
Abstract
While many proteases in articular cartilage have been described, current studies indicate that members of two families of metalloproteases – MMPs and the ADAMTSs – are responsible for the degradation of the major components of this tissue. Collagenases (MMPs) make the first cleavage in triple-helical collagen, allowing its further degradation by other proteases. Aggrecanases (ADAMTSs), in conjunction with other MMPs, degrade aggrecan, a component of the proteoglycan aggregate. Anti-neoepitope antibodies that recognize the cleavage products of collagen and aggrecan generated by these enzymes are now available and are being used to detect the sites of action and to quantitate degradation products.
Similar content being viewed by others


Introduction
Destruction of articular cartilage is an irreversible consequence of arthritis. Cartilage consists of two major components, a type-II-collagen-fibril network with associated small proteoglycans, and proteoglycan aggregates composed of a noncovalent association between aggrecan, hyaluronate, and link protein. In arthritis, proteoglycan degradation is thought to be an early and reversible process, whereas the breakdown of the collagen network is believed to be irreversible, contributing to loss of joint function. While free radical attack and the action of glycosidases may play a role in cartilage deterioration, the most important degradative agents are proteolytic enzymes.
Proteases
Proteolytic cleavage of the major components of the cartilage extracellular matrix is effected by a number of proteases, many of which are synthesized by chondrocytes and synovial cells in response to inflammatory stimuli. Members of each of the four classes of protease – serine/threonine proteases, cysteine proteases, aspartic proteases, and metalloproteases – have been implicated in the degradation of cartilage. However, current data indicate that the initial steps in matrix breakdown are extracellular processes involving metalloproteases. This class of enzyme is characterized by the presence, within the active site, of a metal ion (usually zinc), which is required for catalytic activity. Of the metalloproteases, the members of two families, the matrix metalloproteases (MMPs) and the ADAMTSs (a disintegrin and a metalloprotease with thrombospondin motifs) family, have been implicated in the breakdown of collagen and aggrecan, respectively. These enzymes are members of the M10 and M12 peptidase families as classified in the universal protease database, MEROPS [1].
Cleavage of peptide bonds is a very simple chemical reaction and many proteolytic enzymes are relatively small proteins (~30 kDa) consisting simply of a binding site to accommodate about six amino acid residues of the substrate and the catalytic machinery. Other proteases, in particular the metalloproteases involved in matrix degradation, have a more sophisticated, multidomain composition, in which additional protein elements are present, giving additional functions to the protease. These include assisting in substrate binding by attachment at sites remote from where peptide bond cleavage occurs, and binding to nonsubstrate molecules in the extracellular matrix, allowing appropriate localization of the enzyme.
The MMPs
The MMPs form a multigene family and can be classified into subfamilies on the basis of domain structure and substrate selectivity. A convenient grouping is that of collagenases, stromelysins, gelatinases, and membrane-type MMPs (MT-MMPs) (Fig. 1). Of these enzymes, the collagenases (MMP-1, -8, and -13) are the most specific, since they alone are able to degrade native fibrillar collagens. Cleavage occurs at a single locus, to yield fragments three-quarters and one-quarter the size of the original molecule. Domain substitution experiments have shown that the hemopexin region of collagenase is required for the catalytic domain to cleave triple-helical collagen, but the exact mechanism underlying this interaction is still not clear [2]. The three mammalian collagenases exhibit differing substrate specificities, with MMP-13 preferentially cleaving the major collagen constituent of cartilage, type II collagen [3]. Once the initial cleavage has been made in the collagen fibrils, the triple helix unwinds, rendering the resulting fragments excellent substrates for the gelatinases, MMPs -2 and -9. However, cleavage of the collagen fibril itself may require clearance of small proteoglycans and cleavage of interfibrillar cross-links in order for the collagenases to access triplehelical regions. The additional elements in the gelatinases (the fibronectin-type-II repeats and the hemopexin domain) assist proteolysis by binding to the substrate and also enable the enzyme to attach to other components of the connective tissue matrix [4]. The stromelysins are characterized by a broad substrate specificity and broad optimum pH range and are able to degrade many of the extracellular matrix proteins [5], including gelatins, proteoglycans, fibronectin, and type IX collagen. The MT-MMPs are a more recently identified subgroup of the MMPs, which contain a transmembrane C-terminal domain. It has been shown [6] that MT1-MMP is capable of digesting fibrillar type collagens I, II, and III into the characteristic three-quarter and one-quarter fragments, preferentially cleaving type I collagen, as well as degrading other extracellular components, including gelatin, proteoglycan, fibronectin, and laminin.

Schematic representation of the domain structure of the matrix metalloproteinases (MMPs) associated with cartilage degradation. The sequence HEXXH is a conserved motif in this family of metalloproteases. The two histidine residues (H) are ligands for the essential zinc ion, and the side chain of the glutamic acid (E) acts as a general base for peptide bond cleavage. MMP-1, -8, and -13, collagenases; MMP-3, stromelysin; MMP-2 and -9, gelatinases; MMP-7, matrilysin; MMP-14, membrane type metalloproteinase-1
Control of MMP synthesis, activation, and activity is tightly regulated under physiological conditions. Thus, all the MMPs are synthesized as inactive proenzymes. Enzyme latency is maintained by ligation of a cysteine residue in the prodomain to the active-site Zn2+ ion. Activation occurs via complex, highly regulated intermolecular proteolytic cascades leading to destabilization of the Cys–Zn interaction [7], followed by a second cleavage that results in the release of the prodomain from the active enzyme. While many pathways for MMP activation have been demonstrated using in vitro model systems, in most cases the actual in vivo mechanisms are still not clear. MMP activity is modulated by the naturally occurring endogenous inhibitors of the MMPs, the tissue inhibitors of MMPs (TIMPs). These molecules bind tightly to the active site of activated MMPs with a 1:1 stoichiometry and have Ki values of less than 10-9 M [8].
The MT-MMPs represent an important control point in MMP activation. In contrast to most of the other MMPs, the MT-MMPs are activated intracellularly, in the Golgi apparatus, by the action of the serine protease furin, which cleaves pro-MT-MMPs at a specific site. Once at the cell surface, active MT1-MMP forms a trimolecular complex with pro-MMP-2 and the inhibitor TIMP-2, resulting in the activation of MMP-2.
It is apparent, then, that the accelerated turnover of collagen associated with joint diseases may result from a number of factors, including increased synthesis and activation of MMPs and/or an imbalance in levels of MMPs and their inhibitors, the TIMPs.
The ADAMTS family
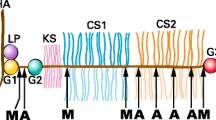
Of particular importance in cartilage turnover are members of a very recently characterized family of metalloproteases termed ADAMTSs [9]. These enzymes are similar in structure to the better-known ADAM ('a disintegrin and a metalloprotease') family of membrane-bound metalloproteases [10], which in addition to the zinc-dependent protease unit contain a disintegrin domain. In some enzymes, this domain interacts with cell-surface integrins to disrupt cell/matrix interactions but more generally can be expected to mediate interactions with other cell and matrix components. Instead of a membrane-spanning domain, the ADAMTS members contain one or more thrombospondin-type-I domains (Fig. 2). While two members of this family (ADAMTS-2 and ADAMTS-3) are collagen N-propeptide-processing enzymes, several other ADAMTSs have been implicated in aggrecan degradation in arthritis. It had been known for many years that in cartilage, aggrecan is cleaved at five unique sites along the core protein after glutamic acid residues [11]. The activities mediating these cleavages were termed 'aggrecanases', and using a direct approach, Elizabeth Arner's group purified two proteases [12,13] that fulfil this role (aggrecanase-1 and -2), showing that they are members of the ADAMTS family (ADAMTS-4 and -5). Subsequently, it was demonstrated that ADAMTS-1, which had previously been characterized as an inflammation-associated protein [14] but which is also expressed in cartilage [15], is an aggrecanase [16]. The type-I-thrombospondin motifs in these proteases bind to the sulfated GAGs in the matrix, thus targeting the enzymes to the site of aggrecan breakdown [17] and increasing their degradative efficiency. This property may also account for the reported association of these activities with the chondrocyte membrane [18]. Although regulation of the synthesis of ADAMTS proteases is not fully understood, upregulation of aggrecanase activity by interleukin 1 is well known. As with the MT-MMPs, the proforms of aggrecanases contain furin-processing motifs and exit from the Golgi apparatus as active proteases. Recent in vitro studies indicate that TIMP-3, but not TIMP-1, -2, or -4 [19,20], is a potent inhibitor of ADAMTS-4 and -5 and thus is a potential endogenous inhibitor of this class of enzyme.

Schematic representation of the domain structure of ADAMTS members involved in aggrecan degradation. The conserved HEXXH motif is as in Figure 1. ADAMTS-4 and -5, aggrecanase-1 and -2; ADAMTS-1, also termed METH-1 (metalloprotease and thrombospondin domains).
Cysteine and other proteases
Members of the cysteine and aspartic protease classes may also contribute to cartilage matrix degradation. Thus the lysosomal enzymes, including cathepsins B, D, and L, are thought to play a secondary role in cartilage degradation, involving intracellular digestion of products released by other proteases. It has also been postulated that at least some of these enzymes may function extracellularly within the cartilage, with cathepsin B, for example, potentially contributing to aggrecan breakdown [21]. The protein is expressed in situ in synovial cells attached to cartilage and bone at sites of erosion, and an enhanced transcription of the protein was observed in these synovial cells obtained from patients with rheumatoid arthritis compared with the transcription in normal fibroblasts [22]. The relative importance of different cysteine proteases in osteoarthritis has been investigated recently, and again cathepsin B appears to be a major factor [23]. In addition, cathepsin K, which is the principal cysteine protease of the osteoclast, has also been found in the synovium and may contribute to cartilage degradation [24].
Evidence for the action of specific proteases in cartilage
An important characteristic of the metalloproteases, which is thought to mediate the initial steps in the cleavage of collagen and aggrecan, is their specificity. Collagenases make an initial cleavage at a specific weak point in the collagen fibril, and aggrecanases cleave the core protein at five well characterized sites. In addition, other MMPs, such as stromelysin (MMP-3), cleave aggrecan at a well-characterized site in the aggrecan core protein [25]. These cleavage events generate terminal neoepitopes that are antigenically distinct from the same sequence in the intact protein [26]. Antibodies produced against such neoepitopes have been used to demonstrate the sites of collagenase and aggrecanase action as well as those of other metalloproteases such as stromelysin in normal and pathological turnover of cartilage [27,28]. In addition, since even after further processing the neoepitopes survive on smaller peptides, they can be detected in body fluids and used to evaluate the levels of ongoing matrix degradation [29].
Conclusions: protease inhibition as a therapeutic option
Evidence for the action of MMPs, aggrecanases, and other proteases in the degradation of cartilage associated with arthritis immediately suggests that their inhibition would be a fruitful therapeutic approach. While to date very few bioavailable cysteine protease inhibitors have been developed, a massive effort to produce MMP inhibitors over the past decade has resulted in several potent compounds. As yet, their use in arthritis therapy has been unsuccessful, in part because of an incomplete understanding of the entire metalloprotease repertoire. One of the problems in assessing the efficacy of protease inhibitor therapy is the difficulty in measuring biological outcome, since changes in cartilage metabolism are not readily apparent, particularly to the patient. The availability of methods to measure specific cartilage matrix fragments in synovial fluid, blood, or urine that are indicative of unique protease action, using specific anti-neoepitope antibodies for aggrecan and collagen fragments, promises to allow direct monitoring of the effect of different therapeutic approaches.
Abbreviations
- Note:
-
While the terms 'protease' and 'proteinase' were once used interchangeably, more recently 'protease' has become predominant. Except in definitions of abbreviations, which were based on the earlier usage, the term 'protease' is used in this review.
- ADAM:
-
a disintegrin and a metalloprotease
- ADAMTS:
-
a disintegrin and a metalloprotease with thrombospondin motifs
- MMP:
-
matrix metalloproteinase
- MT-MMP:
-
membrane-type matrix metalloproteinase
- TIMP:
-
tissue inhibitor of metalloproteinases.
References
The MEROPS database. [http://www.merops.ac.uk/]
Chung L, Shimokawa K, Dinakarpandian D, Grams F, Fields GB, Nagase H: Identification of the 183 RWTNNFREY191 region as a critical segment of matrix metalloproteinase 1 for the expression of collagenolytic activity. J Biol Chem. 2000, 275: 29610-29617. 10.1074/jbc.M004039200.
Knauper V, Lopez-Otin C, Smith B, Knight CG, Murphy G: Biochemical characterization of human collagenase-3. J Biol Chem. 1996, 271: 1544-1550. 10.1074/jbc.271.3.1544.
Steffensen B, Bigg HF, Overall CM: The involvement of the fibronectin type II-like modules of human gelatinase A in cell surface localization and activation. J Biol Chem. 1998, 273: 20622-20628. 10.1074/jbc.273.32.20622.
Nagase H, Woessner JF: Matrix metalloproteinases. J Biol Chem. 1999, 274: 21491-21494. 10.1074/jbc.274.31.21491.
Ohuchi E, Imai K, Fujii Y, Sato H, Seiki M, Okada Y: Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J Biol Chem. 1997, 272: 2446-2451. 10.1074/jbc.272.4.2446.
Springman EB, Angleton EL, Birkedal-Hansen H, Van Wart HE: Multiple modes of activation of latent human fibroblast collagenase: Evidence for the role of a Cys73 active-site zinc complex in latency and a "cysteine switch" mechanism for activation. Proc Natl Acad Sci U S A. 1990, 87: 364-368.
Willenbrock F, Crabbe T, Slocombe PM, Sutton CW, Docherty WJP, Cockett MI, O'Shea M, Brocklehurst K, Phillips IR, Murphy G: The activity of the tissue inhibitor of metalloproteinases is regulated by C-terminal domain interactions. A kinetic analysis of the inhibition of gelatinase A. Biochemistry. 1993, 32: 4330-4337.
Kaushal GP, Shah SV: The new kids on the block: ADAMTSs, potentially multifunctional metalloproteinases of the ADAM family. J Clin Invest. 2000, 105: 1335-1337.
Wolfsberg TG, White JM: ADAMs in fertilization and development. Dev Biol. 1996, 180: 389-401. 10.1006/dbio.1996.0313.
Loulakis P, Shrikhande A, Davis G, Maniglia CA: N-Terminal sequence of proteoglycan fragments isolated from medium of interleukin-1-treated articular-cartilage cultures. Biochem J. 1992, 284: 589-593.
Tortorella MD, Burn TC, Pratta MA, Abbaszade I, Hollis JM, Liu R, Rosenfeld SA, Copeland RA, Decicco CP, Wynn R, Rockwell A, Yang F, Duke JL, Solomon K, George H, Bruckner R, Nagase H, Itoh Y, Ellis DM, Ross H, Wiswall BH, Murphy K, Hillman MCJ, Hollis GF, Newton RC, Magolda RL, Trzaskos JM, Arner EC: Purification and cloning of aggrecanase-1: A member of the ADAMTS family of proteins. Science. 1999, 284: 1664-1666. 10.1126/science.284.5420.1664.
Abbaszade I, Liu RQ, Yang F, Rosenfeld SA, Ross OH, Link JR, Ellis DM, Tortorella MD, Pratta MA, Hollis JM, Wynn R, Duke JL, George HJ, Hillman MC, Murphy K, Wiswall BH, Copeland RA, Decicco CP, Bruckner R, Nagase H, Itoh Y, Newton RC, Magolda RL, Trzaskos JM, Hollis GF, Arner EC, Burn TC: Cloning and characterization of ADAMTS11, an aggrecanase from the ADAMTS family. J Biol Chem. 1999, 274: 23443-23450. 10.1074/jbc.274.33.23443.
Kuno K, Terashima Y, Matsushima K: ADAMTS-1 is an active metalloproteinase associated with the extracellular matrix. J Biol Chem. 1999, 274: 18821-18826. 10.1074/jbc.274.26.18821.
Flannery CR, Little CB, Hughes CE, Caterson B: Expression of ADAMTS homologues in articular cartilage. Biochem Biophys Res Commun. 1999, 260: 318-322. 10.1006/bbrc.1999.0909.
Kuno K, Okada Y, Kawashima H, Nakamura H, Miyasaka M, Ohno H, Matsushima K: ADAMTS-1 cleaves a cartilage proteoglycan, aggrecan. FEBS Lett. 2000, 478: 241-245. 10.1016/S0014-5793(00)01854-8.
Tortorella MD, Pratta M, Liu RQ, Abbaszade I, Ross H, Burn T, Arner E: The thrombospondin motif of aggrecanase-1 (ADAMTS-4) is critical for aggrecan substrate recognition and cleavage. J Biol Chem. 2000, 275: 25791-25797. 10.1074/jbc.M001065200.
Billington CJ, Clark IM, Cawston TE: An aggrecan-degrading activity associated with chondrocyte membranes. Biochem J. 1998, 336: 207-212.
Kashiwagi M, Tortorella M, Nagase H, Brew K: TIMP-3 is a potent inhibitor of ADAM-TS4 (aggrecanase 1) and ADAM-TS5 (aggrecanase 2). J Biol Chem. 2001, 276: 12501-12504. 10.1074/jbc.C000848200.
Hashimoto G, Aoki T, Nakamura H, Tanzawa K, Okada Y: Inhibition of ADAMTS4 (aggrecanase-1) by tissue inhibitors of metalloproteinases (TIMP-1, 2, 3 and 4). FEBS Lett. 2001, 494: 192-195. 10.1016/S0014-5793(01)02323-7.
Mort JS, Magny M-C, Lee ER: Cathepsin B: an alternative protease for the generation of an aggrecan "metalloproteinase" cleavage neoepitope. Biochem J. 1998, 335: 491-494.
Trabandt A, Gay RE, Fassbender H-G, Gay S: Cathepsin B in synovial cells at the site of joint destruction in rheumatoid arthritis. Arthritis Rheum. 1991, 34: 1444-1451.
Lang A, Horler D, Baici A: The relative importance of cysteine peptidases in osteoarthritis. J Rheumatol. 2000, 27: 1970-1979.
Dodds RA, Connor JR, Drake FH, Gowen M: Expression of cathepsin K messenger RNA in giant cells and their precursors in human osteoarthritic synovial tissues. Arthritis Rheum. 1999, 42: 1588-1593. 10.1002/1529-0131(199908)42:8<1588::AID-ANR4>3.0.CO;2-S.
Flannery CR, Lark MW, Sandy JD: Identification of a stromelysin cleavage site within the interglobular domain of human aggrecan. Evidence for proteolysis at this site in vivo in human articular cartilage. J Biol Chem. 1992, 267: 1008-1014.
Mort JS, Buttle DJ: The use of cleavage site specific antibodies to delineate protein processing and breakdown pathways. J Clin Pathol :Mol Pathol. 1999, 52: 11-18.
Stoop R, van der Kraan PM, Buma P, Hollander AP, Billinghurst RC, Poole AR, van den Berg WB: Type II collagen degradation in spontaneous osteoarthritis in C57Bl/6 and BALB/c mice. Arthritis Rheum. 1999, 42: 2381-2389. 10.1002/1529-0131(199911)42:11<2381::AID-ANR17>3.0.CO;2-E.
Lark MW, Bayne EK, Flanagan J, Harper CF, Hoerrner LA, Hutchinson NI, Singer II, Donatelli SA, Weidner JR, Williams HR, Mumford RA, Lohmander LS: Aggrecan degradation in human cartilage. Evidence for both matrix metalloproteinase and aggrecanase activity in normal, osteoarthritic, and rheumatoid joints. J Clin Invest. 1997, 100: 93-106.
Downs JT, Lane CL, Nestor NB, McLellan TJ, Kelly MA, Karam GA, Mezes PS, Pelletier JP, Otterness IG: Analysis of collagenase-cleavage of type II collagen using a neoepitope ELISA. J Immunol Methods. 2001, 247: 25-34. 10.1016/S0022-1759(00)00302-1.
Acknowledgements
We thank Guylaine Bedard for artwork. The authors' research is funded by the Shriners of North America.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mort, J.S., Billington, C.J. Articular cartilage and changes in Arthritis: Matrix degradation. Arthritis Res Ther 3, 337 (2001). https://doi.org/10.1186/ar325
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar325