arthritis. Hum Immunol. 2010, 71: 206-211. 10.1016/j.humimm.2009.11.008.
Alsaleh G, Suffert G, Semaan N, Juncker T, Frenzel L, Gottenberg JE, Sibilia J, Pfeffer S, Wachsmann D: Bruton's tyrosine kinase is involved in miR-346-related regulation of IL-18 release by lipopolysaccharide-activated rheumatoid fibroblast-like synoviocytes. J Immunol. 2009, 182: 5088-5097. 10.4049/jimmunol.0801613.
Niimoto T, Nakasa T, Ishikawa M, Okuhara A, Izumi B, Deie M, Suzuki O, Adachi N, Ochi M: MicroRNA-146a expresses in interleukin-17 producing T cells in rheumatoid arthritis patients. BMC Musculoskelet Disord. 2010, 11: 209-10.1186/1471-2474-11-209.
Li J, Wan Y, Guo Q, Zou L, Zhang J, Fang Y, Fu X, Liu H, Lu L, Wu Y: Altered microRNA expression profile with miR-146a upregulation in CD4+ T cells from patients with rheumatoid arthritis. Arthritis Res Ther. 2010, 12: R81-10.1186/ar3006.
Nakamachi Y, Kawano S, Takenokuchi M, Nishimura K, Sakai Y, Chin T, Saura R, Kurosaka M, Kumagai S: MicroRNA-124a is a key regulator of proliferation and monocyte chemoattractant protein 1 secretion in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Arthritis Rheum. 2009, 60: 1294-1304. 10.1002/art.24475.
Venables PJ: Sjogren's syndrome. Best Pract Res Clin Rheumatol. 2004, 18: 313-329.
Alevizos I, Bajracharya SD, Alexander S, Turner RJ, Illei GG: MicroRNA profiling of minor salivary glands identifies disease and inflammation biomarkers in Sjogren's syndrome patients [abstract]. Arthritis Rheum. 2009, 60: S733-S734.
Michael A, Bajracharya SD, Yuen PS, Zhou H, Star RA, Illei GG, Alevizos I: Exosomes from human saliva as a source of microRNA biomarkers. Oral Dis. 2010, 16: 34-38. 10.1111/j.1601-0825.2009.01604.x.
Alevizos I, Illei GG: MicroRNAs in Sjogren's syndrome as a prototypic autoimmune disease. Autoimmun Rev. 2010, 9: 618-621. 10.1016/j.autrev.2010.05.009.
Alevizos I, Alexander S, Turner RJ, Illei GG: MicroRNA expression profiles as biomarkers of minor salivary gland inflammation and dysfunction in Sjogren's syndrome. Arthritis Rheum. 2011, 63: 535-544.
Pauley KM, Stewart CM, Gauna AE, Dupre LC, Kuklani R, Chan AL, Pauley BA, Reeves WH, Chan EK, Cha S: Altered miR-146a expression in Sjogren's syndrome and its functional role in innate immunity. Eur J Immunol. 2011, 41: 2029-2039.
Mayes MD, Lacey JV, Beebe-Dimmer J, Gillespie BW, Cooper B, Laing TJ, Schottenfeld D: Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003, 48: 2246-2255. 10.1002/art.11073.
Maurer B, Stanczyk J, Jüngel A, Akhmetshina A, Trenkmann M, Brock M, Kowal-Bielecka O, Gay RE, Michel BA, Distler JH, Gay S, Distler O: MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum. 2010, 62: 1733-1743.
Bohan A, Peter JB: Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975, 292: 403-407. 10.1056/NEJM197502202920807.
Eisenberg I, Eran A, Nishino I, Moggio M, Lamperti C, Amato AA, Lidov HG, Kang PB, North KN, Mitrani-Rosenbaum S, Flanigan KM, Neely LA, Whitney D, Beggs AH, Kohane IS, Kunkel LM: Distinctive patterns of microRNA expression in primary muscular disorders. Proc Natl Acad Sci USA. 2007, 104: 17016-17021. 10.1073/pnas.0708115104.
Bohan A, Peter JB: Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975, 292: 344-347. 10.1056/NEJM197502132920706.
Mammen AL: Dermatomyositis and polymyositis: Clinical presentation, autoantibodies, and pathogenesis. Ann N Y Acad Sci. 2010, 1184: 134-153. 10.1111/j.1749-6632.2009.05119.x.
Acknowledgements
This work was supported in part by a grant from the Lupus Research Institute and the National Institutes of Health grant AI47859.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Ceribelli, A., Yao, B., Dominguez-Gutierrez, P.R. et al. MicroRNAs in systemic rheumatic diseases. Arthritis Res Ther 13, 229 (2011). https://doi.org/10.1186/ar3377
Published:
DOI: https://doi.org/10.1186/ar3377