
Abstract
Patients with systemic lupus erythematosus have up to a 50-fold increased risk of developing atherosclerotic cardiovascular disease. Recent advances in the etiology of vascular damage in this disease stress the interplay of lupus-specific inflammatory factors with traditional cardiac risk factors, leading to increased endothelial damage. This review analyzes the putative role that immune dysregulation and lupus-specific factors may play in the pathogenesis of premature vascular damage in this disease. The potential role of various cytokines, in particular type I interferons, in the development of accelerated atherosclerosis is examined. Potential therapeutic targets are discussed.
Similar content being viewed by others
Epidemiology of premature vascular damage in systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune disease with heterogeneous manifestations, including internal organ damage, which can result in severe morbidity and even death and often requires aggressive immunosuppressive treatment. More than 30 years ago, a bimodal peak in mortality was described in lupus patients, with late increases in death commonly seen as secondary to premature cardiovascular disease (CVD) [1]. Indeed, this enhanced atherosclerotic risk increases with each year of disease duration. This is especially the case in young females with SLE, where the CVD risk can be up to 50-fold higher than in age-matched controls [2, 3]. While traditional Framingham risk factors likely contribute to CVD in SLE, they cannot fully account for the increased risk. Instead, the pathogenesis of premature CVD in SLE may rely on factors unique to the disease itself [4].
While systemic inflammation has been linked to atherosclerosis development in the general population and in specific conditions, SLE typically has a lower 'classical inflammatory burden' compared to what would be seen in rheumatoid arthritis or spondyloarthro-pathies; yet, lupus is associated with a higher CVD risk than these other diseases. This observation suggests that factors that trigger accelerated atherosclerosis in lupus differ from the typical proinflammatory factors (that is, high C-reactive protein (CRP)) linked to 'idiopathic' atherosclerosis. Atherosclerosis progression in lupus patients develops or progresses in 10% of SLE patients per year. Among other factors, this progression is associated with older age at diagnosis and with longer disease duration, supporting the hypothesis that chronic exposure to lupus immune dysregulation promotes CVD [5].
Subclinical and clinical vascular damage in SLE
Premature damage in SLE occurs in both the macro- and microvasculature. Vascular functional abnormalities in lupus are present even shortly after disease diagnosis [6]. SLE patients have significantly decreased flow-mediated dilation of the brachial artery and this correlates with increased carotid intima media thickness (IMT) [7]. Additionally, carotid plaque can be detected in 21% of SLE patients under the age of 35 years and in up to 100% of those over the age of 65 years [8]. Aortic atherosclerosis is also increased in SLE [9]. These macrovascular findings correlate with disease activity and disease duration [7–9]. Damage to the coronary circulation is also common in SLE patients, with 54% displaying non-calcified coronary plaque [10]. There is also impairment of coronary microvasculature flow reserve, even in patients with grossly normal coronary arteries. This dysfunction correlates with disease duration and severity, suggesting that microvascular damage and dysfunction are also part of SLE-related CV pathology [11]. Additionally, SLE patients have a higher probability of developing left ventricular hypertrophy, independent of baseline hypertension, again emphasizing the role of lupus-related factors in CVD damage [12].
Mechanisms of atherosclerosis development in the general population
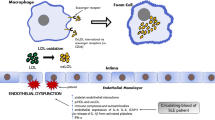
Various groups have proposed that CVD, endothelial dysfunction and atherosclerosis arise from chronic injury to the endothelium, which allows for invasion of inflammatory cells and lipid deposition. Current dogma upholds that chronic inflammation instigates and perpetuates the atherogenic cycle. Factors such as oxidized low density lipoprotein (LDL) activate the endothelium to secrete chemokines, which recruit inflammatory cells, including T lymphocytes, dendritic cells (DCs) and monocytes. These monocytes differentiate into macrophages and foam cells under the influence of locally secreted factors [13]. Various stimuli, including cholesterol crystals, then activate macrophages and foam cells to secrete inflammatory cytokines, reactive oxygen and nitrogen species and proteases, all of which contribute to the atherogenic phenotype in the blood vessel [14]. Invasion of the atherosclerotic plaque by CD4+ T cells also contributes to vascular pathology by recognizing epitopes of various molecules, including oxidized LDL, and by secreting IFN-γ, which then leads to increased inflammatory cytokine production. This chronic production of inflammatory cytokines and proteases may lead to thinning of the plaque wall and eventual rupture, which results in exposure of the blood to phospholipids, tissue factor and platelet-adhesive matrix molecules, eventually promoting thrombosis and acute CVD events [13].
Coupled to this inflammatory injury, a loss of endothelial cells can occur, which, if not repaired, leads to increased inflammatory cell invasion, vascular smooth muscle proliferation and neo-intima formation [15]. Endothelial cell apoptosis is a phenomenon with potentially significant deleterious effects on vascular health, including loss of nitric oxide, generation of phosphatidylserine-rich microparticles with significant tissue factor activity, and potential predisposition to acute coronary events [16, 17].
Under normal conditions, vascular damage triggers a response leading to an attempt to repair the endothelium. Although our understanding of vascular repair is rapidly evolving, it is still unclear how it occurs. Several groups have proposed that repair of the vasculature occurs primarily by bone marrow-derived endothelial progenitor cells (EPCs) and myelomonocytic circulating angiogenic cells (CACs) [18]. Indeed, decreased numbers or dysfunction of these cell types may contribute to CVD as EPC numbers inversely correlate with CVD risk, time to first CVD event, and in-stent restenosis risk [19, 20]. Additionally, functional impairment of EPCs correlates with coronary artery disease risk [21]. Various mechanisms have been implicated in EPC/CAC dysfunction in these conditions, including reactive oxygen species, telomere shortening/senescence and cytokines such as TNF [22–24].
Mechanisms of endothelial injury and atherosclerosis in SLE
Induction of an imbalance of vascular damage and repair by type I IFNs
Patients with SLE have increased numbers of circulating apoptotic endothelial cells, which correlates with endothelial dysfunction and generation of tissue factor [6]. Various soluble adhesion molecules, such as vascular cell adhesion molecule (VCAM), inter-cellular adhesion molecule and E-selectin, which are released after endothelial cell damage, are increased in SLE and correlate with increased coronary calcium scores. Additionally, soluble levels of the antithrombotic endothelial protein C receptor, which is released secondary to inflammatory activation of metalloproteinases, are increased in SLE and correlate with the presence of carotid plaque [25]. These findings suggest that chronic vascular insult and inflammation may be important for atherosclerotic pathology [26]. Despite evidence that accelerated endothelial cell death occurs in lupus, a phenomenon that should trigger enhanced vascular repair, the latter is significantly impaired in lupus patients. SLE patients have decreased circulating EPCs/CACs, and those that persist are characterized by increased apoptosis, even during quiescent disease, decreased proangiogenic molecule synthesis, and decreased capacity to incorporate into formed vascular structures and differentiate into mature endothelial cells [27, 28] (Figure 1). Thus, patients with SLE have compromised repair of the damaged endothelium, likely leading to the establishment of a milieu that promotes the development of plaque.

Endothelial progenitor cells/circulating angiogenic cells from patients with systemic lupus erythematosus are unable to differentiate into mature endothelial cells in culture. Photomicrographs of primary blood mononuclear cells from a healthy control (left) and a patient with systemic lupus erythematosus (right) after 2 weeks of culture in proangiogenic media on fibronectin-coated plates. Cells were imaged via inverted phase microscopy at a total magnification of 100×. Photomicrographs by Seth G Thacker.
Our group has proposed that the mechanism by which vascular repair is impaired in SLE is through increased levels and enhanced effects of type I IFNs. Human and murine studies from various groups indicate that IFN-α may be crucial in the pathogenesis of SLE. SLE patients have an 'IFN signature' in peripheral blood mononuclear cells, kidneys and other tissues that correlates with disease activity [29], and type I IFN levels are increased in lupus serum [30]. Further, lupus cells appear to be more sensitive to the effects of type I IFN [31]. As part of this pathology, we and others have suggested that the development of lupus-related CVD is, at least partially, attributable to IFN-α and, potentially, to other type I IFNs. Our group has reported that dysfunction of EPC/CAC differentiation in SLE is mediated by IFN-α, as neutralization of this cytokine restores a normal EPC/CAC phenotype [28]. This is further reinforced by the observation of abrogated EPC/CAC numbers and function observed in lupus-prone New Zealand black/New Zealand white F1 mice, a strain that depends on type I IFNs for disease development. Additionally, non-lupus-prone mice EPCs are unable to properly differentiate into mature endothelial cells in the presence of IFN-α [32, 33]. The pathways by which IFN-α mediates aberrant vascular repair may depend on repression of the proangiogenic factors IL-1β and vascular endothelial growth factor and on upregulation of the antiangiogenic IL-1 receptor antagonist. Indeed, addition of recombinant human IL-β to SLE EPC/CAC cultures restores normal endothelial differentiation [32]. Further supporting a role for type I IFNs in premature vascular damage in SLE, patients with high type I IFN signatures have decreased endothelial function, as assessed by peripheral arterial tone measurements [34]. Preliminary evidence indicates that type I IFN signatures correlate with carotid IMT in a lupus cohort [35]. Furthermore, there is evidence that an anti-angiogenic phenotype is present in patients with SLE, manifested by decreased vascular density and increased vascular rarefaction in renal blood vessels in vivo, associated with upregulation of the IL-1 receptor antagonist and decreased vascular endothelial growth factor in both the kidney and serum [28, 36].
The cellular source of type I IFNs leading to abnormal vascular repair was recently examined. Depletion of plasmacytoid DCs (the major producers of IFN-α) does not lead to abrogation of abnormal lupus EPC/CAC differentiation in culture [37]; therefore, other cellular sources for this cytokine have been sought. Neutrophil-specific genes are abundant in peripheral blood mononuclear cell microarrays from lupus patients because of the presence of low-density granulocytes (LDGs) in mononuclear cell fractions [38, 39]. The functionality and pathogenicity of these LDGs was recently investigated by our group. Among other findings, these cells are significantly cytotoxic to endothelial cells. In addition, LDGs have the capacity to secrete sufficient amounts of IFN-α to interfere with vascular repair. LDG depletion from lupus peripheral blood mononuclear cells restores the ability of EPC/CACs to differentiate in vitro into endothelial monolayers [37]. This suggests that the presence of these abnormal granulocytes contributes to endothelial dysfunction and vascular damage in SLE.
The above findings suggest that abrogation of the aberrant effects of type I IFNs in SLE may not only decrease disease activity but also lead to decreases in CVD risk. Future clinical trials should assess this possibility.
The potentially deleterious effects of type I IFNs in cardiovascular health are also being explored in non-SLE-related atherosclerosis. For example, IFN-α-producing plasmacytoid DCs have been identified in areas of atheromatous plaque. IFN-α then activates plaque-residing CD4+ T cells to increase TNF-related apoptosis-inducing ligand (TRAIL) expression, which results in killing of plaque stabilizing cells and a potential increase in the risk of plaque rupture. Additionally, IFN-α sensitizes plaque-residing myeloid DCs, which may result in further inflammation and plaque destabilization. This cytokine appears to synergize with bacterial products (such as lipopolysaccharide) to increase the synthesis of various proinflammatory cytokines and metalloproteinases [40, 41]. These findings indicate that type I IFNs could potentially be involved in atherosclerosis development not only in autoimmune disorders but also in the general population in the context of microbial infections. This hypothesis merits further investigation. Additionally, type I IFNs inhibit CRP upregulation [42], which may explain why the CRP response is usually downregulated in SLE flares and why it does not appear to correlate well with atherosclerotic burden in this disease [43].
Other cytokines
The inflammatory cytokine TNF-α appears to play an important role in the initiation and perpetuation of atherosclerotic lesions in the general population. It increases the level of adhesion molecules on the surface of vascular endothelium and promotes enhanced levels of chemotactic proteins, which allows for recruitment of monocytes and T cells into the endothelial wall [44]. In SLE, serum TNF-α levels have been reported to be elevated and correlate with coronary calcium scores [26]. TNF-α levels are also increased in SLE patients with CVD compared to those without CVD, and this correlates with altered lipid profiles [45]. Additionally, it has been postulated that elevated levels of TNF-α may increase soluble VCAM-1 in SLE [46]. However, the exact role this cytokine plays in the development of vascular damage in SLE remains unclear.
IFN-γ, secreted by glycolipid-activated invariant natural killer T-cells, may also contribute to a pathogenic role in SLE-related atherosclerosis [47]. The antiatherogenic cytokine transforming growth factor-β is decreased in SLE and this decrease may potentially play a role in related CVD [48]. The cytokine IL-17, which stimulates production of other pro-inflammatory cytokines, as well as upregulation of chemokines and adhesion molecules, has been linked to atherosclerotic plaque development in non-lupus-prone models. Atherosclerotic-prone mice have reduced plaque burden when transplanted with bone marrow deficient in the IL-17 receptor [49]. SLE patients have elevated levels of IL-17 and Th17 cells are expanded in SLE and can induce endothelial adhesion molecule upregulation [50, 51]. Thus, there is a theoretical role for Th17 T cells and IL-17 in the upregulation of inflammatory mediators and adhesion molecules that contribute to CVD in SLE. Future studies should address if, indeed, any of these cytokines play a prominent role in vascular damage and atherosclerosis progression in this disease.
Adiponectin is an adipocytokine with potential beneficial effects at sites of blood vessel injury through inhibition of monocyte adhesion to endothelial cells and of migration and proliferation of smooth muscle cells. However, this molecule is increased in lupus serum and independently correlates with augmented severity of carotid plaque, but not coronary calcification, in lupus patients [25, 52]. One hypothesis to explain this discrepancy is that chronic vascular damage in SLE leads to positive feedback on adiponectin-secreting cells. While this may lead to increases in levels of this cytokine, its effects are blunted at the site of endothelial damage due to the unique inflammatory milieu in SLE [53]. Supporting a putative protective role for adiponectin in SLE-mediated CVD, this molecule is required for the beneficial effects of rosiglitazone on atherosclerosis development in a mouse model of SLE [54].
T cells
Th1 CD4+ T cells play a pathogenic role in CVD and their differentiation is promoted in atherosclerotic lesions by the increased expression of IFN-γ and IL-12 [44]. Recent evidence suggests that these cells may also play a role in SLE-related CVD, as atherosclerosis-prone LDL receptor-deficient mice have increased vascular inflammation and CD4+ T cell infiltration in their plaques after bone marrow transplant with lupus-susceptible cells [55]. As mentioned above, CD4+ T cells increase TRAIL expression when exposed to IFN-α, which can lead to plaque destabilization [41]. A hypothetical role for autoreactive CD4+ T cells in endothelial damage in SLE also exists. SLE autoreactive T cells can kill antigen presenting cells [56]. Endothelial cells have the ability to act as antigen presenting cells upon activation, and research on transplant rejection suggests that graft endothelial cells are activated to a pro-inflammatory phenotype and killed by host T cells during antigen presentation [57]. Further research into whether interactions between endothelial cells and SLE autoreactive T cells result in endothelial damage and an increased risk of atherosclerosis should be considered.
The roles of other T-cell subsets in atherosclerosis development are being investigated. Invariant natural killer T cells, which recognize glycolipids and increase with the duration of lupus, may be proatherogenic [47]. In addition, whether the abnormalities reported in T regulatory cells in SLE contribute to atherosclerosis development is unknown [58]. A putative role is suggested by the observation that if regulatory T cell function is compromised in mouse models of atherosclerosis, CVD development is significantly more pronounced [59].
Complement and immune complexes
Inhibition of complement regulatory proteins increases atherosclerosis in mice and decreases in the membrane-attack complex attenuate atherosclerotic plaque formation [60]. Complement activated by inflammatory stimuli can interact with immune complexes (ICs), such as seen in SLE, and result in upregulation of endothelial adhesion molecules, including E-selectin and VCAM-1. These molecules may enhance neutrophil recruitment and endothelial damage [61]. High levels of oxidized LDL/β2 glycoprotein 1 complexes and anti-complex IgG or IgM have been reported in SLE. As the titers of these complexes correlate with a number of CVD risk factors [62], it is possible that they could be proatherogenic. The complement component C1q has anti-atherosclerotic effects by facilitating macrophage clearance of oxidized and acetylated LDL. As C1q deficiency is linked to SLE predisposition, its absence may also have a potential role in SLE-mediated atherosclerosis [63]. A role for complement activation in atherogenesis has been proposed [64], but the exact role this phenomenon plays in premature vascular damage in SLE remains unclear. ICs may also potentially play a role in atherosclerosis development. IC formation in rabbits accelerates diet-induced atherosclerosis, and mice deficient in IC receptors have limited atherosclerotic development [65].
Lupus-related dyslipidemias
SLE patients have disturbances in lipoprotein levels and their processing in the bloodstream. High density lipoprotein (HDL) is decreased, while LDL, very low density lipoprotein and triglyceride levels are increased. These alterations may be related to abnormal chylomicron processing secondary to low levels of lipoprotein lipase [66]. Additionally, SLE patients have higher levels of pro-inflammatory HDL, which is unable to protect LDL from oxidation and promotes endothelial injury. Increased pro-inflammatory HDL in SLE is associated with augmented atherosclerosis [67]. In addition, the lipid profile of SLE patients may be more susceptible to environmental effects. Lupus-prone mice exposed to high-fat chow showed increased pro-inflammatory HDL and lipid deposition in vessels when compared to non-lupus mice [68]. A high fat diet administered to LDL receptor-deficient mice, made susceptible to SLE via bone marrow transplantation, resulted in very elevated lipid levels and significant increases in mortality when compared to similar mice fed regular chow [55]. Thus, predisposition to SLE may increase sensitivity to lipid perturbations by diet and other exposures.
Oxidative stress
Endothelial damage and the initiation of the atherogenic cycle may be influenced by the redox environment. SLE patients have increased levels of reactive oxygen and nitrogen species and antibodies to resultant protein adducts, which correlate with disease activity and provide an environment for oxidation of lipoproteins and atherosclerosis development [69]. Homocysteine, a molecule with the capacity to increase reactive oxygen species in the bloodstream, is also increased in SLE patients and correlates with carotid IMT and with coronary calcification [5, 70, 71]. Further, defense mechanisms against an altered redox environment are decreased in SLE. For example, paraoxonase, an enzyme with antioxidant activity that circulates attached to HDL and prevents LDL oxidation, is decreased in this disease. This correlates with the presence of antibodies to HDL and β2-glycoprotein and with enhanced atherosclerosis risk [72].
Antiphospholipid antibodies
The role of antiphospholipid (APL) antibodies in premature CVD remains a matter of debate. β2-glycoprotein I, abundantly found in vascular plaques, has been hypothesized to be protective against atherosclerosis development. Antibodies against this molecule could, in theory, be detrimental to the vessel wall and promote activation of inflammatory cascades by IC formation [73]. APL antibodies may increase the likelihood of abnormal ankle brachial index and anti-cardiolipin anti-body titers correlate with carotid IMT [70, 74]. However, a recent study examining flow-mediated dilation and EPC numbers in primary APL syndrome (APS) did not detect any difference in these early markers of CVD risk compared with age and gender matched healthy controls [75]. This supports previous work in which the presence of APL antibodies did not correlate with endothelial dysfunction or carotid IMT in SLE [7, 76]. Using cardiac MRI to find evidence of subclinical ischemic disease, 26% of patients with APS had occult myocardial scarring compared to 11% of controls. This study, however, enrolled patients with secondary APS from SLE (22% of their APS cohort) and it is unclear whether a significant number of the patients with myocardial damage also had lupus [77]. Thus, the role of APL antibodies in atherosclerosis development in SLE remains unclear. Nevertheless, because of the arterial thrombosis associated with APS itself, there remains a putative role for these antibodies in the triggering of unstable angina and acute coronary syndromes.
Other autoantibodies
Autoantibodies against regulatory proteins in the atherogenic cycle in SLE may potentially contribute to CVD. Antibodies to the anti-atherogenic HDL and one of its components, Apo A-1, are increased in SLE and rise with disease flares [78]. SLE patients have increased levels of anti-lipoprotein lipase antibodies. These also increase with disease activity and may contribute to increased levels of triglycerides [79]. Antibodies to endothelial cells are common in SLE and have been proposed to mediate endothelial injury [80]; however, various groups have shown that these antibodies may not correlate with other markers of endothelial dysfunction [81]. Additionally, antibodies to oxidized LDL, lipoprotein lipase, CRP and annexin V may have a putative role in CVD in SLE [82, 83]. Antibodies to heat shock proteins enhance atherosclerotic development in various non-lupus models and are increased in SLE serum [84, 85]. Whether this class of antibodies contributes specifically to SLE-related atherosclerosis is unknown.
Preventive measures for cardiovascular disease in SLE
Various studies indicate that early and appropriate treatment of immune dysregulation in SLE could be key to hampering CVD development and progression in SLE. Patients treated with lower doses of cyclophosphamide, azathioprine or corticosteroids had greater progression of CVD than those treated with higher doses [5]. Further, aortic atherosclerosis risk is lower in SLE patients who have undergone treatment with cyclophosphamide when compared to SLE patients who have not received this medication [9]. The role of corticosteroid treatment is complex and poorly understood, with potentially dual effects on CVD risk that may depend on dose and time of exposure [8].
While no studies have shown a reduced incidence of CVD in patients taking antimalarials, these drugs have positive effects on glucose tolerance, lipid profiles, and thrombosis potential [86]. Studies using surrogate markers for CVD have provided mixed results. Antimalarials were significantly associated with decreased presence of carotid plaque in patients with SLE [87]. A correlation between lack of antimalarial use and increased vascular stiffness in SLE patients has been demonstrated, but no association between their use and coronary calcification was found [88, 89]. A cohort study suggested a clear survival benefit in SLE patients taking antimalarials, but the mechanisms for this effect remain to be determined [90]. Because antimalarials can weakly inhibit IFN-α production through inhibition of IC formation and toll-like receptor-7 and -9 signaling [91], modulation of IFN-α levels with a potential improvement in endothelial function and vascular repair may contribute to the survival benefit. More research into the vascular effects of antimalarials is needed to understand their benefits and whether they have an impact on atherosclerotic development.
Mycophenolate mofetil (MMF), an immunosuppressive medication commonly used in SLE, may be potentially beneficial in atherosclerosis. MMF has a protective effect on the development of both transplant and diet-mediated atherosclerosis in animals and is also beneficial in preventing coronary pathology in cardiac transplant patients [92]. MMF decreases atherosclerotic plaque inflammation in patients treated for 2 weeks prior to carotid endarterectomy [93]. Whether this drug has a CVD benefit in SLE patients remains to be determined, and future studies will hopefully address this question.
The role of novel biologics in CVD prevention in SLE remains unknown. Currently, studies targeting type I IFNs, IL-17 and the various anti-B cell therapies are underway in SLE and other diseases. Long-term follow-up to assess atherosclerosis progression in these groups would be important to identify if favorable effects are identified. Given the recent observation that impairment in IL-1 pathways in SLE may mediate abnormal vascular repair in this disease [32], a note of caution is added with regards to the use of anakinra and other anti-IL-1 therapies, particularly in SLE, but also in other diseases where aberrant vasculogenesis is observed.
Other non-disease modifying medications may also have a benefit in SLE-related CVD. SLE patients have a higher incidence of metabolic syndrome and insulin resistance, and this correlates with increases in homocysteine and high sensitivity CRP [94]. Treatment of insulin-resistant states may improve CVD profiles in SLE. Our group reported that treatment of SLE-prone mice with the peroxisome proliferator-activated receptor γ (PPAR-γ) agonist pioglitazone, which is used to treat type II diabetes in humans, resulted in improved insulin sensitivity, improved endothelial function and restored EPC differentiation [94]. Additionally, rosiglitazone, another PPAR-γ agonist, decreased aortic atherosclerosis in lupus- and atherosclerosis-prone Gld.apoeE-/- mice [54]. How this class of medications would benefit CVD in SLE patients warrants additional studies.
Guidelines for CVD prevention in SLE remain nebulous. The latest European League Against Rheumatism (EULAR) recommendations suggest yearly monitoring of traditional and/or non-lupus-specific CVD risk factors, including smoking, activity level, oral contraceptive use, hormonal therapies and family history of CVD. Monitoring of blood pressure, lipids and glucose is also recommended [95]. One group has proposed treating SLE as a coronary heart disease equivalent, targeting recommendations as suggested by the Adult Treatment Panel guidelines (ATPIII) [96]. However, whether these guidelines will be sufficient to abrogate CVD risk in SLE remains to be determined. The use of statins in SLE has not been systematically or extensively studied, but they have been shown to improve endothelium-dependent flow-mediated dilation and possibly slow progression of carotid IMT in adult lupus as well as increase EPC numbers in other conditions, including diabetes mellitus [97–99]. While trending toward a protective effect for carotid IMT thickness in pediatric SLE, prophylactic statin use in children did not show a statistically significant difference compared to placebo [100]. A murine lupus/atherosclerosis model displayed decreased atherosclerosis and amelioration of renal disease when treated with simvastatin [101]. Statins can also block IFN-α production in peripheral blood from healthy controls in response to exposure to SLE patients' serum. Th is blockade occurs through inhibition of the Rho kinase, likely in plasmacytoid DCs [102]. Future research will hopefully clarify the role of statin use in SLE patients.
Finally, diet may be an important modifiable risk factor that can alter predisposition to atherosclerotic lesions. LDL receptor-deficient mice that underwent bone marrow transplant with SLE-prone cells had increased sensitivity to dietary fat. A Western-style diet containing 21% fat increased atherosclerotic lesions, pathogenic antibody formation and severity of renal disease when compared to mice fed a regular diet [55]. A different model of lupus-prone mice fed high-fat chow or administered leptin had accelerated and increased proteinuria, suggesting an interplay between diet and lupus [68]. Certainly, some murine lupus models have decreased life spans when fed a high-fat diet [103]. Thus, further understanding of the role of diet on immune modulation and CVD risk in SLE may be key in vascular damage prevention.
Conclusion
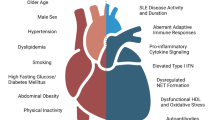
The CVD risk in SLE patients stems from a combination of traditional risk factors and SLE-specific mechanisms that incorporate chronic inflammation, endothelial dysfunction, decreased vascular repair through a type I IFN effect, antibody formation and perturbed lipid homeostasis and redox environment (Figure 2). Continued research into the mechanisms of lupus-related CVD will hopefully provide effective tools and targets to improve their survival and overall quality of life. Additionally, it is crucial that future clinical trials in SLE include biomarkers of vascular damage, functional studies of vascular health and assessment of subclinical and clinical CVD as endpoints in their efficacy analysis.

The interplay of various inflammatory mediators increases vascular damage and plaque formation in systemic lupus erythematosus. IFN-α contributes to endothelial dysfunction and decreased repair of endothelial damage by decreasing numbers and function of endothelial progenitor cells (EPCs) and circulating angiogenic cells (CACs). In addition to synthesizing type I IFNs, low density granulocytes (LDGs) present in systemic lupus erythematosus patients are directly toxic to the endothelium. Altered lipid profiles secondary to abnormal chylomicron processing, increased pro-inflammatory high density lipoprotein (pi-HDL) and increased oxidized low density lipoprotein (ox-LDL) also promote atherosclerosis development. The abnormal redox environment in systemic lupus erythematosus also promotes endothelial dysfunction and modulates lipid profiles. Antibodies to lipoproteins or endothelial targets may also contribute to vascular damage. Cytokines such as TNF-α, IL-17 and IFN-γ may also have pro-atherogenic effects on blood vessels. The combination of some or all of these factors in an individual patient results in endothelial dysfunction, increased plaque burden and an increased risk of cardiovascular events. IC, immune complex; PDC, plasmacytoid dendritic cell; RNS, reactive nitrogen species; ROS, reactive oxygen species.
Note
Autoimmune Basis of Rheumatic Diseases
This article is part of a series on Systemic lupus erythematosus, edited by David Pisetsky, which can be found online at http://arthritis-research.com/series/lupus
This series forms part of a special collection of reviews covering major autoimmune rheumatic diseases, available at: http://arthritis-research.com/series/abrd
Abbreviations
- APL:
-
antiphospholipid
- APS:
-
APL syndrome
- CAC:
-
circulating angiogenic cell
- CRP:
-
C-reactive protein
- CVD:
-
cardiovascular disease
- DC:
-
dendritic cell
- EPC:
-
endothelial progenitor cell
- HDL:
-
high density lipoprotein
- IC:
-
immune complex
- IFN:
-
interferon
- IL:
-
interleukin
- IMT:
-
intima media thickness
- LDG:
-
low-density granulocyte
- LDL:
-
low density lipoprotein
- MMF:
-
mycophenolate mofetil
- PPAR-γ:
-
peroxisome proliferator-activated receptor γ
- SLE:
-
systemic lupus erythematosus
- TNF:
-
tumor necrosis factor
- TRAIL:
-
tumor necrosis factor-related apoptosis-inducing ligand
- VCAM:
-
vascular cell adhesion molecule.
References
Urowitz MB, Bookman AA, Koehler BE, Gordon DA, Smythe HA, Ogryzlo MA: The bimodal mortality pattern of systemic lupus erythematosus. Am J Med. 1976, 60: 221-225. 10.1016/0002-9343(76)90431-9.
Manzi S, Meilahn EN, Rairie JE, Conte CG, Medsger TA, Jansen-McWilliams L, D'Agostino RB, Kuller LH: Age-specific incidence rates of myocardial infarction and angina in women with systemic lupus erythematosus: comparison with the Framingham Study. Am J Epidemiol. 1997, 145: 408-415.
Hak AE, Karlson EW, Feskanich D, Stampfer MJ, Costenbader KH: Systemic lupus erythematosus and the risk of cardiovascular disease: Results from the nurses' health study. Arthritis Rheum. 2009, 61: 1396-1402. 10.1002/art.24537.
Esdaile JM, Abrahamowicz M, Grodzicky T, Li Y, Panaritis C, du Berger R, Côte R, Grover SA, Fortin PR, Clarke AE, Senécal JL: Traditional Framingham risk factors fail to fully account for accelerated atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 2001, 44: 2331-2337. 10.1002/1529-0131(200110)44:10<2331::AID-ART395>3.0.CO;2-I.
Roman MJ, Crow MK, Lockshin MD, Devereux RB, Paget SA, Sammaritano L, Levine DM, Davis A, Salmon JE: Rate and determinants of progression of atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 2007, 56: 3412-3419. 10.1002/art.22924.
Rajagopalan S, Somers EC, Brook RD, Kehrer C, Pfenninger D, Lewis E, Chakrabarti A, Richardson BC, Shelden E, McCune WJ, Kaplan MJ: Endothelial cell apoptosis in systemic lupus erythematosus: a common pathway for abnormal vascular function and thrombosis propensity. Blood. 2004, 103: 3677-3683. 10.1182/blood-2003-09-3198.
El-Magadmi M, Bodill H, Ahmad Y, Durrington PN, Mackness M, Walker M, Bernstein RM, Bruce IN: Systemic lupus erythematosus: an independent risk factor for endothelial dysfunction in women. Circulation. 2004, 110: 399-404. 10.1161/01.CIR.0000136807.78534.50.
Manzi S, Selzer F, Sutton-Tyrrell K, Fitzgerald SG, Rairie JE, Tracy RP, Kuller LH: Prevalence and risk factors of carotid plaque in women with systemic lupus erythematosus. Arthritis Rheum. 1999, 42: 51-60. 10.1002/1529-0131(199901)42:1<51::AID-ANR7>3.0.CO;2-D.
Roldan C, Joson J, Sharrar J, Qualls C, Sibbitt W: Premature aortic atherosclerosis in systemic lupus erythematosus: a controlled transesophageal echocardiographic study. J Rheumatol. 2010, 37: 71-78. 10.3899/jrheum.090665.
Kiani AN, Vogel-Claussen J, Magder LS, Petri M: Noncalcified coronary plaque in systemic lupus erythematosus. J Rheumatol. 2010, 37: 579-584. 10.3899/jrheum.090824.
Recio-Mayoral A, Mason JC, Kaski JC, Rubens MB, Harari OA, Camici PG: Chronic inflammation and coronary microvascular dysfunction in patients without risk factors for coronary artery disease. Eur Heart J. 2009, 30: 1837-1843. 10.1093/eurheartj/ehp205.
Pieretti J, Roman MJ, Devereux RB, Lockshin MD, Crow MK, Paget SA, Schwartz JE, Sammaritano L, Levine DM, Salmon JE: Systemic lupus erythematosus predicts increased left ventricular mass. Circulation. 2007, 116: 419-426. 10.1161/CIRCULATIONAHA.106.673319.
Hansson GK: Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005, 352: 1685-1695. 10.1056/NEJMra043430.
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nuñez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E: NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010, 464: 1357-1361. 10.1038/nature08938.
Lindner V, Fingerle J, Reidy MA: Mouse model of arterial injury. Circ Res. 1993, 73: 792-796.
Mallat Z, Benamer H, Hugel B, Benessiano J, Steg PG, Freyssinet JM, Tedgui A: Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation. 2000, 101: 841-843.
Chironi GN, Boulanger CM, Simon A, Dignat-George F, Freyssinet JM, Tedgui A: Endothelial microparticles in diseases. Cell Tissue Res. 2009, 335: 143-151. 10.1007/s00441-008-0710-9.
Briasoulis A, Tousoulis D, Antoniades C, Stefanadis C, Papageorgiou N: The role of endothelial progenitor cells in vascular repair after arterial injury and atherosclerotic plaque development. Cardiovasc Ther. 2010,
Schmidt-Lucke C, Rössig L, Fichtlscherer S, Vasa M, Britten M, Kämper U, Dimmeler S, Zeiher AM: Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: proof of concept for the clinical importance of endogenous vascular repair. Circulation. 2005, 111: 2981-2987. 10.1161/CIRCULATIONAHA.104.504340.
George J, Herz I, Goldstein E, Abashidze S, Deutch V, Finkelstein A, Michowitz Y, Miller H, Keren G: Number and adhesive properties of circulating endothelial progenitor cells in patients with in-stent restenosis. Arterioscler Thromb Vasc Biol. 2003, 23: e57-60. 10.1161/01.ATV.0000107029.65274.db.
Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, Zeiher AM, Dimmeler S: Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001, 89: E1-E7. 10.1161/hh1301.093953.
MacEneaney OJ, Kushner EJ, Westby CM, Cech JN, Greiner JJ, Stauffer BL, DeSouza CA: Endothelial progenitor cell function, apoptosis, and telomere length in overweight/obese humans. Obesity. 2010, 18: 1677-1682. 10.1038/oby.2009.494.
Zhang Y, Herbert BS, Rajashekhar G, Ingram DA, Yoder MC, Clauss M, Rehman J: Premature senescence of highly proliferative endothelial progenitor cells is induced by tumor necrosis factor-alpha via the p38 mitogen-activated protein kinase pathway. FASEB J. 2009, 23: 1358-1365. 10.1096/fj.08-110296.
Brunetti ND, Munno I, Pellegrino PL, Ruggero V, Correale M, De Gennaro L, Cuculo A, Campanale EG, Di Biase M: Inflammatory cytokines imbalance in the very early phase of acute coronary syndrome: correlations with angiographic findings and in-hospital events. Inflammation. 2011, 34: 58-66. 10.1007/s10753-010-9208-1.
Reynolds HR, Buyon J, Kim M, Rivera TL, Izmirly P, Tunick P, Clancy RM: Association of plasma soluble E-selectin and adiponectin with carotid plaque in patients with systemic lupus erythematosus. Atherosclerosis. 2010, 210: 569-574. 10.1016/j.atherosclerosis.2009.12.007.
Rho YH, Chung CP, Oeser A, Solus J, Raggi P, Gebretsadik T, Shintani A, Stein CM: Novel cardiovascular risk factors in premature coronary atherosclerosis associated with systemic lupus erythematosus. J Rheumatol. 2008, 35: 1789-1794.
Westerweel PE, Luijten RK, Hoefer IE, Koomans HA, Derksen RH, Verhaar MC: Haematopoietic and endothelial progenitor cells are deficient in quiescent systemic lupus erythematosus. Ann Rheum Dis. 2007, 66: 865-870. 10.1136/ard.2006.065631.
Denny MF, Thacker S, Mehta H, Somers EC, Dodick T, Barrat FJ, McCune WJ, Kaplan MJ: Interferon-alpha promotes abnormal vasculogenesis in lupus: a potential pathway for premature atherosclerosis. Blood. 2007, 110: 2907-2915. 10.1182/blood-2007-05-089086.
Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW: Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. 2003, 100: 2610-2615. 10.1073/pnas.0337679100.
Kim T, Kanayama Y, Negoro N, Okamura M, Takeda T, Inoue T: Serum levels of interferons in patients with systemic lupus erythematosus. Clin Exp Immunol. 1987, 70: 562-569.
Kariuki SN, Kirou KA, MacDermott EJ, Barillas-Arias L, Crow MK, Niewold TB: Cutting edge: autoimmune disease risk variant of STAT4 confers increased sensitivity to IFN-alpha in lupus patients in vivo. J Immunol. 2009, 182: 34-38.
Thacker SG, Berthier CC, Mattinzoli D, Rastaldi MP, Kretzler M, Kaplan MJ: The detrimental effects of IFN-α on vasculogenesis in lupus are mediated by repression of IL-1 pathways: potential role in atherogenesis and renal vascular rarefaction. J Immunol. 2010, 185: 4457-4469. 10.4049/jimmunol.1001782.
Thacker SG, Duquaine D, Park J, Kaplan MJ: Lupus-prone New Zealand Black/New Zealand White F1 mice display endothelial dysfunction and abnormal phenotype and function of endothelial progenitor cells. Lupus. 2010, 19: 288-299. 10.1177/0961203309353773.
Lee PY, Li Y, Richards HB, Chan FS, Zhuang H, Narain S, Butfiloski EJ, Sobel ES, Reeves WH, Segal MS: Type I interferon as a novel risk factor for endothelial progenitor cell depletion and endothelial dysfunction in systemic lupus erythematosus. Arthritis Rheum. 2007, 56: 3759-3769. 10.1002/art.23035.
Zhao W, Somers E, McCune WJ, Kaplan MJ: Type I Interferon signatures are associated with vascular risk and atherosclerosis in systemic lupus erythematosus. Arthritis Rheum. 2009, 59: S582-
Thacker SG, Berthier CC, Mattinzoli D, Rastaldi MP, Kretzler M, Kaplan MJ: The detrimental effects of IFN-{alpha} on vasculogenesis in lupus are mediated by repression of IL-1 pathways: potential role in atherogenesis and renal vascular rarefaction. J Immunol. 2010, 185: 4457-4469. 10.4049/jimmunol.1001782.
Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, McCune WJ, Kaplan MJ: A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol. 2010, 184: 3284-3297. 10.4049/jimmunol.0902199.
Hacbarth E, Kajdacsy-Balla A: Low density neutrophils in patients with systemic lupus erythematosus, rheumatoid arthritis, and acute rheumatic fever. Arthritis Rheum. 1986, 29: 1334-1342. 10.1002/art.1780291105.
Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V: Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003, 197: 711-723. 10.1084/jem.20021553.
Niessner A, Shin MS, Pryshchep O, Goronzy JJ, Chaikof EL, Weyand CM: Synergistic proinflammatory effects of the antiviral cytokine interferon-alpha and Toll-like receptor 4 ligands in the atherosclerotic plaque. Circulation. 2007, 116: 2043-2052. 10.1161/CIRCULATIONAHA.107.697789.
Niessner A, Weyand CM: Dendritic cells in atherosclerotic disease. Clin Immunol. 2010, 134: 25-32. 10.1016/j.clim.2009.05.006.
Enocsson H, Sjöwall C, Skogh T, Eloranta ML, Rönnblom L, Wetterö J: Interferon-alpha mediates suppression of C-reactive protein: explanation for muted C-reactive protein response in lupus flares?. Arthritis Rheum. 2009, 60: 3755-3760. 10.1002/art.25042.
Nikpour M, Gladman DD, Ibaez D, Urowitz MB: Variability and correlates of high sensitivity C-reactive protein in systemic lupus erythematosus. Lupus. 2009, 18: 966-973. 10.1177/0961203309105130.
McMahon M, Hahn BH: Atherosclerosis and systemic lupus erythematosus - mechanistic basis of the association. Curr Opin Immunol. 2007, 19: 633-639. 10.1016/j.coi.2007.11.001.
Svenungsson E, Fei GZ, Jensen-Urstad K, de Faire U, Hamsten A, Frostegard J: TNF-alpha: a link between hypertriglyceridaemia and inflammation in SLE patients with cardiovascular disease. Lupus. 2003, 12: 454-461. 10.1191/0961203303lu412oa.
Svenungsson E, Cederholm A, Jensen-Urstad K, Fei GZ, de Faire U, Frostegård J: Endothelial function and markers of endothelial activation in relation to cardiovascular disease in systemic lupus erythematosus. Scand J Rheumatol. 2008, 37: 352-359. 10.1080/03009740802007514.
Major AS, Singh RR, Joyce S, Van Kaer L: The role of invariant natural killer T cells in lupus and atherogenesis. Immunol Res. 2006, 34: 49-66. 10.1385/IR:34:1:49.
Becker-Merok A, Eilertsen GØ, Nossent JC: Levels of transforming growth factor-β are low in systemic lupus erythematosus patients with active disease. J Rheumatol. 2010, 37: 2039-2045. 10.3899/jrheum.100180.
van Es T, van Puijvelde GH, Ramos OH, Segers FM, Joosten LA, van den Berg WB, Michon IM, de Vos P, van Berkel TJ, Kuiper J: Attenuated atherosclerosis upon IL-17R signaling disruption in LDLr deficient mice. Biochem Biophys Res Commun. 2009, 388: 261-265. 10.1016/j.bbrc.2009.07.152.
Mok MY, Wu HJ, Lo Y, Lau CS: The relation of interleukin 17 (IL-17) and IL-23 to Th1/Th2 cytokines and disease activity in systemic lupus erythematosus. J Rheumatol. 2010, 37: 2046-2052. 10.3899/jrheum.100293.
Yang J, Chu Y, Yang X, Gao D, Zhu L, Yang X, Wan L, Li M: Th17 and natural Treg cell population dynamics in systemic lupus erythematosus. Arthritis Rheum. 2009, 60: 1472-1483. 10.1002/art.24499.
Chung CP, Long AG, Solus JF, Rho YH, Oeser A, Raggi P, Stein CM: Adipocytokines in systemic lupus erythematosus: relationship to inflammation, insulin resistance and coronary atherosclerosis. Lupus. 2009, 18: 799-806. 10.1177/0961203309103582.
Clancy R, Ginzler E: Endothelial function and its implications for cardiovascular and renal disease in systemic lupus erythematosus. Rheum Dis Clin North Am. 2010, 36: 145-160. 10.1016/j.rdc.2009.12.011.
Aprahamian T, Bonegio RG, Richez C, Yasuda K, Chiang LK, Sato K, Walsh K, Rifkin IR: The peroxisome proliferator-activated receptor {gamma} agonist rosiglitazone ameliorates murine lupus by induction of adiponectin. J Immunol. 2009, 182: 340-346.
Braun N, Wade N, Wakeland E, Major A: Accelerated atherosclerosis is independent of feeding high fat diet in systemic lupus erythematosus- susceptible LDLr-/- mice. Lupus. 2008, 17: 1070-1078. 10.1177/0961203308093551.
Kaplan MJ, Lewis EE, Shelden EA, Somers E, Pavlic R, McCune WJ, Richardson BC: The apoptotic ligands TRAIL, TWEAK, and Fas ligand mediate monocyte death induced by autologous lupus T cells. J Immunol. 2002, 169: 6020-6029.
Al-Lamki R, Bradley J, Pober J: Endothelial cells in allograft rejection. Transplantation. 2008, 86: 1340-1348. 10.1097/TP.0b013e3181891d8b.
Bonelli M, Smolen JS, Scheinecker C: Treg and lupus. Ann Rheum Dis. 2010, 69: i65-i66. 10.1136/ard.2009.117135.
Nilsson J, Wigren M, Shah P: Regulatory T cells and the control of modified lipoprotein autoimmunity-driven atherosclerosis. Trends Cardiovasc Med. 2009, 19: 272-276. 10.1016/j.tcm.2010.02.010.
Wu G, Hu W, Shahsafaei A, Song W, Dobarro M, Sukhova GK, Bronson RR, Shi GP, Rother RP, Halperin JA, Qin X: Complement regulator CD59 protects against atherosclerosis by restricting the formation of complement membrane attack complex. Circ Res. 2009, 104: 550-558. 10.1161/CIRCRESAHA.108.191361.
Clancy RM: Circulating endothelial cells and vascular injury in systemic lupus erythematosus. Curr Rheumatol Rep. 2000, 2: 39-43. 10.1007/s11926-996-0067-6.
Bassi N, Zampieri S, Ghirardello A, Tonon M, Zen M, Beggio S, Matsuura E, Doria A: Oxldl/2gpi complex and anti-oxldl/2gpi In Sle: prevalence and correlates. Autoimmunity. 2009, 42: 289-291. 10.1080/08916930902828247.
Fraser DA, Tenner AJ: Innate immune proteins C1q and mannan-binding lectin enhance clearance of atherogenic lipoproteins by human monocytes and macrophages. J Immunol. 2010, 185: 3932-3939. 10.4049/jimmunol.1002080.
Haskard DO, Boyle JJ, Mason JC: The role of complement in atherosclerosis. Curr Opin Lipidol. 2008, 19: 478-482. 10.1097/MOL.0b013e32830f4a06.
Mayadas TN, Tsokos GC, Tsuboi N: Mechanisms of immune complex-mediated neutrophil recruitment and tissue injury. Circulation. 2009, 120: 2012-2024. 10.1161/CIRCULATIONAHA.108.771170.
Borba EF, Bonf E, Vinagre CG, Ramires JA, Maranho RC: Chylomicron metabolism is markedly altered in systemic lupus erythematosus. Arthritis Rheum. 2000, 43: 1033-1040. 10.1002/1529-0131(200005)43:5<1033::AID-ANR11>3.0.CO;2-B.
McMahon M, Grossman J, Skaggs B, Fitzgerald J, Sahakian L, Ragavendra N, Charles-Schoeman C, Watson K, Wong WK, Volkmann E, Chen W, Gorn A, Karpouzas G, Weisman M, Wallace DJ, Hahn BH: Dysfunctional proinflammatory high-density lipoproteins confer increased risk of atherosclerosis in women with systemic lupus erythematosus. Arthritis Rheum. 2009, 60: 2428-2437. 10.1002/art.24677.
Hahn BH, Lourencço EV, McMahon M, Skaggs B, Le E, Anderson M, Iikuni N, Lai CK, La Cava A: Pro-inflammatory high-density lipoproteins and atherosclerosis are induced in lupus-prone mice by a high-fat diet and leptin. Lupus. 2010, 19: 913-917. 10.1177/0961203310364397.
Wang G, Pierangeli SS, Papalardo E, Ansari GA, Khan MF: Markers of oxidative and nitrosative stress in systemic lupus erythematosus: correlation with disease activity. Arthritis Rheum. 2010, 62: 2064-2072.
Ames PR, Margarita A, Delgado Alves J, Tommasino C, Iannaccone L, Brancaccio V: Anticardiolipin antibody titre and plasma homocysteine level independently predict intima media thickness of carotid arteries in subjects with idiopathic antiphospholipid antibodies. Lupus. 2002, 11: 208-214. 10.1191/0961203302lu165oa.
Kiani AN, Magder L, Petri M: Coronary calcium in systemic lupus erythematosus is associated with traditional cardiovascular risk factors, but not with disease activity. J Rheumatol. 2008, 35: 1300-1306.
Delgado Alves J, Ames PR, Donohue S, Stanyer L, Nourooz-Zadeh J, Ravirajan C, Isenberg DA: Antibodies to high-density lipoprotein and beta2-glycoprotein I are inversely correlated with paraoxonase activity in systemic lupus erythematosus and primary antiphospholipid syndrome. Arthritis Rheum. 2002, 46: 2686-2694. 10.1002/art.10542.
George J, Harats D, Gilburd B, Afek A, Levy Y, Schneiderman J, Barshack I, Kopolovic J, Shoenfeld Y: Immunolocalization of beta2-glycoprotein I (apolipoprotein H) to human atherosclerotic plaques: potential implications for lesion progression. Circulation. 1999, 99: 2227-2230.
Baron MA, Khamashta MA, Hughes GRV, D'Cruz DP: Prevalence of an abnormal ankle-brachial index in patients with primary antiphospholipid syndrome: preliminary data. Ann Rheuma Dis. 2005, 64: 144-146. 10.1136/ard.2003.016204.
Gresele P, Migliacci R, Vedovati MC, Ruffatti A, Becattini C, Facco M, Guglielmini G, Boscaro E, Mezzasoma AM, Momi S, Pengo V: Patients with primary antiphospholipid antibody syndrome and without associated vascular risk factors present a normal endothelial function. Thromb Res. 2009, 123: 444-451. 10.1016/j.thromres.2008.05.015.
Farzaneh-Far A, Roman MJ, Lockshin MD, Devereux RB, Paget SA, Crow MK, Davis A, Sammaritano L, Levine DM, Salmon JE: Relationship of antiphospholipid antibodies to cardiovascular manifestations of systemic lupus erythematosus. Arthritis Rheum. 2006, 54: 3918-3925. 10.1002/art.22265.
Sacré K, Brihaye B, Hyafil F, Serfaty JM, Escoubet B, Zennaro MC, Lidove O, Laissy JP, Papo T: Asymptomatic myocardial ischemic disease in antiphospholipid syndrome: a controlled cardiac magnetic resonance imaging study. Arthritis Rheum. 2010, 62: 2093-2100. 10.1002/art.27304.
O'Neill SG, Giles I, Lambrianides A, Manson J, D'Cruz D, Schrieber L, March LM, Latchman DS, Isenberg DA, Rahman A: Antibodies to apolipoprotein A-I, high-density lipoprotein, and C-reactive protein are associated with disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2010, 62: 845-854. 10.1002/art.27286.
Rodrigues CEM, Bonfá E, Carvalho JF: Review on anti-lipoprotein lipase antibodies. Clin Chimi Acta. 2010, 411: 1603-1605. 10.1016/j.cca.2010.07.028.
Domiciano D, Carvalho J, Shoenfeld Y: Pathogenic role of anti-endothelial cell antibodies in autoimmune rheumatic diseases. Lupus. 2009, 18: 1233-1238. 10.1177/0961203309346654.
Duval A, Helley D, Capron L, Youinou P, Renaudineau Y, Dubucquoi S, Fischer AM, Hachulla E: Endothelial dysfunction in systemic lupus patients with low disease activity: evaluation by quantification and characterization of circulating endothelial microparticles, role of anti-endothelial cell antibodies. Rheumatology. 2010, 49: 1049-1055. 10.1093/rheumatology/keq041.
Elliott J, Manzi S: Cardiovascular risk assessment and treatment in systemic lupus erythematosus. Best Pract Res Clin Rheumatol. 2009, 23: 481-494. 10.1016/j.berh.2009.03.005.
Meyer O: Anti-CRP antibodies in systemic lupus erythematosus. Joint Bone Spine. 2010, 77: 384-389. 10.1016/j.jbspin.2010.04.010.
George J, Harats D, Gilburd B, Levy Y, Langevitz P, Shoenfeld Y: Atherosclerosis-related markers in systemic lupus erythematosus patients: the role of humoral immunity in enhanced atherogenesis. Lupus. 1999, 8: 220-226. 10.1191/096120399678847597.
Mandal K, Foteinos G, Jahangiri M, Xu Q: Role of antiheat shock protein 60 autoantibodies in atherosclerosis. Lupus. 2005, 14: 742-746. 10.1191/0961203305lu2212oa.
Petri M: Hydroxychloroquine use in the Baltimore Lupus Cohort: effects on lipids, glucose and thrombosis. Lupus. 1996, 5 (Suppl 1): S16-22. 10.1177/096120339600500105.
Roman MJ, Shanker BA, Davis A, Lockshin MD, Sammaritano L, Simantov R, Crow MK, Schwartz JE, Paget SA, Devereux RB, Salmon JE: Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. N Engl J Med. 2003, 349: 2399-2406. 10.1056/NEJMoa035471.
Selzer F, Sutton-Tyrrell K, Fitzgerald S, Tracy R, Kuller L, Manzi S: Vascular stiffness in women with systemic lupus erythematosus. Hypertension. 2001, 37: 1075-1082.
Asanuma Y, Oeser A, Shintani AK, Turner E, Olsen N, Fazio S, Linton MF, Raggi P, Stein CM: Premature coronary-artery atherosclerosis in systemic lupus erythematosus. N Engl J Med. 2003, 349: 2407-2415. 10.1056/NEJMoa035611.
Shinjo SK, Bonfá E, Wojdyla D, Borba EF, Ramirez LA, Scherbarth HR, Brenol JC, Chacón-Diaz R, Neira OJ, Berbotto GA, De La Torre IG, Acevedo-Vázquez EM, Massardo L, Barile-Fabris LA, Caeiro F, Silveira LH, Sato EI, Buliubasich S, Alarcón GS, Pons-Estel BA, Grupo Latino Americano de Estudio del Lupus Eritematoso (Gladel): Antimalarial treatment may have a time-dependent effect on lupus survival: data from a multinational Latin American inception cohort. Arthritis Rheum. 2010, 62: 855-862. 10.1002/art.27300.
Kalia S, Dutz JP: New concepts in antimalarial use and mode of action in dermatology. Dermatol Ther. 2007, 20: 160-174. 10.1111/j.1529-8019.2007.00131.x.
Gibson WT, Hayden MR: Mycophenolate mofetil and atherosclerosis: results of animal and human studies. Ann N Y Acad Sci. 2007, 1110: 209-221. 10.1196/annals.1423.023.
van Leuven SI, van Wijk DF, Volger OL, de Vries JP, van der Loos CM, de Kleijn DV, Horrevoets AJ, Tak PP, van der Wal AC, de Boer OJ, Pasterkamp G, Hayden MR, Kastelein JJ, Stroes ES: Mycophenolate mofetil attenuates plaque inflammation in patients with symptomatic carotid artery stenosis. Atherosclerosis. 2010, 211: 231-236. 10.1016/j.atherosclerosis.2010.01.043.
Mok CC, Poon WL, Lai JP, Wong CK, Chiu SM, Wong CK, Lun SW, Ko GT, Lam CW, Lam CS: Metabolic syndrome, endothelial injury, and subclinical Page 10 of 10 atherosclerosis in patients with systemic lupus erythematosus. Scand J Rheumatol. 2010, 39: 42-49. 10.3109/03009740903046668.
Mosca M, Tani C, Aringer M, Bombardieri S, Boumpas D, Brey R, Cervera R, Doria A, Jayne D, Khamashta MA, Kuhn A, Gordon C, Petri M, Rekvig OP, Schneider M, Sherer Y, Shoenfeld Y, Smolen JS, Talarico R, Tincani A, van Vollenhoven RF, Ward MM, Werth VP, Carmona L: European League Against Rheumatism recommendations for monitoring patients with systemic lupus erythematosus in clinical practice and in observational studies. Ann Rheum Dis. 2010, 69: 1269-1274. 10.1136/ard.2009.117200.
Wajed J, Ahmad Y, Durrington PN, Bruce IN: Prevention of cardiovascular disease in systemic lupus erythematosus - proposed guidelines for risk factor management. Rheumatology. 2004, 43: 7-12. 10.1093/rheumatology/keg436.
Ferreira GA, Navarro TP, Telles RW, Andrade LEC, Sato EI: Atorvastatin therapy improves endothelial-dependent vasodilation in patients with systemic lupus erythematosus: an 8 weeks controlled trial. Rheumatology. 2007, 46: 1560-1565. 10.1093/rheumatology/kem186.
Dimmeler S, Aicher A, Vasa M, Mildner-Rihm C, Adler K, Tiemann M, Rütten H, Fichtlscherer S, Martin H, Zeiher AM: HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001, 108: 391-397.
Petri M: Lupus Atherosclerosis Prevention Study (LAPS): a randomized double blind placebo controlled trial of atorvastatin versus placebo. Arthritis Rheum. 2006, 54: S520-
Sandborg CI, Barnhart HX, Ardoin SP, Yow E, Evans GW, Mieszkalski KL, Ilowite NT, Eberhard A, Imundo LF, Kimura Y, Von Scheven E, Silverman E, Bowyer SL, Punaro L, Singer NG, Sherry DD, McCurdy D, Klein-Gittelman M, Wallace CA, Silver R, Wagner-Weiner L, Higgins GC, Brunner HI, Jung L, Soep JB, Reed A, Tegler C, APPLE Investigators: Does atorvastatin reduce progression of carotid intimal medial thickening in childhood SLE? Results from the Atherosclerosis Prevention in Pediatric Lupus (APPLE) trial: a multicenter, randomized, double-blind placebo-controlled study [abstract]. Arthritis Rheum. 2010, 62: S1677-
Aprahamian T, Bonegio R, Rizzo J, Perlman H, Lefer DJ, Rifkin IR, Walsh K: Simvastatin treatment ameliorates autoimmune disease associated with accelerated atherosclerosis in a murine lupus model. J Immunol. 2006, 177: 3028-3034.
Amuro H, Ito T, Miyamoto R, Sugimoto H, Torii Y, Son Y, Nakamichi N, Yamazaki C, Hoshino K, Kaisho T, Ozaki Y, Inaba M, Amakawa R, Fukuhara S: Statins, inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A reductase, function as inhibitors of cellular and molecular components involved in type I interferon production. Arthritis Rheum. 2010, 62: 2073-2085.
Lin BF, Jeng SJ, Chiang BL, Huang CC: Dietary fat affects lipids and anti-cardiolipin antibody levels in autoimmune-prone NZB/W F1 mice. Br J Nutr. 1997, 77: 657-669. 10.1079/BJN19970063.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Kahlenberg, J.M., Kaplan, M.J. The interplay of inflammation and cardiovascular disease in systemic lupus erythematosus. Arthritis Res Ther 13, 203 (2011). https://doi.org/10.1186/ar3264
Published:
DOI: https://doi.org/10.1186/ar3264