
Abstract
Introduction
Cytochrome P-450 2E1 (CYP2E1) is an important member of the CYP superfamily, which is involved in the metabolism and activation of many low molecular weight toxic compounds. We tried to investigate the possible association of CYP2E1 tag single nucleotide polymorphisms (SNPs) with susceptibility to systemic lupus erythematosus (SLE) in a Chinese Han population.
Methods
The coding and flanking regions of the CYP2E1 gene were scanned for polymorphisms and tag SNPs were selected. A two-stage case-control study was performed to genotype a total of 876 SLE patients and 680 geographically matched healthy controls (265 cases and 288 controls in stage I and 611 cases and 392 controls in stage II). SLE associations of alleles, genotypes and haplotypes were tested by age and sex adjusted logistic regression. The gene transcription quantitation was carried out for peripheral blood mononuclear cell (PBMC) samples from 120 healthy controls.
Results
Tag SNP rs2480256 was found significantly associated with SLE in both stages of the study. The "A" allele was associated with slightly higher risk (odds ratio (OR) = 1.165, 95% confidence interval (CI) 1.073 to 1.265, P = 2.75E-4) and "A/A" genotype carriers were with even higher SLE risk (OR = 1.464 95% CI 1.259 to 1.702, P = 7.48E-7). When combined with another tag SNP rs8192772, we identified haplotype "rs8192772-rs2480256/TA" over presented in SLE patients (OR 1.407, 95% CI 1.182 to 1.675, P = 0.0001) and haplotype "TG" over presented in the controls (OR 0.771, 95% CI 0.667 to 0.890, P = 0.0004). The gene transcription quantitation analysis further proved the dominant effect of rs2480256 as the "A/A" genotype showed highest transcription.
Conclusions
Our results suggest the involvement of CYP2E1 as a susceptibility gene for SLE in the Chinese population.
Similar content being viewed by others

Introduction
Systemic lupus erythematosus (SLE (OMIM 152700)) is a chronic autoimmune disease with heterogeneous clinical features ranging from mild forms to progressive end organ damage. It is characterized by humoral and cellular immunity to self antigens. Approximately 90% of SLE patients are female. Epidemiological studies suggest the contribution of multiple factors including genetics, environmental exposure and gene-environmental interactions in the etiology of SLE [1–4] The significant difference of SLE prevalence between African descents in industrialized countries and residents in West African countries [5] suggested the combined effects of genetic basis and environmental effect(s) for the clinical manifestation of SLE. Potential environmental triggers for SLE include radiation and exposure to a wide range of exogenous and endogenous xenobiotic compounds [6, 7]. Exposure to trichloroethylene (TCE), a volatile organic solvent, was found in several epidemiological studies to induce SLE and some other autoimmune diseases such as scleroderma [8, 9]. Autoimmune effects including activation of CD4+ T-cells and an increase of autoantibodies were detected in MRL+/+ mice subjected to TCE exposure [10–12]. Recently, silica exposure from a variety of industrial occupations has been associated with an increased risk of SLE [13].
The cytochrome P-450 enzymes (CYPs) are responsible for the oxidative metabolism and biotransformation of a variety of substrates. The marked inter-individual variability in the expression of CYP genes contributes to the difference in the disposition of many endo- and xeno-biotics, including the metabolites of steroids, environmental toxins and therapeutics [14]. CYP2E1, an important member of CYP superfamily, is involved in the metabolism and activation of many low molecular weight toxic compounds, including ethanol, benzene, nitrosamine and TCE [15]. In MRL +/+ mice, inhibition of CYP2E1 reversed immune-related effects induced by TCE [16]. We undertook this study to evaluate any association between CYP2E1 genetic polymorphisms and SLE in a Chinese study population.
Materials and methods
Subjects and study strategy
A total of 876 patients with SLE (89% female, mean age 24.9 ± 9.9 years) and 680 healthy individuals (58% female, mean age 51.0 ± 17.6 years) were recruited in the present two-stage study from July 2004 to September 2006 (the first stage) and from October 2006 to August 2007 (the second stage). Unrelated Chinese subjects with SLE according to the American College of Rheumatology (ACR) 1982 revised criteria for the classification of SLE [17] were recruited from multiple medical centers in Zhejiang, Shangdong and Liaoning provinces. All medical records were reviewed to confirm the patient's eligibility and to gather relevant clinical data at the time of diagnosis. Clinical features of the disease were recorded in standardized questionnaires and written informed consent from each participant was also received. The clinical and immunological features of the SLE patients are shown in Supplementary Table S1 in Additional file 1. The controls are area-matched unrelated healthy individuals visiting hospitals. Sample handling and confidentiality protection strictly adhered to the protocols approved by Ethics Committee of The Hong Kong University of Science and Technology. Genomic DNA was extracted from EDTA-treated whole blood using the GFX™ Genomic Blood DNA Purification Kit (Amersham Biosciences, Piscataway, NJ, USA).
In this study, we first screened CYP2E1 gene polymorphisms in a panel of 96 healthy individuals. Then the association study for tag SNPs and haplotypes was carried out in a two-stage study approach. During the stage I study, we recruited and genotyped 265 SLE cases (86% female) and 288 controls (81% female) to identify SLE associated SNPs and haploty pes. For the stage II study, we recruited and genotyped 611 SLE patients (90% female) and 392 healthy controls (48% female).
CYP2E1sequence variations screening and tag SNP selection
For the sequence variation screening the CYP2E1 gene region was amplified as eight polymerase chain reaction (PCR) fragments covering a total of 7.5 kb, including the promoter region, exons, exon-intron junctions, as well as the 5' and 3' untranslated regions. Supplementary Table S2 in Additional file 1 shows the primer sequences and PCR conditions used in this study. Direct DNA sequencing was employed to identify the sequence variations. PCRs were performed using hot-start AmpliTaq® Gold DNA polymerase kit (Applied Biosystems, Foster City, CA, USA) and a MJ PTC-200 (Bio-Rad, Hercules, CA, USA) thermal cycler. PCR reaction was carried out in 20 μl solution containing 1.5 mM Mg2+, 200 μM dNTP, 0.3 μM each primer, 10 ng genomic DNA as template, and 0.5 U polymerase. Five percent DMSO is included in the PCR reaction mixture, particularly for the amplification of exon 2 and exon 9. After confirming the purity and mobility by agarose gel electrophoresis, each PCR product was purified and subjected to DNA sequencing using BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) and the ABI Prism® 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Each sample was sequenced for both strands to confirm the results and the SNPs were identified by using SeqScape (ABI, Foster City, CA, USA).
Genotyping for tag SNPs rs8192772, rs2070672 and rs2480256 in stage I and II were performed using direct sequencing described above, while tag SNP rs2031920 was detected using previously described Restriction Fragment Length Polymorphism methods [18].
Statistical analysis
The Hardy-Weinberg equilibrium test was done for each polymorphism detected in the control population. The software HaploView 3.32 (Massachusetts Institute of Technology, Cambridge, MA, USA) [19] was used to calculate linkage disequilibrium (LD) and software PHASE v 2.1 (Department of Statistics, University of Chicago, Chicago, IL, USA) [20] was used for inferring haplotypes as well as analyzing haplotype association. Tag SNPs were selected using Carlson's method [21]. Using this method, bins of the common SNPs that are in very strong LD with a specified r2 threshold were identified and then one SNP was selected to represent the remaining SNPs in each bin. We used an r2 threshold of 0.8 and a minimum minor allele frequency of 0.05. When multiple SNPs were assigned as tag SNPs for a particular bin, the SNP was selected for that bin based on LD pattern, ease of assay design and probability of being functionally important. The alleles and genotypes of the SNPs were counted, and their distributions between the case and control groups were compared by the χ2 test to test the hypothesis of association between polymorphism and SLE. Considering the multiple tests involved in this study, a P-value < 0.01 was set as significant level. The P-value and odd ratios (OR) with 95% confidence intervals (CI) were estimated using conditional logistic regression models adjusted for potential confounders. All the analysis was performed with program SPSS 12.0 (SPSS Inc., Chicago, IL, USA).
CYP2E1transcription quantification
One hundred and twenty healthy individuals were assessed for the CYP2E1 mRNA levels in peripheral blood. The QuantiGene® assay (Affymetrix Inc., Santa Clara, CA, USA) was used to quantify the mRNA considering the accuracy and precision of this technology without RNA purification and RT-PCR [22, 23]. According to the manufacture's design and instruction, 10 ul EDTA anti-coagulant blood was lysed at the day of collection. Blood lysates and specific probe sets were incubated in the capture plate and hybridized overnight at 55°C. Pre-amplifier and amplifier reagents were incubated in the capture plate for 60 minutes at 55°C followed by 50°C incubation of label probe with washing between each incubation. Substrate reagents were added in the end and plates were read on a chemiluminescent plate reader. The CYP2E1 signal was normalized to an averaged signal from two reference genes GAPDH and ACTB. All participants were genotyped by sequencing for SNPs rs8192772 and rs2480256. ANOVA test (SPSS 12.0) was used to compare the normalized transcription level among different genotype groups.
Results
Sequence variations in the CYP2E1gene
A total of 32 sequence polymorphisms (minor allele frequency MAF >/= 1%) were identified in 96 healthy control samples (12 in the putative promoter region, 2 in the coding region, 16 in the introns and 2 in the 3' UTR). Genotypes were in Hardy-Weinberg equilibrium at each polymorphic locus. Supplementary Table S3 in Additional file 1 and Figure 1 shows the location and frequency of these SNPs. Thirty of them were previously reported in the dbSNP [24] and in the literature [18, 25, 26], whereas the -1381C/T and -760T/G were newly identified in our study. A high density of SNPs was observed in the putative promoter region, where 12 SNPs were identified within 1.8 kb sequence. Compared with the putative promoter region and the intron regions, the frequency of variations in the coding region is relatively low. Only two synonymous variations, rs28371746 and rs2515641, were detected in exon 6 and exon 8 respectively. We excluded five rare SNPs with MAF less than 5% from LD analysis, haplotype reconstruction and association analysis because rare alleles do not have sufficient statistic power [27, 28].

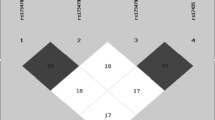
LD structure of CYP2E1 gene region in Chinese population. LD structure is determined by using the confidence intervals option of Haploview. D prime values are displayed in the squares (values of 1.0 are not shown). Square background colors showed the pairwise r2 value (r2 = 1 for black color and gradually coloring down to white as r2 = 0).
Tag SNP selection
Pairwise LD across the CYP2E1 gene (Figure 1) was relatively strong in this Chinese population and without clear LD block subdivision. All the 27 common SNPs were classified into four bins using Carlson's method [21]. Four tag SNPs, rs2031920, rs2070672, rs8192772 and rs2480256, were selected from each bin for further study based on their allele frequencies, LD pattern and probability of being functionally important. The rs2031920 and rs2070672 were located in the promoter region, rs8192772 was in the intron 2 and rs2480256 was in the 3'UTR.
Stage I: association of tag SNPs and haplotypes with SLE
In stage I, we recruited 265 SLE patients and 288 controls for genotyping the four tag SNPs. Table 1 showed the distribution of both allele and genotype frequencies of the case and control groups. Association analysis revealed that two tag SNPs were associated with SLE. For tag SNP rs8192772, frequency of "C/C" genotype in cases (5.3%) was almost two-fold higher than in the control (1.7%). Therefore, at genotypic level the homozygous "C/C" genotype was associated with elevated risk of SLE compared with T/T and T/C carriers (OR = 1.912, 95% CI 1.131 to 3.324, P = 0.016). In the case of tag SNP rs2480256, association analysis at both allelic and genotypic levels suggested that this SNP contributes to the susceptibility of SLE. The frequency of "A/A" genotype was significantly higher in SLE patients (22.6%) than in the healthy controls (11.8%). The "A/A" genotype carriers have significantly elevated risk of SLE compared with "G/G" and "A/G" carriers (OR = 1.475, 95% CI 1.170 to 1.860, P = 0.001). At allelic level, a marginal association was found that the "A" allele carriers showed increased risk to SLE (OR = 1.128, 95% CI 0.999 to 1.274, P = 0.053).
The common haplotypes and their frequency in the patients and controls inferred from PHASE program were listed in the Table 1. Only three haplotypes were presented with frequency greater than 0.05 and all the other minor haplotypes were combined for association study. The frequency of haplotype "TATA" in SLE patients is higher than that in the controls and revealed a marginal association (P = 0.081). Since this haplotype "TATA" could be identified from other common haplotypes by genotyping SNPs rs8192772 and rs2480256, we then only genotyped these two haplotype tag SNPs in stage II study with enlarged sample pool.
Stage II: verifying the association of tag SNPs rs8192772 and rs2480256 with SLE
During the stage II study, we recruited another 611 SLE patients and 392 controls for the genotyping of SNPs rs8192772 and rs2480256. The result confirmed the association tendency revealed in the stage I study (Table 2). For SNP rs8192772, the frequency of "C/C" genotype is higher in the SLE patients than in controls (OR = 1.433, 95% CI 1.023 to 2.009, P = 0.037). For SNP rs2480256, both "A" allele (OR = 1.115, 95% CI 1.016 to 1.223, P = 0.022) and "A/A" genotype (OR = 1.256, 95% CI 1.062 to 1.485, P = 0.008) are associated with SLE.
When all genotyping data from both stage I and II studies were combined (Table 2), tag SNP rs8192772 was associated with SLE at genotypic level and the "C/C" carrier showed a 1.820-fold increase in SLE risk (95% CI 1.33 to 2.49 P = 1.08E-4). For tag SNP rs2480256, it associated with SLE at both allelic and genotypic level. The "A" allele was associated with slightly higher risk (OR = 1.165, 95% CI 1.073 to 1.265, P = 2.75E-4) and "A/A" genotype carriers were with even higher SLE risk (OR = 1.464, 95% CI 1.259 to 1.702, P = 7.48E-7). Haplotypes comprising tag SNPs rs8192772 and rs2480256 and their frequencies in case and controls were also estimated. Three haplotypes were observed with frequency higher than 5% (Table 2). The frequency of haplotype "TA" was 24.5% in SLE patients, compared with 18.8% in controls (OR = 1.407, 95% CI 1.182 to 1.675, P = 0.0001). In contrast, haplotype "TG" is more common in the controls (60.4%) than in the patients (54.0%) (OR = 0.771, 95% CI 0.667 to 0.890, P = 0.0004). We also analyzed the association between SNP genotypes and various clinical features adjusted by both sex and age. However, no genotype correlation with any clinical feature was identified.
Transcription quantification of CYP2E1in PBMC
In order to study the effect of rs2480256 on CYP2E1 gene expression, we further recruited 120 healthy individuals to explore the CYP2E1 mRNA level in PBMC. By sequencing genotyping of these 120 individuals, 22 were found as "A/A" genotype, 45 were "G/G" and 53 were heterozygous "G/A". As shown in Figure 2A, the normalized relative mRNA levels from genotype "A/A", "G/G" and "G/A" were 2.01(95% CI 1.79 to 2.23), 1.59 (95% CI 1.43 to 1.76) and 1.74 (95% CI 1.62 to 1.86) respectively. The multiple group test showed the transcription levels among these three groups were significant different (P-value = 0.006). The risky genotype "A/A" showed the highest transcription while the lowest is genotype "G/G". We also genotyped SNP rs8192772 and analyzed the transcription difference in different genotypes (Figure 2B). Only "C/C" (2.35, 95% CI 1.68 to 3.03) genotype showed significant higher transcription (P < 0.05) comparing to the other two genotypes. The transcription levels between "T/T" (1.78, 95% CI 1.65 to 1.91) and "T/C" (1.60, 95% CI 1.47 to 1.73) genotype did not show statistical significant difference.

Relative transcription level of CYP2E1on SNP rs2480256 (A) and rs8192772 (B).
Discussion
After conducting a two-stage study involving the genotyping and clinical evaluation of 876 SLE patients and 680 healthy controls recruited among Han Chinese, we provide the first time evidence for the statistically significant association for SNP rs2480256, of CYP2E1 with SLE. The "A/A" carriers for SNP rs2480256 showed higher SLE risk (OR = 1.464 95% CI 1.259 to 1.702, P = 7.48E-7). We also detected that haplotype "rs8192772-rs2480256/TG" was overrepresented in controls (OR = 0.771, 95% CI 0.667 to 0.890, P = 0.0004) whereas haplotype "TA" was overrepresented in SLE patients (OR = 1.407, 95% CI 1.182 to 1.675, P = 0.0001). Though the "C/C" genotype of rs8192772 showed a 1.820-fold higher SLE risk (95% CI 1.33 to 2.49, P = 1.08E-4), the haplotype analysis suggested the dominant effect of SNP rs2480256 in SLE susceptibility.
SNP rs2480256 is located in the 3' UTR, a region with proven role(s) in the regulation of gene expression. Many disease-associated variants have been mapped in the 3' UTR of human protein-coding genes [29]. A recent publication on SLE susceptibility exploring also revealed a significant association of a variation located in 3'UTR of ETS1 gene [30, 31]. The significant association of SNP rs2480256 to SLE maybe due to its effect on CYP2E1 gene expression and/or its LD with a functional variant residing in either CYP2E1 or neighboring genes. In order to clarify whether this SNP can affect the CYP2E1 transcription, we examined the CYP2E1 mRNA level in PBMC sample collected from individuals with different genotype. The statistical analysis showed that CYP2E1 mRNA levels are significantly different among different rs2480256 genotypes. The most risky genotype "A/A" showed highest transcription levels, while the protective genotype "G/G" is the lowest. It is also interesting that, for SNP rs8192772, the only risky genotype "C/C" showed significantly higher gene transcription but not "T/C" heterozygous. By examining the LD between these two SNPs, we noticed that only the C allele of rs8192772 is in very strong LD with the A allele of rs2480256 (D' = 1). This explained why the rs8192772 "C/C" genotype showed higher CYP2E1 transcription but not in the "C/T" genotype. This transcriptional analysis confirmed the genotyping haplotype analysis proposed dominant effect of rs2480256.
Xenobiotic exposures often trigger the onset of autoimmune diseases including SLE, human CYP genes have significant bearing on individual susceptibility to chemical toxicity and were speculated to be involved in several 'lupus-like' disorders [32]. The inactive CYP2D6*4A allele may be a contributory factor for SLE [33]. After genotyping 90 SLE patients and 94 healthy controls, Yen et al. [34] concluded that CYP1A1 4887A may be a precipitating factor for SLE, and they detected a synergistic effect for SLE susceptibility between CYP1A1 4887C/A and Mn SOD 1183T/T. Recent studies in Chinese and Korean further identified Glutathione S-transferase family and CYP1A1 are involved in SLE development [35, 36]. As a phase I enzyme, CYP2E1 encodes N,N-dimethylnitrosamino N-demethylase which can catalyze xenobiotics into a more reactive and toxic form. In addition to its ability to metabolize a wide variety of low molecular weight compounds, it is also an effective generator of reactive oxygen species (ROS) [37]. Among the wide substrate spectrum of CYP2E1, TCE has been implicated in the development of autoimmune disorders and immune system dysfunction both in human and animal studies [12, 16, 38]. TCE is a widely used industrial solvent for degreasing and paint stripping. This volatile organic compound is a common pollutant found in soil and water. Recently, ROS and oxidative stress were implicated in the pathogenesis of SLE [39]. It has been demonstrated that DNA damaged by ROS is highly immunogenic [40, 41]. Therefore, it is possible that the higher expression of CYP2E1 contributed to SLE development through the production of more ROS during the compound metabolization with/or particular toxic intermediates produced, for example, metabolites of TCE.
CYP2E1 is located in the chromosome 10q24.3-qter, the gene spans over 11 kb and contains 9 exons. When we started this project, the tag SNPs for CYP2E1 was not available in the HAPMAP database and literature. We were able to identify four tag SNPs (rs2031920, rs2070672, rs8192772 and rs2480256) by pairwise LD analysis for haplotype inferring. Such detailed information is a good complementary to the released Phase III Hapmap data. And by searching the most updated public database, no other putative or known genes showed significant LD with the SNP rs2480256. In recent years, several genome-wide association studies have been conducted to identify SLE susceptibility markers in different ethnic groups, including the Chinese Han population [31, 42–46]. Though validity of these findings has been shown in genes such as ETS1 and SLC15A4, people also noticed that every study revealed some unique markers. And in these published Genome Wide Association Study data, none of the susceptible markers has a physical linkage with the markers we identified in this study. On the other hand, several genomic scans detecting SLE predisposition genes were performed in extended multicase families or sib-pairs [47–54]. The few overlaps detected among these studies indicate that multiple predisposing genes, ethnic diversity, and genetic heterogeneity are likely. After conducting a 10-cM full genome scan with approximately 400 multiallelic markers for 238 individuals from a multiethnic panel of 62 multiplex SLE families, Cantor et al. [47] confirmed the linkage to four previously reported sites (1q23, 2q33, 16q12 to 13 and 17q21 to 23) and revealed four novel sites (3p24, 10q23 to 24, 13q32 and 18q22 to 23). One of these sites, 10q23 to 24, is very close to the CYP2E1 locus. These projects exhibit the complexity of identifying susceptibility genes in a case-control study. The elucidation of disease mechanism and the functional importance of markers involved may eventually guide us to clinical and therapeutic applications.
In this project, we adopted a two-stage association study strategy. In the first stage, all tag SNPs were screened and only the most promising markers were then genotyped in the second stage in other samples. Compared to one-stage designs, two-stage designs can reduce the amount of genotyping and provide near-optimal power to detect the true marker conferring disease risk [55]. In conclusion, the present study suggests that genetic variations in the CYP2E1 gene may contribute to SLE susceptibility in the Chinese Han population. One potential concern of the current study is that we only tested limited variants in the functional region of CYP2E1 gene and left a relatively large region of introns unexplored, which is also not available in a chip-based genotyping assay. It is possible that, by employing a denser marker map, we may observe some other significantly associated markers which may be involved in regulating gene expression. In addition, replications in other populations and further functional studies are also required to confirm and interpret the association of CYP2E1 gene with SLE.
Conclusions
CYP2E1 gene contributes to the SLE susceptibility in Han Chinese population. The haplotype "rs8192772-rs2480256/TG" was overrepresented in controls (OR 0.771, 95% CI 0.667 to 0.890, P = 0.0004), whereas haplotype "TA" was overrepresented in SLE patients (OR = 1.407, 95% CI 1.182 to 1.675, P = 0.0001). SNP rs2480256 showed the dominant effect by significantly affecting the CYP2E1 transcription.
Abbreviations
- ACR:
-
American College of Rheumatology
- CI:
-
confidence interval
- CYP2E1:
-
Cytochrome P-450 2E1
- LD:
-
linkage disequilibrium
- OR:
-
odds ratio
- PBMC:
-
peripheral blood mononuclear cell
- ROS:
-
reactive oxygen species
- SLE:
-
systemic lupus erythematosus
- SNP:
-
single nucleotide polymorphism
- TCE:
-
trichloroethylene
- UTR:
-
untranslated region.
References
Deapen D, Escalante A, Weinrib L, Horwitz D, Bachman B, Roy-Burman P, Walker A, Mack TM: A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum. 1992, 35: 311-318. 10.1002/art.1780350310.
Hochberg MC: Epidemiology of systemic lupus erythematosus. Dubois' Lupus Erythematosus. Edited by: Wallace DJ, Hahn BH. 1997, Baltimore: Lippincott Williams & Wilkins, 49-65.
Shen N, Tsao BP: Current advances in the human lupus genetics. Curr Rheumatol Rep. 2004, 6: 391-398. 10.1007/s11926-004-0014-3.
Wakeland EK, Liu K, Graham RR, Behrens TW: Delineating the genetic basis of systemic lupus erythematosus. Immunity. 2001, 15: 397-408. 10.1016/S1074-7613(01)00201-1.
Bae SC, Fraser P, Liang MH: The epidemiology of systemic lupus erythematosus in populations of African ancestry: a critical review of the "prevalence gradient hypothesis". Arthritis Rheum. 1998, 41: 2091-2099. 10.1002/1529-0131(199812)41:12<2091::AID-ART2>3.0.CO;2-D.
Balluz L, Philen R, Ortega L, Rosales C, Brock J, Barr D, Kieszak S: Investigation of systemic lupus erythematosus in Nogales, Arizona. Am J Epidemiol. 2001, 154: 1029-1036. 10.1093/aje/154.11.1029.
Cooper GS, Parks CG, Treadwell EL, St Clair EW, Gilkeson GS, Dooley MA: Occupational risk factors for the development of systemic lupus erythematosus. J Rheumatol. 2004, 31: 1928-1933.
Kilburn KH, Warshaw RH: Prevalence of symptoms of systemic lupus erythematosus (SLE) and of fluorescent antinuclear antibodies associated with chronic exposure to trichloroethylene and other chemicals in well water. Environ Res. 1992, 57: 1-9. 10.1016/S0013-9351(05)80014-3.
Iavicoli I, Marinaccio A, Carelli G: Effects of occupational trichloroethylene exposure on cytokine levels in workers. J Occup Environ Med. 2005, 47: 453-457. 10.1097/01.jom.0000161728.23285.66.
Gilbert KM, Griffin JM, Pumford NR: Trichloroethylene activates CD4+ T cells: potential role in an autoimmune response. Drug Metab Rev. 1999, 31: 901-916. 10.1081/DMR-100101945.
Khan MF, Kaphalia BS, Prabhakar BS, Kanz MF, Ansari GA: Trichloroethene-induced autoimmune response in female MRL +/+ mice. Toxicol Appl Pharmacol. 1995, 134: 155-160. 10.1006/taap.1995.1179.
Griffin JM, Gilbert KM, Lamps LW, Pumford NR: CD4(+) T-cell activation and induction of autoimmune hepatitis following trichloroethylene treatment in MRL+/+ mice. Toxicol Sci. 2000, 57: 345-352. 10.1093/toxsci/57.2.345.
Finckh A, Cooper GS, Chibnik LB, Costenbader KH, Watts J, Pankey H, Fraser PA, Karlson EW: Occupational silica and solvent exposures and risk of systemic lupus erythematosus in urban women. Arthritis Rheum. 2006, 54: 3648-3654. 10.1002/art.22210.
Hanioka N, Tanaka-Kagawa T, Miyata Y, Matsushima E, Makino Y, Ohno A, Yoda R, Jinno H, Ando M: Functional characterization of three human cytochrome p450 2E1 variants with amino acid substitutions. Xenobiotica. 2003, 33: 575-586. 10.1080/0049825031000086400.
Lipscomb JC, Garrett CM, Snawder JE: Cytochrome P450-dependent metabolism of trichloroethylene: interindividual differences in humans. Toxicol Appl Pharmacol. 1997, 142: 311-318. 10.1006/taap.1996.8040.
Griffin JM, Gilbert KM, Pumford NR: Inhibition of CYP2E1 reverses CD4+ T-cell alterations in trichloroethylene-treated MRL+/+ mice. Toxicol Sci. 2000, 54: 384-389. 10.1093/toxsci/54.2.384.
Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, Schaller JG, Talal N, Winchester RJ: The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982, 25: 1271-1277. 10.1002/art.1780251101.
Watanabe J, Hayashi S, Nakachi K, Imai K, Suda Y, Sekine T, Kawajiri K: PstI and RsaI RFLPs in complete linkage disequilibrium at the CYP2E gene. Nucleic Acids Res. 1990, 18: 7194-10.1093/nar/18.23.7194.
Barrett JC, Fry B, Maller J, Daly MJ: Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005, 21: 263-265. 10.1093/bioinformatics/bth457.
Stephens M, Smith NJ, Donnelly P: A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001, 68: 978-989. 10.1086/319501.
Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA: Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004, 74: 106-120. 10.1086/381000.
Canales RD, Luo Y, Willey JC, Austermiller B, Barbacioru CC, Boysen C, Hunkapiller K, Jensen RV, Knight CR, Lee KY, Ma Y, Maqsodi B, Papallo A, Peters EH, Poulter K, Ruppel PL, Samaha RR, Shi L, Yang W, Zhang L, Goodsaid FM: Evaluation of DNA microarray results with quantitative gene expression platforms. Nat Biotechnol. 2006, 24: 1115-1122. 10.1038/nbt1236.
Zheng Z, Luo Y, McMaster GK: Sensitive and quantitative measurement of gene expression directly from a small amount of whole blood. Clin Chem. 2006, 52: 1294-1302. 10.1373/clinchem.2005.065078.
The NCBI dbSNP. [http://www.ncbi.nlm.nih.gov/snp]
Hu Y, Oscarson M, Johansson I, Yue QY, Dahl ML, Tabone M, Arinco S, Albano E, Ingelman-Sundberg M: Genetic polymorphism of human CYP2E1: characterization of two variant alleles. Mol Pharmacol. 1997, 51: 370-376.
Solus JF, Arietta BJ, Harris JR, Sexton DP, Steward JQ, McMunn C, Ihrie P, Mehall JM, Edwards TL, Dawson EP: Genetic variation in eleven phase I drug metabolism genes in an ethnically diverse population. Pharmacogenomics. 2004, 5: 895-931. 10.1517/14622416.5.7.895.
Goddard KA, Hopkins PJ, Hall JM, Witte JS: Linkage disequilibrium and allele-frequency distributions for 114 single-nucleotide polymorphisms in five populations. Am J Hum Genet. 2000, 66: 216-234. 10.1086/302727.
Lewontin RC: The detection of linkage disequilibrium in molecular sequence data. Genetics. 1995, 140: 377-388.
Chen JM, Ferec C, Cooper DN: A systematic analysis of disease-associated variants in the 3' regulatory regions of human protein-coding genes I: general principles and overview. Hum Genet. 2006, 120: 1-21. 10.1007/s00439-006-0180-7.
Shen N, Fu Q, Deng Y, Qian X, Zhao J, Kaufman KM, Wu YL, Yu CY, Tang Y, Chen JY, Yang W, Wong M, Kawasaki A, Tsuchiya N, Sumida T, Kawaguchi Y, Howe HS, Mok MY, Bang SY, Liu FL, Chang DM, Takasaki Y, Hashimoto H, Harley JB, Guthridge JM, Grossman JM, Cantor RM, Song YW, Bae SC, Chen S, et al: Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc Natl Acad Sci USA. 2010, 107: 15838-15843. 10.1073/pnas.1001337107.
Yang W, Shen N, Ye DQ, Liu Q, Zhang Y, Qian XX, Hirankarn N, Ying D, Pan HF, Mok CC, Chan TM, Wong RW, Lee KW, Mok MY, Wong SN, Leung AM, Li XP, Avihingsanon Y, Wong CM, Lee TL, Ho MH, Lee PP, Chang YK, Li PH, Li RJ, Zhang L, Wong WH, Ng IO, Lau CS, Sham PC, et al: Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet. 2010, 6: e1000841-10.1371/journal.pgen.1000841.
McKinnon RA, Nebert DW: Possible role of cytochromes P450 in lupus erythematosus and related disorders. Lupus. 1994, 3: 473-478. 10.1177/096120339400300608.
Sabbagh N, Marez D, Queyrel V, Lo Guidice JM, Spire C, Vanhille P, Jorgensen C, Hachulla E, Broly F: Genetic analysis of the cytochrome P450 CYP2D6 polymorphism in patients with systemic lupus erythematosus. Pharmacogenetics. 1998, 8: 191-194. 10.1097/00008571-199806000-00001.
Yen JH, Chen CJ, Tsai WC, Lin CH, Ou TT, Hu CJ, Liu HW: Cytochrome P450 and manganese superoxide dismutase genes polymorphisms in systemic lupus erythematosus. Immunol Lett. 2003, 90: 19-24. 10.1016/S0165-2478(03)00157-3.
Kang TY, El-Sohemy A, Comelis MC, Eny KM, Bae SC: Glutathione S-transferase genotype and risk of systemic lupus erythematosus in Koreans. Lupus. 2005, 14: 381-384. 10.1191/0961203305lu2100oa.
Zhang J, Deng J, Zhang C, Lu Y, Liu L, Wu Q, Shao Y, Zhang J, Yang H, Yu B, Wan J: Association of GSTT1, GSTM1 and CYP1A1 polymorphisms with susceptibility to systemic lupus erythematosus in the Chinese population. Clin Chim Acta. 2010, 411: 878-881. 10.1016/j.cca.2010.03.007.
Kessova I, Cederbaum AI: CYP2E1: biochemistry, toxicology, regulation and function in ethanol-induced liver injury. Curr Mol Med. 2003, 3: 509-518. 10.2174/1566524033479609.
Griffin JM, Blossom SJ, Jackson SK, Gilbert KM, Pumford NR: Trichloroethylene accelerates an autoimmune response by Th1 T cell activation in MRL +/+ mice. Immunopharmacology. 2000, 46: 123-137. 10.1016/S0162-3109(99)00164-2.
Wang G, Cai P, Ansari GA, Khan MF: Oxidative and nitrosative stress in trichloroethene-mediated autoimmune response. Toxicology. 2007, 229: 186-193. 10.1016/j.tox.2006.10.014.
Cooke MS, Mistry N, Wood C, Herbert KE, Lunec J: Immunogenicity of DNA damaged by reactive oxygen species--implications for anti-DNA antibodies in lupus. Free Radic Biol Med. 1997, 22: 151-159. 10.1016/S0891-5849(96)00283-3.
Ashok BT, Ali R: Binding of human anti-DNA autoantibodies to reactive oxygen species modified-DNA and probing oxidative DNA damage in cancer using monoclonal antibody. Int J Cancer. 1998, 78: 404-409. 10.1002/(SICI)1097-0215(19981109)78:4<404::AID-IJC2>3.0.CO;2-Y.
Graham RR, Hom G, Ortmann W, Behrens TW: Review of recent genome-wide association scans in lupus. J Intern Med. 2009, 265: 680-688. 10.1111/j.1365-2796.2009.02096.x.
Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z, Xu JH, Cai ZM, Huang W, Zhao GP, Xie HF, Fang H, Lu QJ, Xu JH, Li XP, Pan YF, Deng DQ, Zeng FQ, Ye ZZ, Zhang XY, Wang QW, Hao F, Ma L, Zuo XB, Zhou FS, Du WH, Cheng YL, Yang JQ, Shen SK, Li J, et al: Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. 2009, 41: 1234-1237. 10.1038/ng.472.
International Consortium for Systemic Lupus Erythematosus Genetics (SLEGEN), Harley JB, Alarcón-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ, Langefeld CD, Nath SK, Guthridge JM, Cobb BL, Mirel DB, Marion MC, Williams AH, Divers J, Wang W, Frank SG, Namjou B, Gabriel SB, Lee AT, Gregersen PK, Behrens TW, Taylor KE, Fernando M, Zidovetzki R, Gaffney PM, Edberg JC, Rioux JD, Ojwang JO, et al: Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008, 40: 204-210. 10.1038/ng.81.
Hom G, Graham RR, Modrek B, Taylor KE, Ortmann W, Garnier S, Lee AT, Chung SA, Ferreira RC, Pant PV, Ballinger DG, Kosoy R, Demirci FY, Kamboh MI, Kao AH, Tian C, Gunnarsson I, Bengtsson AA, Rantapaa-Dahlqvist S, Petri M, Manzi S, Seldin MF, Ronnblom L, Syvanen AC, Criswell LA, Gregersen PK, Behrens TW: Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med. 2008, 358: 900-909. 10.1056/NEJMoa0707865.
Kariuki SN, Franek BS, Kumar AA, Arrington J, Mikolaitis RA, Utset TO, Jolly M, Crow MK, Skol AD, Niewold TB: Trait-stratified genome-wide association study identifies novel and diverse genetic associations with serologic and cytokine phenotypes in systemic lupus erythematosus. Arthritis Res Ther. 2010, 12: R151-10.1186/ar3101.
Cantor RM, Yuan J, Napier S, Kono N, Grossman JM, Hahn BH, Tsao BP: Systemic lupus erythematosus genome scan: support for linkage at 1q23, 2q33, 16q12-13, and 17q21-23 and novel evidence at 3p24, 10q23-24, 13q32, and 18q22-23. Arthritis Rheum. 2004, 50: 3203-3210. 10.1002/art.20511.
Gaffney PM, Kearns GM, Shark KB, Ortmann WA, Selby SA, Malmgren ML, Rohlf KE, Ockenden TC, Messner RP, King RA, Rich SS, Behrens TW: A genome-wide search for susceptibility genes in human systemic lupus erythematosus sib-pair families. Proc Natl Acad Sci USA. 1998, 95: 14875-14879. 10.1073/pnas.95.25.14875.
Gaffney PM, Ortmann WA, Selby SA, Shark KB, Ockenden TC, Rohlf KE, Walgrave NL, Boyum WP, Malmgren ML, Miller ME, Kearns GM, Messner RP, King RA, Rich SS, Behrens TW: Genome screening in human systemic lupus erythematosus: results from a second Minnesota cohort and combined analyses of 187 sib-pair families. Am J Hum Genet. 2000, 66: 547-556. 10.1086/302767.
Gray-McGuire C, Moser KL, Gaffney PM, Kelly J, Yu H, Olson JM, Jedrey CM, Jacobs KB, Kimberly RP, Neas BR, Rich SS, Behrens TW, Harley JB: Genome scan of human systemic lupus erythematosus by regression modeling: evidence of linkage and epistasis at 4p16-15.2. Am J Hum Genet. 2000, 67: 1460-1469. 10.1086/316891.
Johansson CM, Zunec R, Garcia MA, Scherbarth HR, Tate GA, Paira S, Navarro SM, Perandones CE, Gamron S, Alvarellos A, Graf CE, Manni J, Berbotto GA, Palatnik SA, Catoggio LJ, Battagliotti CG, Sebastiani GD, Migliaresi S, Galeazzi M, Pons-Estel BA, Alarcón-Riquelme ME: Chromosome 17p12-q11 harbors susceptibility loci for systemic lupus erythematosus. Hum Genet. 2004, 115: 230-238. 10.1007/s00439-004-1145-3.
Lindqvist AK, Steinsson K, Johanneson B, Kristjansdottir H, Arnasson A, Grondal G, Jonasson I, Magnusson V, Sturfelt G, Truedsson L, Svenungsson E, Lundberg I, Terwilliger JD, Gyllensten UB, arcon-Riquelme ME: A susceptibility locus for human systemic lupus erythematosus (hSLE1) on chromosome 2q. J Autoimmun. 2000, 14: 169-178. 10.1006/jaut.1999.0357.
Moser KL, Neas BR, Salmon JE, Yu H, Gray-McGuire C, Asundi N, Bruner GR, Fox J, Kelly J, Henshall S, Bacino D, Dietz M, Hogue R, Koelsch G, Nightingale L, Shaver T, Abdou NI, Albert DA, Carson C, Petri M, Treadwell EL, James JA, Harley JB: Genome scan of human systemic lupus erythematosus: evidence for linkage on chromosome 1q in African-American pedigrees. Proc Natl Acad Sci USA. 1998, 95: 14869-14874. 10.1073/pnas.95.25.14869.
Shai R, Quismorio FP, Li L, Kwon OJ, Morrison J, Wallace DJ, Neuwelt CM, Brautbar C, Gauderman WJ, Jacob CO: Genome-wide screen for systemic lupus erythematosus susceptibility genes in multiplex families. Hum Mol Genet. 1999, 8: 639-644. 10.1093/hmg/8.4.639.
Satagopan JM, Venkatraman ES, Begg CB: Two-stage designs for gene-disease association studies with sample size constraints. Biometrics. 2004, 60: 589-597. 10.1111/j.0006-341X.2004.00207.x.
Acknowledgements
We acknowledge the financial support of Grant 2010J05071 from Fujian Provincial Department of Science & Technology, 471 HKUST6117/01M from RGC, Grant CMI03/04.SC03 from HKUST472 and Grant UPHMSGF 06/07.SC01 from Mochtar Riady Institute for 473 nanotechnology.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors have no competing interests as defined by Arthritis Research & Therapy, or other interests that might be perceived to influence the results and/or discussion reported in this article.
Authors' contributions
LHL participated in the sample collection and genotyping, carried out the data analysis and drafted the manuscript. HZ and MPL participated in the sample collection and genotyping. SLC and NS participated in study design and data interpretation. MW participated in the study design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
13075_2010_2988_MOESM1_ESM.DOC
Additional File 1: Supplementary Tables S1-S3. Supplementary Table S1: Clinical and immunological features of cases. Supplementary Table S2: Primers for CYP2E1 genotyping. Supplementary Table S3: Sequence variations identified in 96 healthy people in CYP2E1. (DOC 103 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Liao, Lh., Zhang, H., Lai, MP. et al. Single-nucleotide polymorphisms and haplotype of CYP2E1gene associated with systemic lupus erythematosus in Chinese population. Arthritis Res Ther 13, R11 (2011). https://doi.org/10.1186/ar3232
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar3232