
Abstract
Interleukin-11 (IL-11) is a pleiotropic cytokine that regulates the growth and development of hematopoietic stem cells and decreases the proinflammatory mediators of cytokine and nitric oxide production. In animal models of arthritis, treatment with recombinant human IL-11 (rhIL-11) reduces both the level of synovitis and the histologic lesion scores in the joints.
The goal of this phase-I/II study in adults with rheumatoid arthritis (RA) was to evaluate the safety and clinical activity of different doses and schedules of rhIL-11 in patients with active RA for whom treatment with at least one disease-modifying antirheumatic drug had failed.
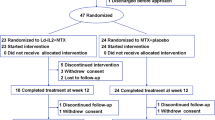
This was a multicenter, randomized, placebo-controlled trial that evaluated the safety and tolerability of rhIL-11 in 91 patients with active RA. rhIL-11 was administered subcutaneously; patients were randomized into one of five treatment groups (ratio of rhIL-11 to placebo, 4:1). Patients were treated for 12 weeks with either 2.5 or 7.5 μg/kg of rhIL-11 or placebo twice per week or 5 or 15 μg/kg of rhIL-11 or placebo once per week. The status of each subject's disease activity in accordance with the American College of Rheumatology (ACR) criteria was assessed before, during, and after completion of administration of the study drug.
Administration of rhIL-11 was well tolerated at all doses and schedules. The most frequent adverse event was a reaction at the injection site. The data suggest a statistically significant reduction in the number of tender joints (P < 0.008) at the 15 μg/kg once-weekly dose schedule but showed no overall significant benefit at the ACR criterion of a 20% response.
The trial showed rhIL-11 to be safe and well tolerated at a variety of doses and schedules over a 12-week treatment period in patients with active RA. The only adverse event clearly associated with rhIL-11 administration was reaction at the injection site.
Similar content being viewed by others


Introduction
Recombinant human interleukin-11 (rhIL-11) is a pleiotropic cytokine that regulates the growth and development of hematopoietic stem cells and decreases the proinflammatory mediators of cytokine and nitric oxide production [1]. Treatment with rhIL-11 decreases the production of proinflammatory cytokines, including tumor necrosis factor (TNF)-α and interleukin (IL)-1β, and inhibits NF-κB binding activity [2,3,4]. In animal models both of collagen-induced and adjuvant-induced arthritis, rhIL-11 reduced both the level of synovitis and the histologic lesion score in the joints [5].
Rheumatoid arthritis (RA) is a chronic inflammatory disease of unknown etiology. Its worldwide prevalence is approximately 1% [6]. There appears to be no single therapeutic regimen or combination of therapies that has consistently been associated with sustained improvement (i.e. remission) [7]. A number of disease-modifying drugs (e.g. methotrexate) are available to treat RA but are limited by significant toxicity requiring adjunct therapy, or are of variable efficacy. There are currently a number of anticytokine and other immune-modulating therapies in clinical trials [8]. The proinflammatory cytokine TNF has been shown to play a role in the pathogenesis of RA [9,10]. Etanercept (sTNFR:Fc) and infliximab (chimeric anti-TNF monoclonal antibody) have been shown to be efficacious in the treatment of patients with RA for whom treatment with at least one disease-modifying antirheumatic drug had previously failed [11,12]; both of these biologicals have been approved by regulatory agencies for the treatment of patients with RA.
rhIL-11 has previously been assessed in a Phase-I/II, masked, placebo-controlled trial in patients with active Crohn's disease. The data suggested clinical benefit as assessed from the mean change from baseline in the Crohn's Disease Severity Index after 21 days at doses of 16 and 40 μg/kg per week (P < 0.05). The researchers concluded that rhIL-11 was safe at the doses and schedules used and had a therapeutic effect [13,14]. The findings supported the initiation of a phase-I/II clinical trial in patients with RA.
Materials and Methods
Patients
Men and women at least 18 years of age were eligible to participate if they had met the American College of Rheumatology (ACR) criteria for RA for at least one year and were in functional Class I, II, or III [15]. All patients had to have had at least one failed trial of a disease-modifying antirheumatic drug (DMARD) and had to have at least 10 painful and 10 swollen joints at entry. No DMARD therapy was allowed within 4 weeks of randomization. Patients receiving nonsteroidal anti-inflammatory drugs, corticosteroids (≤ 10 mg per day), or both were allowed in the trial if they had been on stable doses for at least four weeks before randomization. Required baseline laboratory values included serum creatinine and blood urea nitrogen ≤ 1.5 × the upper limit of normal and alanine aminotranferase and aspartate aminotransferase ≤ 2.0 × the upper limit of normal. Exclusionary laboratory values included a platelet count of > 500,000/mm3, hematocrit <30%, and a white-blood-cell count < 3000/mm3. Other exclusionary criteria included use of intravenous or intra-articular corticosteroids within four weeks of randomization, any prior use of cyclosporine or cyclophosphamide, use of any investigational agent within 30 days of randomization, severe extra-articular manifestations of RA, acute infection requiring antibiotic therapy within two weeks of randomization, other autoimmune disease (e.g. systemic lupus erythematosus), or any other condition that the investigator thought might have placed the patient at undue risk if they had participated in the trial. Men and women of childbearing potential were required to use approved methods of birth control. Women had to have a negative result on a test of serum beta human chorionic gonadotropin at screening.
Medications including cyclosporine, cyclophosphamide, and any DMARD were not allowed. The use of intravenous or intra-articular corticosteroids was not permitted. The following analgesics were permitted: acetaminophen, acetaminophen with codeine, acetaminophen with oxycodone, and propoxyphene. Patients were instructed not to take analgesics within 12 hours of their planned study visit.
Study protocol
An Institutional Review Board approved the protocol at each of the 12 participating centers. Patients gave prior written, informed consent to any study-related procedures. Within 14 days before randomization, patients had a complete medical history taken and a complete physical examination. A standard hematology profile was prepared, including white-blood-cell count with differential count, and a serum-chemistry profile was prepared, consisting of sodium, potassium, chloride, blood urea nitrogen, creatinine, glucose, phosphorus, total bilirubin, alkaline phosphatase, aspartate aminotransferase, alanine aminotransferase, albumin, uric acid, total protein, C-reactive protein (CRP), fibrinogen, and a microscopic urinalysis. An additional serum specimen was taken on days 1, 28, 84, and 112 to measure for the presence of IL-6. Screening clinical assessments included vital signs, counts of tender and swollen joints, the patient's global assessment and assessment of their pain, and a physician's global assessment using a 7-point Likert scale (0 = none, or good, to 7 = severe, or very bad). Rheumatoid factor was also measured. A serum pregnancy test was done in all female patients of childbearing potential. Disease activity and clinical laboratory values were assessed again on day 1, and then every 2 weeks for 12 weeks, and at 4 weeks post dosing. Day-1 procedures were performed before the first dose of study drug was administered.
Rheumatologists or trained study coordinators performed clinical assessments. To minimize variability, the same person assessed disease activity during the trial whenever possible.
Patients could withdraw from the trial at any time for any of the following reasons: at the patient's request, if a serious adverse event occurred, if a concomitant medication not permitted by the protocol was required, or if a patient became pregnant, did not comply with study-related procedures, or needed surgery.
Treatment
Twelve centers randomized patients, using a central randomization to one of five treatment groups: placebo, 2.5 or 7.5 μg/kg of rhIL-11 twice per week, or 5.0 or 15 μg/kg of rhIL-11 given subcutaneously once per week. The maximum dose in any single injection was 1000 μg. The study coordinator taught patients how to administer the study drug subcutaneously themselves. Either rhIL-11 or a placebo (saline) was supplied as 1 mg lyophilized powder in a phosphate/glycine formulation and reconstituted with 1 ml water for injection.
Statistical analysis
The primary objective of this trial was to evaluate the safety and tolerability of rhIL-11 given to patients with active RA. The safety endpoints were the incidence and severity of reported adverse events and abnormal laboratory findings. Adverse events are reported without regard to causality and are described for the study as a whole and by treatment group. Descriptive statistics summarizing the incidence and severity of adverse events are presented.
The secondary objective of this trial was to evaluate and characterize the clinical activity of rhIL-11. The endpoint used to characterize clinical activity was the proportion of patients whose disease improved according to the 20% ACR criteria [16]. The overall proportion of patients whose disease improved within each individual category of the ACR criteria is also presented. Exploratory comparative tests on differences in the proportions of improvement between the rhIL-11-treated groups and the placebo group were done, using a two-sided Fisher's exact test. Logistic regression was used to explore the effect of total weekly dose, dosing schedule, treatment duration, and outcome on clinical activity. Missing data were handled by both the ad hoc last-observation-carried-forward method and the worst-case-scenario method. In the last-observation-carried-forward approach, when an observation for a 12-week time point was lacking for a patient, the last available observed value was carried forward. In the worst-case-scenario approach, dropouts were treated as failures. The sample size was based on an assumed 10% placebo success rate. The sample size had a >80% power to detect a 0.51 difference from the placebo success rate in at least one of the rhIL-11 treatment groups at the α = 0.05 level of significance.
Results
Characteristics of patients
The pretherapy characteristics of the patients are summarized in Table 1. Sixty-seven women and 24 men were randomized to the study and 90 patients received at least one dose of study drug. One patient was randomized but never received the study drug as they withdrew consent prior to treatment. The mean age of all patients was 51 years (range 21–83) and the mean time since diagnosis was 12.6 years (range 1.2-40.3). No significant differences between the pretherapy characteristics of men and women were noted. The mean numbers of tender and swollen joints in all patients at screening were 27 and 20, respectively. Forty-nine of the 90 patients (54.4%) who were dosed completed 12 weeks of dosing. The majority of patients (24 of 41 or 58.5%) who discontinued early withdrew due to lack of clinical effect. Eleven patients (12.2%) withdrew because of adverse events. Of the remaining six patients, four withdrew their consent, one was lost to follow-up, and one received a prohibited medication. Patients who discontinued from the trial were evenly distributed among all the treatment groups.
Safety
rhIL-11 was well tolerated. No dose-limiting toxicities were observed. In the 11 patients who withdrew because of an adverse event, none of these events were considered to be drug related. One patient died during the study, from complications associated with a spontaneous pneumothorax; this patient was in the placebo group. The only adverse events clearly related to rhIL-11 were reactions at the injection site, which were seen in 60.6% (43 of 71) of patients who received rhIL-11 and none of the 19 placebo recipients. The incidence of injection-site reactions was not related to any dose or schedule. These reactions (erythema or erythema with induration) were mild and resolved within days after the study drug was discontinued. There were no statistically significant differences in the incidences of respiratory infections, non-respiratory infections, or edema. A higher incidence of infection in the rhIL-11 treated group (7.8%) than in the placebo group (0%) was not statistically significant (P = 0.34). Clinical trials with other agents that affect levels of circulating TNF-α have also found more frequent, nonstatistical differences in the incidence of upper-respiratory infections [11]. Adverse events occurring in more than 5% of patients are summarized in Table 2.
No significant changes in hematology or chemistry parameters were noted during the study. There were no statistically significant changes in fibrinogen levels or platelet counts in rhIL-11 treated patients compared with placebo during the study at all doses and schedules.
Disease activity
No clear therapeutic effect associated with the administration of rhIL-11 was observed. Figure 1 describes the data using the 20% ACR criteria for response at the various doses or schedules tested. The placebo response rate at the 20% improvement criteria was unusually high (32%) for a placebo-controlled trial. Several (6/19) patients in the placebo group experienced a significant (>50%) improvement in their tender- and swollen-joint counts during the study. These findings in the placebo group obscure any comparative rhIL-11 benefit. Values for placebo-treated patients regressed towards baseline during the follow-up period in all analyses performed.

Mean percentage of improvement in tender-joint counts (with last value carried forward [LVCF]).
The group treated with rhIL-11 at 15 μg/kg once per week produced a statistically significant improvement in tender-joint counts (P = 0.007) at 12 weeks (Figure 1). The mean percentage of improvement in the tender-joint count at this dose and schedule was 31.8 (Figure 2). This trend was consistent when an intent-to-treat (ITT) analysis was used to evaluate changes from baseline in tender-joint count in all rhIL-11 treated cohorts (P < 0.015). There is no obvious reason why an improvement in tender-joint count were not mirrored by changes in other clinical measurements.

Proportion of patients with 20% improvement in ACR criteria (with last value carried forward [LVCF]).
Comparative assessment (unadjusted for multiple comparisons) at the end-of-treatment time point revealed statistically significant correlations between serum levels of IL-6 and CRP to each of the six clinical outcomes (tender-joint count, swollen-joint count, patient's global assessment, patient's assessment of pain, physician's global assessment, score on health-assessment questionnaire). Correlation coefficients were moderate (r = 0.24–0.42) and all corresponding, unadjusted, P-values were less than 0.03. These were: tender-joint count versus IL-6 (P = 0.003); swollen-joint count versus IL-6 (P = 0.026); tender-joint count versus CRP (P = 0.001); swollen-joint count versus CRP (P = 0.002). P-values of comparisons between the other four clinical assessments and IL-6/CRP were stronger, with all = 0.001.
Discussion
In this study, no therapeutic benefit of rhIL-11 (at the doses and frequencies of administration studied) was found. Some individual measurements of therapeutic effect (tender-joint counts) and biologic activity (IL-6 levels) were observed. rhIL-11 was well tolerated at all doses.
Except for the tender-joint count, none of the observed disease activities was statistically different from placebo according to the ACR criteria at the 20% improvement level. The inability of rhIL-11 to bring about a statistically significant improvement in the ACR 20% criteria may also be the result of an inability of rhIL-11 to stimulate the synthesis of acute-phase proteins, of the significant clinical response of several placebo-treated patients, or of a lack of adequate power of the study to show efficacy. Larger clinical trails of rhIL-11 at higher or more frequent dosing intervals and for a longer dosing period may provide more definitive data on the safety and efficacy of rhIL-11 in the treatment of patients with RA.
The mechanism of action of rhIL-11 in the treatment of active RA most likely results from the ability of rhIL-11 to reduce levels of TNF-α, IL-1β, and IL-12 p40. The anti-inflammatory activity of rhIL-11 is mediated in part by inhibition of NF-κB-dependent transcriptional activation of proinflammatory cytokine genes. The ability of rhIL-11 to affect levels of IL-6 is also probably mediated through this mechanism. An analysis of pharmacodynamic markers in skin biopsies obtained from patients with moderate psoriasis treated with rhIL-11 revealed lower levels of INF-γ, TNF-α, and inducible nitric oxide than before treatment.
Conclusion
These preliminary data suggest that rhIL-11 may be safely administered to patients with active RA at a variety of doses and schedules. No dose-limiting adverse event was observed. The dose of 15 μg/kg of rhIL-11 once per week suggested a minimal therapeutic effect, an improvement in tender-joint counts. Further studies are warranted to find out if higher or more frequent doses of rhIL-11 may be safe and efficacious in treating RA.
Abbreviations
- ACR:
-
American College of Rheumatology
- CRP:
-
C-reactive protein
- DMARD:
-
disease-modifying antirheumatic drug
- IL:
-
interleukin
- NF:
-
nuclear factor
- RA:
-
rheumatoid arthritis
- rhIL-11:
-
recombinant human IL-11
- TNF:
-
tumor necrosis factor.
References
Trepecchio WL, Bozza M, Pednault G, Dorner AJ: Recombinant human IL-11 attenuates the inflammatory response through down-regulation of proinflammatory cytokine release and nitric oxide production. J Immunol. 1996, 157: 3627-3634.
Leng SX, Elias JA: Interleukin-11 inhibits macrophage interleukin-12 production. J Immunol. 1997, 159: 2161-2168.
Hermann JA, Hall MA, Maini RN, Feldmann M, Brennan F: Important immunoregulatory role of interleukin-11 in the inflammatory process in rheumatoid arthritis. Arthritis Rheum. 1998, 41: 1388-97. 10.1002/1529-0131(199808)41:8<1388::AID-ART7>3.0.CO;2-F.
Trepecchio WL, Wang L, Bozza M, Dorner AJ: IL-11 regulates macrophange effector function through inhibition of nuclear factor-κB. J Immunol. 1997, 159: 5661-5670.
Walmsley M, Butler DM, Marinova-Mutafchieva L, Feldmann M: An anti-inflammatory role for interleukin-11 in established murine collagen-induced arthritis. Immunology. 1998, 95: 31-37. 10.1046/j.1365-2567.1998.00568.x.
Hochberg MC, Spector TD: Epidemiology of rheumatoid arthritis: update. Epidemiol Rev. 1990, 12: 247-252.
Ferraccioli G, Salaffi F, Nervetti A, Cavalieri F: Slow-acting drugs - outcome is no different than 15 years ago. J Rheumatol. 1990, 17: 1249-1250.
Moreland L, Heck L, Koopman W: Biologic agents for treating rheumatoid arthritis. Arthritis Rheum. 1997, 40: 397-409.
Thorton SC, Por SB, Penny R, Richter M, Shelley L, Breit SN: Identification of the major fibroblast growth factors released spontaneously in inflammatory arthritis as platelet derived growth factor and tumor necrosis factor-alpha. Clin Exp Immunol. 1991, 86: 79-86.
Beckham JC, Caldwell DS, Peterson BL, Pippen AM, Currie MS, Keefe FJ, Weinberg JB: Disease severity in rheumatoid arthritis: relationships of plasma tumor necrosis factor-alpha, soluble interleukin 2-receptor, soluble CD4/CD8 ratio, neopterin, and fibrin D-dimer to traditional severity and functional measures. J Clin Immunol. 1992, 12: 353-361.
Moreland LW, Schiff MH, Baumgartner SW, Tindall EA, Fleischmann RM, Bulpitt KJ, Weaver AL, Keystone EC, Furst DE, Mease PJ, Ruderman EM, Horwitz DA, Arkfeld DG, Garrison L, Burge DJ, Blosch CM, Lange ML, McDonnell ND, Weinblatt ME: Etanercept therapy in rheumatoid arthritis. A randomized, controlled trial. Ann Intern Med. 1999, 130: 478-486.
Lipsky PE, van der Heijde DMFM, St Clair EW, Furst DE, Breedveld FC, Kalden JR, Smolen JS, Weisman M, Emery P, Feldmann M, Harriman GR, Maini RN: Infliximab and methotrexate in the treatment of rheumatoid arthritis. N Engl J Med. 2000, 343: 1594-602. 10.1056/NEJM200011303432202.
Sands B, Bank S, Sninsky C, Robinson M, Katz S, Singleton J, Miner P, Safdi M, Galandiuk S, Hanauer S, Varilek G, Buchman A, Rogers V, Salzberg B, Cai B, Rogge H, Schwertschlag U: Safety and activity evaluation of rh-IL-11 in subjects with active Crohn's disease. Gastroenterology. 1999, 117: 59-64.
Sands B, Winston B, Salzberg B, Barish C, Safdi M, Wruble L, Varilek G, Singleton J, Katz S, Miner P, Shapiro M, Schwertschlag US: A randomized, double-masked, placebo-controlled study of rh-IL-11 in Crohn's disease subjects not receiving prednisone. American Gastrologists Association, Orlando,. 1999
Hochberg MC, Chang RW, Dwosh I, Lindsey S, Pincus T, Wolfe F: The American College of Rheumatology 1991 Revised Criteria for the Classification of Global Functional Status in Rheumatoid Arthritis. ArthritisRheum. 1992, 35: 498-502.
Felson DT, Anderson JJ, Boers M, Bombardier C, Furst D, Goldsmith C, Katz LM, Lightfoot R, Paulus H, Strand V: American College of Rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum. 1995, 38: 727-735.
Acknowledgements
The secretarial assistance of Bettie Stone in preparing the manuscript and the expertise of the clinical coordinators involved with this study are greatly appreciated.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moreland, L., Gugliotti, R., King, K. et al. Results of a phase-I/II randomized, masked, placebo-controlled trial of recombinant human interleukin-11 (rhIL-11) in the treatment of subjects with active rheumatoid arthritis. Arthritis Res Ther 3, 247 (2001). https://doi.org/10.1186/ar309
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar309