
Abstract
Anti-neutrophil cytoplasmic antibody (ANCA) - associated vasculitis (AAV) is a life-threatening autoimmune disease characterized by an antibody-mediated glomerulonephritis and necrotizing vasculitis. Apart from antibodies, T cells are also involved in disease pathogenesis. This review stresses the hallmarks of T cell-mediated pathology in AAV and highlights the characteristics of lesional and circulating T cells in the immune response in AAV. Circulating effector T-cell populations are expanded and are in a persistent state of activation. Circulating regulatory T-cell subsets are less well characterized but seem to be impaired in function. Lesional effector T cells are present in granulomas, vasculitic lesions, and nephritis. Lesional T cells usually show pro-inflammatory properties and promote granuloma formation. Apart from T cells, dendritic cells are abundantly present at the sites of inflammation and locally orchestrate the immune response. Targeting the above-mentioned T cell-mediated disease mechanisms will potentially provide powerful therapeutic tools for AAV.
Similar content being viewed by others


Introduction
Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) is a life-threatening form of autoimmune small-vessel vasculitis. AAV comprises three similar disease entities: Churg-Strauss syndrome (CSS), microscopic polyangiitis (MPA), and Wegener's granulomatosis (WG). These three entities have the presence of ANCA in common. CSS and MPA patients usually have ANCA directed to myeloperoxidase (MPO), whereas in WG, ANCA is in most cases directed to proteinase-3 (PR3) [1]. All three diseases are associated with systemic vasculitis and pauci-immune crescentic, necrotizing glomerulonephritis [2]. In addition, in many patients with WG or CSS, necrotizing granulomatous inflammation of the airways is observed. However, the histological findings vary slightly between the disease entities; tissue infiltration with eosinophils is observed mainly in CSS, whereas neutrophils are found in WG. Often, T cells are present within these granulomas [3]. In accordance with these findings, serum levels of markers of T-cell activity such as soluble interleukin-2-receptor (sIL-2r), neopterin, and soluble CD30 are elevated in AAV and associated with disease activity [4–6]. Increased numbers of T cells reactive to the sense or complementary Wegener auto-antigen PR3 are detectable in some but not all WG patients [7, 8]. In addition, ANCAs have an IgG isotype, suggesting that a T cell-mediated class switch has taken place. Importantly, Ruth and colleagues [9] demonstrated in an animal model of AAV that T-helper-cell depletion ameliorates the course of the disease. Specific T cell-targeted therapy is occasionally used in refractory cases with AAV and this has been demonstrated to have a beneficial effect on the course of the disease [10]. Altogether, these findings suggest that T cells are involved in the pathogenesis of AAV. The newest findings obtained during the last two decades will be discussed in this review. Hallmarks of T-cell pathology such as deficient regulation of T cells, memory T-cell expansion, persistent T-cell activation as well as T-cell polarization are dealt with; subsequently, T cell-driven granuloma and tertiary lymphoid organ (TLO) formation is discussed.
Regulatory versus effector memory T cells
Two major subsets of T cells are thought to dominate the adaptive T-cell immune response: regulatory T cells (Tregs) and CD4+ effector T cells (Teffs). Tregs limit and regulate the immune response, whereas Teffs are 'executors' controlled by Tregs. If this control is not regulated properly, excessive immune responses and loss of self-tolerance will follow (Figure 1). According to this concept, Treg and Teff subsets are the focus of research in AAV. Indeed, numerous reports have confirmed an expanded circulating CD4+CD25+ T-cell population in AAV [4, 11–13]. This T-cell subset usually contains activated Teffs as well as Tregs [14] (Table 1).

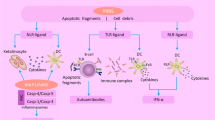
Pathways contributing to disease mechanisms in anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV). The 'classic neutrophil pathway' has been studied and confirmed by several groups. This pathway causes necrotizing vasculitis [87]. We propose an additional 'T-cell pathway' that mainly causes granulomatous inflammation and promotes necrotizing vasculitis. Infections are the starting point of both pathways; infections trigger priming of neutrophils, upregulation of adhesion molecules on endothelial cells, and expansion of circulating effector T cells (Teff s). Primed neutrophils show increased surface expression of ANCA antigens and adhesion molecules. ANCA binding activates the neutrophil in the following ways: 1) enhancing vessel wall adherence and transmigration capacity, 2) production and release of oxygen radicals, and 3) degranulation and release of enzymes, including myeloperoxidase (MPO) and proteinase-3 (PR3). Transient immune complexes are formed locally by binding of ANCA to PR3/MPO sticking to endothelial cells. Subsequently, complement is activated. This all adds to the development of necrotizing vasculitis. The expanded effector memory T cells (Tems) are not sufficiently regulated by regulatory T cells (Tregs), leading to dysbalance in the homeostasis of Tregs and Tems and resulting in further release of pro-inflammatory cytokines promoting neutrophil priming; moreover, ANCA production is enhanced by further T cell/B cell interaction. Expanded circulating Tems migrate into target organs such as the lungs or the kidney. Within tissues, Tems drive formation of granuloma, which is considered an 'executioner' of tissue destruction. Granulomas are composed of numerous cell types such as T cells, B cells, giant cells, and dendritic cells. Moreover, ANCA production occurs in granulomas. Possibly, tertiary lymphoid organs (TLOs) are 'local controllers' of tissue inflammation since induction of Tregs is thought to take place in TLOs. ICX, immune complex.
Abdulahad and colleagues [13, 15] differentiated between CD25low (activated Teff) and CD25high (Treg) CD4+ T cells and found an increase of both populations in patients with WG (Table 1). Moreover, an increase of both FoxP3+ Tregs and FoxP3- effector memory T cells (Tems) in patients with remission was described [13], whereas in a previous study by Marinaki and colleagues [11], no increase of circulating FoxP3+ Tregs was observed. Abdulahad and colleagues [15] demonstrated a functional impairment of CD25high Tregs in WG. These Tregs failed to inhibit proliferation or cytokine production of responder T cells. However, selection criteria of Tregs and their influence on the test system assessing Treg functionality have been recently studied [16]. When Tregs were sorted by CD25 and CD127, Tregs showing CD25high expression first appeared to be impaired in function; excluding CD127+ CD25high T cells restored functionality [16]. Thus, the defect of CD25high Tregs observed in WG might be biased by Treg selection criteria. In addition, there is an ongoing dispute on the specificity of the anti-FoxP3 clone PCH101 commonly used for determining Treg frequency as well as purity [15, 17, 18]. Therefore, additional studies are necessary to confirm whether there is a defect of Treg function in WG. Furthermore, functional and descriptive Treg studies using new markers such as CD127, CD39, and CD73 have not yet been published and are necessary [19, 20].
Factors influencing Treg homeostasis and function might be involved in the pathogenesis of AAV. For instance, leptin has recently emerged as a controller of immunity and especially as a controller of Tregs. Leptin itself is considered to have pro-inflammatory effects, whereas neutralization of leptin leads to Treg expansion in vitro [21]. Surprisingly, Kümpers and colleagues [22] recently found decreased levels of leptin in patients with WG and MPA during active disease. Leptin levels normalized again when entering remission. Low levels of leptin observed during active disease might lead to Treg expansion, restoration of tolerance, and attenuation of the inflammatory response [23].
Apart from an obvious CD25+ T-cell expansion both in patients with active disease and in those in remission [4], memory T cells are expanded in AAV [11] (Table 1). Marinaki and colleagues [24] described low numbers of naïve T cells in AAV as compared with healthy controls (HCs). In line with this study, Abdulahad and colleagues [15] reported an increase of circulating Tems in WG patients in remission. In active WG, the numbers of circulating Tems dropped and could be detected in urine, suggesting that Tems migrate from the circulation to inflammatory sites during active states of the disease [25].
Recently, we confirmed the findings by Abdulahad and colleagues [12, 13] and found an expansion of specific circulating CD25+ Tem subpopulations in WG. These CD25+ Tems were CD134+ and GITR+ and did not show a regulatory phenotype (Table 1). CD134+ T cells were also found in inflammatory lesions. Interestingly, the expansion of specific CD134+ and GITR+ Tem subsets was closely associated with disease activity [12]. However, these findings are in contrast to the study of Abdulahad and colleagues [13] on the overall Tem population that decreases during active disease. We postulate that specific Tem subsets increase during active disease despite a total decrease of Tems. Indeed, Tems are a heterogenous T-cell population and might show varying proliferation, survival, and migratory capacities [26].
Of special interest is the Treg/Tem balance in vivo. In the field of transplant immunology, ratios between Tregs and Tems are regarded as important for immune tolerance. Recently, Kreijveld and colleagues [27] assessed the value of these ratios before and after kidney transplantation and found that the ratio was predictive of rejection. It was demonstrated that a rejection was preceded by a shift from Tregs to Tems. One could speculate that these ratios are also skewed toward Tems in WG, especially in case of a relapse. However, this has been scarcely investigated so far. In one study, Treg/Tem ratios were assessed in WG and no differences were found when compared with HCs [13]. Furthermore, an association with disease activity was not found. Further studies are needed to see whether monitoring the Treg/Tem ratio could be an option for predicting relapses. In AAV, the balance of Tregs and Tems might be altered, leading to less strict control of the immune response (Figure 1). This might promote loss of self-tolerance and autoimmunity (Figure 1).
Aberrant T-cell activation and senescence in AAV
Aberrant T-cell costimulation and T-cell senescence might have a role in the expansion of Tems in AAV and this might facilitate the breakdown of self-tolerance. Indeed, T cells in AAV show an altered expression pattern of costimulatory molecules. This is well studied with regard to the CD28/CD80 pathway [28–30]. Normally, CD28 is present on T cells whereas CD80/86 is found on antigen-presenting cells (APCs). Antigen presentation of APCs to T cells requires additional signaling via the CD28/CD80 pathway, resulting in T-cell activation.
Strikingly, CD28 is downregulated on circulating and lesional CD4+ T-helper cells in AAV [28, 29]. These CD28null T cells are described as a major source of pro-inflammatory cytokines such as interferon-gamma (IFNγ) and tumor necrosis factor-alpha (TNFα) [31]. Moreover, these T cells have killer cell capacities and express perforin as well as NKG2D [30, 32]. These latter features are usually confined to cytolytic natural killer-like T cells (Table 1). In AAV, CD28null T cells have shortened telomeres and they express CD57, which is indicative of replicative senescence [31, 33]. CD28null T cells are supposed to have a role in several other autoimmune diseases and are less responsive to immunoregulation [34]. Although there is a positive correlation with disease severity and the presence of CD28null T-helper cells in AAV patients, a direct pathogenic role has not been proven yet but seems to be likely. Therefore, the presence of CD28null T-helper cells might also be considered to be secondary to latent viral herpes infections such as cytomegalovirus (CMV) since these T cells have antigen specificity for CMV but not to autoantigens [34, 35]. The definitive role of CD28null T-helper cells in AAV needs to be further investigated.
The costimulatory molecules CD80 and CD86 are upregulated on T cells in WG after in vitro stimulation with mitogens and polyclonal stimuli [29]. The functional properties of CD80+ T-helper cells have not been assessed in AAV so far. However, it is known that CD80 expression on T-helper cells in general is accompanied by major histocompatibility complex class II expression [36, 37]. Therefore, CD80+ T cells might be able to present antigen to other T cells. This presentation might lead to activation or - as recently described in a mouse model - inhibition of T-cell responses [37]. Thus, it is not known whether the aberrant CD80 expression observed in AAV favors persistent stimulation of T cells or limits the immune response.
An increased expression of CTLA-4 on CD4+ T cells has been reported in WG [30]. CTLA-4 is a negative costimulator that binds to CD80/CD86 on APCs and thereby inhibits CD28-dependent T-cell activation. CTLA-4 might counterbalance T-cell activation in AAV. Overexpression of CTLA-4 is accompanied by upregulation of anti-apoptotic BCL-2 [38]. Thus, these T cells seem to be protected against activation-induced cell death [38]. Other negative costimulators such as PD-1 have not been assessed in AAV so far.
Genetic polymorphisms of CTLA-4 have also been studied in AAV [39]. Some polymorphisms of the negative costimulator CTLA-4 have a higher prevalence in AAV than in HCs [40–42]. Dinucleotide repeats within the gene encoding CTLA-4 were linked to AAV by several authors [41, 42]. Elongated repeats lead to instability of mRNA and also contribute to hyper-reactivity of T cells [43]. Other associated polymorphisms in the CTLA-4 gene are known to reduce the availability and surface expression of this negative costimulator. Thus, associated polymorphisms might limit the efficacy of CTLA-4 upregulation. Moreover, we studied polymorphisms of another negative costimulator, the PDCD (PD-1) gene [42]. Genetic variants of PD-1 were not directly linked to WG or AAV, but specific combinations of CTLA-4 and PD-1 polymorphisms were less prevalent in AAV [42].
Besides changes in the expression of costimulatory molecules, other markers of T-cell activation are either upregulated or downregulated in AAV patients. For instance, HLA-DR expression of CD4+ T-helper-cells is elevated in WG but is reported to mirror disease activity poorly [4]. In addition, levels of soluble T-cell activation markers such as sIL-2r, sCD4, sCD8, sCD30, and neopterin are increased in AAV [6, 44–46]. Some studies showed an association of these markers and disease activity, while others failed to demonstrate a correlation with disease activity [4–6, 44, 46, 47]. Taken together, T cells in AAV are in a state of persistent activation, not only during active disease but also during remission. This might be caused or followed by an aberrant expression of costimulatory molecules. This aberrant expression pattern might favor Tem expansion and thus contribute to the dysbalance of Tregs and Tems (Figure 1).
The myriad of T-helper-cell polarization in AAV
In AAV, there are differences in Teff type 1/type 2 polarization, depending on disease type and disease stage [48, 49]. As suggested by features of CSS such as asthma and eosinophilia, CSS seems to be associated with Th2 polarization. Kiene and colleagues [50] analyzed T-cell lines of CSS and WG patients. Higher levels of IL-4 and IL-13 were detected in CSS than in WG, whereas elevated levels of IFNγ were produced by T cells in both CSS and WG. Thus, a stronger Th2 polarization was seen in CSS [50] (Table 1).
T-cell polarization in MPA is not well studied and data are conflicting. Some hints point at Th1 polarization where as others indicate Th2 polarization in MPA. It has been shown that, during active disease, soluble CX3CL1 levels are elevated and that CX3CR1 expression on circulating T cells correlates with disease activity. CX3CL1 is a chemokine belonging to the fractalkine family and is suggested to be associated with a preferential Th1 response [51]. In addition, levels of the Th2- associated marker sCD30 were not elevated in MPA during active disease [52]. However, the Th1 marker sCD26 was found to be decreased in active MPA and increased during remission [52]. There are only limited data on lesional T cells in MPA; interestingly, these T cells were not capable of producing Th1 cytokines like IFNγ but instead secreted Th2 cytokines [53].
In WG, it is claimed that localized disease is associated with a preferential Th1 response whereas Th2 responses are common in systemic disease. Plasma levels of sCD26 as a marker for Th1 response are elevated in patients with localized disease [52]. Lamprecht and colleagues [54, 55] compared CCR5 (Th1) and CCR3 (Th2) expression on circulating T cells from patients with WG. In localized disease, there was a higher expression of CCR5 indicative for Th1 response whereas the two markers were expressed at equal levels in systemic disease (Table 1). Furthermore, some studies have characterized lesional T cells derived from granulomas of nasal biopsies. Interestingly, data from two studies suggest that the polarization of tissue-resident T cells depends on disease extent. In localized WG, T cells were mainly positive for CD26 and IFNγ, suggesting a preferential Th1 response, whereas in systemic WG, lesional T cells show a Th2-like response with little IFNγ and high IL-4 expression [48, 56]. In contrast to these studies, however, Csernok and colleagues [49] detected high levels of IFNγ mRNA and low levels of IL-4 mRNA produced by lesional T cells derived from nasal biopsies of patients with systemic disease. In line with this study, Komocsi and colleagues [31] characterized lesional T cells in granulomas by immunohistochemistry and found them to be main producers of IFNγ and TNFα. It remains to be confirmed whether there is a shift in T-cell polarization along with progression from localized to systemic disease.
Another T-helper-cell subset that was recently shown to be of major importance in autoimmunity is the IL-17- producing T-cell subset (Th17) [57]. There are some hints indicating that a skewed Th17 response might also contribute to the disease pathogenesis of AAV [8]. It was recently reported that WG patients in remission bear an increased number of circulating Th17 cells reactive to the autoantigen PR3 [8]. In addition, CSS patients with active disease have an increased number of circulating Th17 cells [58]. Moreover, AAV patients harbor an expanded CD45RC T-helper-cell population that is a source of IL-17 [59] (Table 1). IL-17 also facilitates the migration and activation of neutrophils by promoting the secretion of TNFα and IL-1β [60]. Since the influx of neutrophils is a hallmark of AAV, IL-17 might enable the migration of neutrophils during active vasculitis. The data on T-helper-cell polarization are inconclusive at some points and the evidence on Th17 subsets is limited, so the importance of T-cell polarization needs to be further studied in AAV patients.
Local inflammation in AAV: T cells, granuloma formation, and tissue damage
Circulating T cells migrate into tissue during the active phase of disease. These T cells, present in lesions within organs affected by AAV, have multiple important functions in driving the disease (Figure 1). In general, the following lesions are commonly found: granuloma formation with central necrosis, vasculitis of small- and medium-sized vessels, infiltration with mononuclear cells, and necrotizing crescentic glomerulonephritis without deposits of immune complexes.
Granulomas, which can be detected in WG and CSS but not in MPA, have been demonstrated to be composed of T cells, B cells, giant cells, and dendritic cells (DCs) (Figure 1). Granulomas are observed in the upper airways, the lungs, and occasionally in other organs such as the kidneys [61]. The cellular subsets participating in granulomatous inflammation are best characterized by immunohistochemistry of nasal or lung biopsies [28, 32]. In these granulomatous lesions, a dense T-cell infiltrate is usually found. CD4+ T cells slightly outnumber CD8+ T cells [56]. The T cells express CD45RO and show a CD28- phenotype. Further analysis reveals CD28null T cells as major producers of IFNγ and TNFα [31]. These phenotypic features imply that most of the T cells within the inflamed tissue belong to the senescent memory T-cell population [62]. As mentioned above, the numbers of circulating Tems are found to be decreased in patients with active disease. Given the data from biopsy studies, it is possible that memory T cells migrate from the circulation to the tissue to act as effector cells [13].
Presumably, these migrating Teffs act in different ways. It is well accepted that granuloma formation is driven by Teffs [63]. For instance, granuloma formation is severely impaired after CD4+ T-cell depletion in mice models of tuberculosis. Similarly, low CD4+ T-cell counts in HIV result in an increased susceptibility to tuberculosis and poorly organized granulomas. In addition, the importance of T cell-derived cytokines such as TNFα and INFγ to granuloma formation has been demonstrated [63]. Interestingly, Csernok and colleagues [53] reported that lesional T cells in MPA lack production of IFNγ, which is thought to be essential for granuloma formation. This finding might provide an explanation of why granulomatous inflammation is absent in MPA. Thus, the T-cell infiltrate in AAV seems to be indispensable for initiation and sustaining of granulomatous inflammation, contributing in a direct way to the tissue damage observed in AAV.
Cytotoxic CD8+ T cells are also present in inflamed tissue in AAV. These cells are capable of inducing apoptosis and may be cytotoxic for endothelial cells. Furthermore, it has been shown that CD4+CD28null T cells have cytolytic properties [34]. As these T cells are abundantly present in inflammatory lesions in AAV, a destructive role for these cells has to be considered as well [28]. In line with these findings, a recent report demonstrated that CD4+ Tems can damage endothelial cells and it was found that this process was independent of the CD28 costimulatory pathway, supporting a role for CD4+CD28null Tems in mediating tissue damage [64]. Next to CD4+CD28null T cells, CD4+CD134+ Teffs also are present in vasculitic and renal lesions [12]. It has been reported that the ligand for CD134 is expressed on endothelial cells [65]. Ligation of CD134 promotes T cells to adhere to endothelial cells and this interaction subsequently stimulates T cells to produce pro-inflammatory cytokines [66]. Indeed, pro-inflammatory cytokines are expressed in kidneys and other tissues affected by AAV [31, 67]. Although an exact mechanism of tissue damage has not yet been demonstrated for CD4+CD134+ T cells, it can be hypothesized that pro-inflammatory cytokines such as IFNγ cause direct damage to endothelial cells [68]. However, pro-inflammatory cytokines may also mediate damage by promoting neutrophil-endothelial adhesion and neutrophil activation, finally leading to tissue damage [69]. Thus, lesional T cells bear the potential to destroy tissue integrity by driving granuloma formation, by cytokine production, or by direct effects on target cells (Figure 1).
Local inflammation in AAV: tertiary lymphoid organ formation
As discussed above, T cells act as effectors in inflamed organs. The questions arises of whether there is any 'tissue-resident' surveillance on this inflammatory process. At present, it is unknown whether local activation or control of the immune response within the affected tissue itself occurs in AAV. Local control of immune responses is linked to the development of TLOs and this is also known as lymphoid neogenesis [70]. This has been described for several chronic inflammatory conditions (for example, rejection in the context of organ transplantation and inflammation in several autoimmune diseases) [70]. TLOs resemble the structure of secondary lymphoid organs and consist of B-cell follicles with a surrounding mantle zone with T cells and DCs. Within these TLOs, T-cell activation by APCs such as DCs and B-cell stimulation take place. It is likely that local, tissue-specific (auto) antigens are presented there. Whereas secondary lymphoid organs have organized lymph flow and APC trafficking, TLOs lack these features, resulting in an unrestricted access of antigens, APCs, and lymphocytes. These conditions might promote persistent and non-physiological T-cell activation in autoimmunity [70]. But is there any evidence for the presence of similar structures in AAV?
At present, granulomas are regarded as some form of TLO in which immune responses are modified [61, 71]. For instance, Csernok and colleagues [72] revealed that PR3 is abundantly present in granulomas and renders DCs to powerful Th1-cell activators. Moreover, Voswinkel and colleagues [73] demonstrated that affinity maturation of B cells, as is commonly observed in lymphoid tissue, takes place in granulomas. It is suggested that the production of ANCA takes place locally within these granulomas. Granuloma formation in a classical manner is rarely present in the kidney, but some form of lymphoid neogenesis has been frequently observed in renal biopsies of patients with AAV [74–76]. Immature DCs and T cells form aggregates suggesting a cell-cell interaction. Strikingly, these DCs display costimulatory capability by expressing CD80 [75]. We hypothesize that, in the kidney, activation of Teffs and aggravation of the immune response take place. However, a local induction of Tregs and thus an attenuation of the inflammatory process seem to be possible, too [75, 77]. Our own data indicate that FoxP3+ T cells are present in inflammatory lesions (unpublished data). The induction of Tregs is especially confined to places where abundant immature DCs bearing costimulatory properties are present [78]. Therefore, both local control of tissue inflammation and activation of Teffs at the site of inflammation are likely to occur in AAV (Figure 1).
Conclusion and implications for therapy
Circulating T cells in AAV show several abnormalities. T cells are disturbed in homeostasis and skewed toward memory and pro-inflammatory T-cell types. This knowledge might allow specific and targeted therapies in the future. At present, nearly all available treatment options for AAV are toxic and have severe side effects. Alternative treatment strategies such as cell-based therapy with ex vivo expansion of Tregs and subsequent infusion of these cells could potentially restore tolerance and T-cell homeostasis in AAV patients [79]. However, it has recently been reported that human Tregs can convert to pro-inflammatory IL-17- producing T cells in the presence of a specific cytokine environment (IL-1β, IL-21, IL-23) [80]. Thus, administration of Tregs to patients might not restore tolerance but rather enhance autoimmunity under certain not-yet-known conditions. In light of these recent findings on Treg plasticity, cell-based approaches have to be considered with caution. In addition, control of T-cell activation might pose an attractive therapeutic option. In this regard, blockade of costimulatory pathways such as CD28/CD80 has successfully been used in rheumatoid arthritis and might be a therapeutic opportunity in AAV [81]. However, since an important T-cell population (CD28null T cells) involved in AAV pathogenesis is acting independently of the CD28/CD80 pathway, this novel strategy might not be applicable to all patient cohorts. As IL-2 is also involved in T-cell activation and is regarded as a 'T-cell growth factor', IL-2Rα (CD25)-blocking agents could be of use. These agents are already used in the field of organ transplantation [82]. There is some evidence that anti-CD25 antibody therapy is effective in autoimmune diseases such as multiple sclerosis and vasculitis [83, 84]. Nevertheless, there are no reliable data available in AAV yet. Based upon the finding that CD25+ T cells are expanded in AAV and show an association with disease activity, there is at least a rationale to assume that anti-CD25 agents might have a beneficial effect. However, IL-2 has a role not only in maintaining but also in limiting immune responses. IL-2 shows homeostatic effects on Tregs and promotes activation-induced cell death; thus, adverse events facilitating autoimmunity cannot be excluded with certainty [85]. Next to the blockade of T-cell activation, it might be beneficial to interfere with T-cell trafficking into tissues and thereby dampening inflammation as well as granuloma formation. Natalizumab targets α4-integrin, which has a role in the adhesion of leukocytes to vascular endothelial cells, and thus prevents leukocyte migration [86]. It has been administered in patients suffering from inflammatory bowel disease and in cohorts with multiple sclerosis. In both cases, it was reported to be effective. However, severe side effects such as multifocal leukoencephalopathy have been reported in some cases [84]. Although the safety profile of this drug needs further clarification, the basic mechanism of blocking leukocyte adhesion might be a charming approach in AAV patients to dampen inflammation and tissue destruction. Unraveling T cell-mediated pathology in AAV will provide further insights into disease mechanisms and will potentially result in promising new therapeutic options. It is hoped that further efforts in this field will allow a sustained restoration of self-tolerance in AAV patients.
Abbreviations
- AAV:
-
anti-neutrophil cytoplasmic antibody
- ANCA:
-
associated vasculitis
- ANCA:
-
anti-neutrophil cytoplasmic antibody
- APC:
-
antigen-presenting cell
- CMV:
-
cytomegalovirus
- CSS:
-
Churg-Strauss syndrome
- DC:
-
dendritic cell
- HC:
-
healthy control
- IFNγ:
-
interferon-gamma
- IL:
-
interleukin
- MPA:
-
microscopic polyangiitis
- PR3:
-
proteinase-3
- sIL-2r:
-
soluble interleukin-2-receptor
- Teff:
-
effector T cell
- Tem:
-
effector memory T cell
- TLO:
-
tertiary lymphoid organ
- TNFα:
-
tumor necrosis factor-alpha
- Treg:
-
regulatory T cell
- WG:
-
Wegener granulomatosis.
References
Cohen Tervaert JW, Limburg PC, Elema JD, Huitema MG, The TH, Kallenberg CGM, Horst G: Detection of autoantibodies against myeloid lysosomalenzymes - a useful adjunct to classification of patients with biopsy-proven necrotizing arteritis. Am J Med. 1991, 91: 59-66.
Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, Hagen EC, Hoffman GS, Hunder GG, Kallenberg CGM, McCluskey RT, Sinico RA, Rees AJ, Van Es LA, Waldherr R, Wiik A: Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994, 37: 187-192.
Tipping PG, Holdsworth SR: T cells in crescentic glomerulonephritis. J Am Soc Nephrol. 2006, 17: 1253-1263.
Popa ER, Stegeman CA, Bos NA, Kallenberg CGM, Tervaert JWC: Differential B- and T-cell activation in Wegener's granulomatosis. J Allergy Clin Immunol. 1999, 103: 885-894.
Stegeman CA, Tervaert JWC, Huitema MG, Kallenberg CGM: Serum markers of T-cell activation in relapses of Wegeners granulomatosis. Clin Exp Immunol. 1993, 91: 415-420.
Schmitt WH, Heesen C, Csernok E, Rautmann A, Gross WL: Elevated serum levels of soluble interleukin-2 receptor in patients with Wegener's granulomatosis. Association with disease activity. Arthritis Rheum. 1992, 35: 1088-1096.
Yang JJ, Bautz DJ, Lionaki S, Hogan SL, Chin H, Tisch RM, Schmitz JL, Pressler BM, Jennette JC, Falk RJ, Preston GA: ANCA patients have T cells responsive to complementary PR-3 antigen. Kidney Int. 2008, 74: 1159-1169.
Abdulahad WH, Stegeman CA, Limburg PC, Kallenberg CG: Skewed distribution of Th17 lymphocytes in patients with Wegener's granulomatosis in remission. Arthritis Rheum. 2008, 58: 2196-2205.
Ruth A-J, Kitching AR, Kwan RYQ, Odobasic D, Ooi JDK, Timoshanko JR, Hickey MJ, Holdsworth SR: Anti-neutrophil cytoplasmic antibodies and effector CD4+ cells play nonredundant roles in anti-myeloperoxidase crescentic glomerulonephritis. J Am Soc Nephrol. 2006, 17: 1940-1949.
Schmitt WH, Hagen EC, Neumann I, Nowack R, Flores-Suarez LF, Woude van der FJ: Treatment of refractory Wegener's granulomatosis with antithymocyte globulin (ATG): an open study in 15 patients. Kidney Int. 2004, 65: 1440-1448.
Marinaki S, Neumann I, Kalsch AI, Grimminger P, Breedijk A, Birck R, Schmitt W, Waldherr R, Yard BA, Woude Van Der FJ: Abnormalities of CD4 T cell subpopulations in ANCA-associated vasculitis. Clin Exp Immunol. 2005, 140: 181-191.
Wilde B, Dolff S, Cai X, Specker C, Becker J, Tötsch M, Costabel U, Dürig J, Kribben A, Tervaert JW, Schmid KW, Witzke O: CD4+CD25+ T-cell populations expressing CD134 and GITR are associated with disease activity in patients with Wegener's granulomatosis. Nephrol Dial Transplant. 2009, 24: 161-171.
Abdulahad WH, Geld van der YM, Stegeman CA, Kallenberg CG: Persistent expansion of CD4+ effector memory T cells in Wegener's granulomatosis. Kidney Int. 2006, 70: 938-947.
Sakaguchi S, Setoguchi R, Yagi H, Nomura T: Naturally arising Foxp3- expressing CD25+CD4+ regulatory T cells in self-tolerance and autoimmune disease. Curr Top Microbiol Immunol. 2006, 305: 51-66.
Abdulahad WH, Stegeman CA, Geld van der YM, Doornbos-van der Meer B, Limburg PC, Kallenberg CG: Functional defect of circulating regulatory CD4+ T cells in patients with Wegener's granulomatosis in remission. Arthritis Rheum. 2007, 56: 2080-2091.
Michel L, Berthelot L, Pettre S, Wiertlewski S, Lefrere F, Braudeau C, Brouard S, Soulillou JP, Laplaud DA: Patients with relapsing-remitting multiple sclerosis have normal Treg function when cells expressing IL-7 receptor alpha-chain are excluded from the analysis. J Clin Invest. 2008, 118: 3411-3419.
Tran DQ, Ramsey H, Shevach EM: Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-{beta} dependent but does not confer a regulatory phenotype. Blood. 2007, 110: 2983-2990.
Tran DQ, Shevach EM: Response: anti human FOXP3 mAb PCH101 stains activated human naive T cells nonspecifically. Blood. 2008, 111: 464-466.
Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, Höpner S, Centonze D, Bernardi G, Dell'Acqua ML, Rossini PM, Battistini L, Rötzschke O, Falk K: Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007, 110: 1225-1232.
Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, Gottlieb PA, Kapranov P, Gingeras TR, Fazekas de St Groth B, Clayberger C, Soper DM, Ziegler SF, Bluestone JA: CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med. 2006, 203: 1701-1711.
De Rosa V, Procaccini C, Calì G, Pirozzi G, Fontana S, Zappacosta S, La Cava A, Matarese G: A key role of leptin in the control of regulatory T cell proliferation. Immunity. 2007, 26: 241-255.
Kümpers P, Horn R, Brabant G, Woywodt A, Schiffer M, Haller H, Haubitz M: Serum leptin and ghrelin correlate with disease activity in ANCA-associated vasculitis. Rheumatology. 2008, 47: 484-487.
Thewissen M, Cohen Tervaert JW: Leptin and ANCA-associated vasculitis: a healthy link?. Rheumatology. 2008, 47: 387-389.
Marinaki S, Kalsch AI, Grimminger P, Breedijk A, Birck R, Schmitt WH, Weiss C, Woude van der FJ, Yard BA: Persistent T-cell activation and clinical correlations in patients with ANCA-associated systemic vasculitis. Nephrol Dial Transplant. 2006, 21: 1825-1832.
Abdulahad WH, Kallenberg C, Limburg P, Stegeman C: Urinary CD4 effector memory T-cells as a measure of renal disease activity in ANCA-associated vasculitis. Arthritis Rheum. 2008, 58: S851-S851.
Stockinger B, Bourgeois C, Kassiotis G: CD4(+) memory T cells: functional differentiation and homeostasis. Immunol Rev. 2006, 211: 39-48.
Kreijveld E, Koenen HJPM, van Cranenbroek B, van Rijssen E, Joosten I, Hilbrands LB: Immunological monitoring of renal transplant recipients to predict acute allograft rejection following the discontinuation of tacrolimus. PLoS ONE. 2008, 3: e2711-
Lamprecht P, Moosig F, Csernok E, Seitzer U, Schnabel A, Mueller A, Gross WL: CD28 negative T cells are enriched in granulomatous lesions of the respiratory tract in Wegener's granulomatosis. Thorax. 2001, 56: 751-757.
Moosig F, Csernok E, Wang G, Gross WL: Costimulatory molecules in Wegener's granulomatosis (WG): lack of expression of CD28 and preferential up-regulation of its ligands B7-1 (CD80) and B7-2 (CD86) on T cells. Clin Exp Immunol. 1998, 114: 113-118.
Steiner K, Moosig F, Csernok E, Selleng K, Gross WL, Fleischer B, Broker BM: Increased expression of CTLA-4 (CD152) by T and B lymphocytes in Wegener's granulomatosis. Clin Exp Immunol. 2001, 126: 143-150.
Komocsi A, Lamprecht P, Csernok E, Mueller A, Holl-Ulrich K, Seitzer U, Moosig F, Schnabel A, Gross WL: Peripheral blood and granuloma CD4(+)CD28(-) T cells are a major source of interferon-gamma and tumor necrosis factor-alpha in Wegener's granulomatosis. Am J Pathol. 2002, 160: 1717-1724.
Capraru D, Müller A, Csernok E, Gross WL, Holl-Ulrich K, Northfield J, Klenerman P, Herlyn K, Holle J, Gottschlich S, Voswinkel J, Spies T, Fagin U, Jabs WJ, Lamprecht P: Expansion of circulating NKG2D+ effector memory T-cells and expression of NKG2D-ligand MIC in granulomaous lesions in Wegener's granulomatosis. Clin Immunol. 2008, 127: 144-150.
Vogt S, Iking-Konert C, Hug F, Andrassy K, Hansch GM: Shortening of telomeres: Evidence for replicative senescence of T cells derived from patients with Wegener's granulomatosis. Kidney Int. 2003, 63: 2144-2151.
Thewissen M, Somers V, Hellings N, Fraussen J, Damoiseaux J, Stinissen P: CD4+CD28null T cells in autoimmune disease: pathogenic features and decreased susceptibility to immunoregulation. J Immunol. 2007, 179: 6514-6523.
Berg van de PJ, van Leeuwen EM, ten Berge IJ, van Lier R: Cytotoxic human CD4+ T cells. Curr Opin Immunol. 2008, 20: 339-343.
Vallejo AN: CD28 extinction in human T cells: altered functions and the program of T-cell senescence. Immunol Rev. 2005, 205: 158-169.
Holling TM, Schooten E, Elsen van Den PJ: Function and regulation of MHC class II molecules in T-lymphocytes: of mice and men. Hum Immunol. 2004, 65: 282-290.
Pandiyan P, Gartner D, Soezeri O, Radbruch A, Schulze-Osthoff K, Brunner-Weinzierl MC: CD152 (CTLA-4) determines the unequal resistance of Th1 and Th2 cells against activation-induced cell death by a mechanism requiring PI3 kinase function. J Exp Med. 2004, 199: 831-842.
Spriewald BM, Witzke O, Wassmuth R, Wenzel RR, Arnold ML, Philipp T, Kalden JR: Distinct tumour necrosis factor alpha, interferon gamma, interleukin 10, and cytotoxic T cell antigen 4 gene polymorphisms in disease occurrence and end stage renal disease in Wegener's granulomatosis. Ann Rheum Dis. 2005, 64: 457-461.
Zhou YH, Huang DR, Paris PL, Sauter CS, Prock KA, Hoffman GS: An analysis of CTLA-4 and proinflammatory cytokine genes in Wegener's granulomatosis. Arthritis Rheum. 2004, 50: 2645-2650.
Huang D, Giscombe R, Zhou YH, Lefvert K: Polymorphisms in CTLA-4 but not tumor necrosis factor-alpha or interleukin 1 beta genes are associated with Wegener's granulomatosis. J Rheumatol. 2000, 27: 397-401.
Slot MC, Sokolowska MG, Savelkouls KG, Janssen R, Damoiseaux J, Tervaert JWC: Immunoregulatory gene polymorphisms are associated with ANCA-related vasculitis. Clin Immunol. 2008, 128: 39-45.
Huang DR, Giscombe R, Zhou YH, Pirskanen R, Lefvert AK: Dinucleotide repeat expansion in the CTLA-4 gene leads to T cell hyper-reactivity via the CD28 pathway in myasthenia gravis. J Neuroimmunol. 2000, 105: 69-77.
Schmitt WH, Csernok E, Kobayashi S, Klinkenborg A, Reinhold-Keller E, Gross WL: Churg-Strauss syndrome - Serum markers of lymphocyte activation and endothelial damage. Arthritis Rheum. 1998, 41: 445-452.
Nassonov EL, Samsonov MY, Tilz GP, Beketova TV, Semenkova EN, Baranov A, Wachter H, Fuchs D: Serum concentrations of neopterin, soluble interleukin 2 receptor, and soluble tumor necrosis factor receptor in Wegener's granulomatosis. J Rheumatol. 1997, 24: 666-670.
Kobold ACM, Kallenberg CGM, Tervaert JWC: Monocyte activation in patients with Wegener's granulomatosis. Ann Rheum Dis. 1999, 58: 237-245.
Sanders JSF, Huitma MG, Kallenberg CGM, Stegeman CA: Plasma levels of soluble interleukin 2 receptor, soluble CD30, interleukin 10 and B cell activator of the tumour necrosis factor family during follow-up in vasculitis associated with proteinase 3-antineutrophil cytoplasmic antibodies: associations with disease activity and relapse. Ann Rheum Dis. 2006, 65: 1484-1489.
Balding CEJ, Howie AJ, Drake-Lee AB, Savage COS: Th2 dominance in nasal mucosa in patients with Wegener's granulomatosis. Clin Exp Immunol. 2001, 125: 332-339.
Csernok E, Trabandt A, Muller A, Wang GCC, Moosig F, Paulsen J, Schnabel A, Gross WL: Cytokine profiles in Wegener's granulomatosis - Predominance of type 1 (Th1) in the granulomatous inflammation. Arthritis Rheum. 1999, 42: 742-750.
Kiene A, Csernok E, Mulller A, Metzler C, Trabandt A, Gross WL: Elevated interleukin-4 and interleukin-13 production by T cell lines from patients with Churg-Strauss syndrome. Arthritis Rheum. 2001, 44: 469-473.
Fraticelli P, Sironi M, Bianchi G, D'Ambrosio D, Albanesi C, Stoppacciaro A, Chieppa M, Allavena P, Ruco L, Girolomoni G, Sinigaglia F, Vecchi A, Mantovani A: Fractalkine (CX3CL1) as an amplification circuit of polarized Th1 responses. J Clin Invest. 2001, 107: 1173-1181.
Schonermarck U, Csernok E, Trabandt A, Hansen H, Gross WL: Circulating cytokines and soluble CD23, CD26 and CD30 in ANCA-associated vasculitides. Clin Exp Rheumatol. 2000, 18: 457-463.
Csernok E, Muller A, Gross WL: Immunopathology of ANCA-associated vasculitis. Intern Med. 1999, 38: 759-765.
Lamprecht P, Bruhl H, Erdmann A, Holl-Ulrich K, Csernok E, Seitzer U, Mack M, Feller AC, Reinhold-Keller E, Gross WL, Muller A: Differences in CCR5 expression on peripheral blood CD4+CD28- T-cells and in granulomatous lesions between localized and generalized Wegener's granulomatosis. Clin Immunol. 2003, 108: 1-7.
Lamprecht P, Erdmann A, Mueller A, Csernok E, Reinhold-Keller E, Holl-Ulrich K, Feller AC, Bruehl H, Gross WL: Heterogeneity of CD4 and CD8+ memory T cells in localized and generalized Wegener's granulomatosis. Arthritis Res Ther. 2003, 5: R25-31.
Muller A, Trabandt A, Gloeckner-Hofmann K, Seitzer U, Csernok E, Schonermarck U, Feller AC, Gross WL: Localized Wegener's granulomatosis: predominance of CD26 and IFN-gamma expression. J Pathol. 2000, 192: 113-120.
Bettelli E, Oukka M, Kuchroo VK: TH-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007, 8: 345-350.
Saito H, Tsurikisawa N, Tsuburai T, Oshikata C, Akiyama K: Cytokine production profile of CD4+ T cells from patients with active Churg-Strauss syndrome tends toward Th17. Int Arch Allergy Immunol. 2009, 149: 61-65.
Ordonez L, Bernard I, L'Faqihi-Olive F, Cohen Tervaert JW, Damoiseaux J, Saoudi A: CD45RC isoform expression identifies functionally distinct T cell subsets differentially distributed between healthy individuals and AAV patients. PLoS ONE. 2009, 4: e5287-
Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, Mineau F, Pelletier JP: IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. 1998, 160: 3513-3521.
Voswinkel J, Muller A, Lamprecht P: Is PR3-ANCA formation initiated in Wegener's granulomatosis lesions? Granulomas as potential lymphoid tissue maintaining autoantibody production. Ann N Y Acad Sci. 2005, 1051: 12-19.
Sallusto F, Geginat J, Lanzavecchia A: Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004, 22: 745-763.
Co DO, Hogan LH, Il-Kim S, Sandor M: T cell contributions to the different phases of granuloma formation. Immunol Lett. 2004, 92: 135-142.
Shiao SL, Kirkiles-Smith NC, Shepherd BR, McNiff JM, Carr EJ, Pober JS: Human effector memory CD4+ T cells directly recognize allogeneic endothelial cells in vitro and in vivo. J Immunol. 2007, 179: 4397-4404.
Mestas J, Crampton SP, Hori T, Hughes CCW: Endothelial cell co-stimulation through OX40 augments and prolongs T cell cytokine synthesis by stabilization of cytokine mRNA. Int Immunol. 2005, 17: 737-747.
Imura A, Hori T, Imada K, Ishikawa T, Tanaka Y, Maeda M, Imamura S, Uchiyama T: The human OX40/gp34 system directly mediates adhesion of activated T cells to vascular endothelial cells. J Exp Med. 1996, 183: 2185-2195.
Noronha IL, Kruger C, Andrassy K, Ritz E, Waldherr R: In situ production of TNF-alpha, IL-1 beta and IL-2R in ANCA-positive glomerulonephritis. Kidney Int. 1993, 43: 682-692.
Yamaoka J, Kabashima K, Kawanishi M, Toda K-I, Miyachi Y: Cytotoxicity of IFN-[gamma] and TNF-[alpha] for vascular endothelial cell is mediated by nitric oxide. Biochem Biophys Res Commun. 2002, 291: 780-786.
Bratt J, Palmblad J: Cytokine-induced neutrophil-mediated injury of human endothelial cells. J Immunol. 1997, 159: 912-918.
Aloisi F, Pujol-Borrell R: Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol. 2006, 6: 205-217.
Mueller A, Holl-Ulrich K, Lamprecht P, Gross WL: Germinal centre-like structures in Wegener's granuloma: the morphological basis for autoimmunity?. Rheumatology. 2008, 47: 1111-1113.
Csernok E, Ai M, Gross WL, Wicklein D, Petersen A, Lindner B, Lamprecht P, Holle JU, Hellmich B: Wegener autoantigen induces maturation of dendritic cells and licenses them for Th1 priming via the protease-activated receptor-2 pathway. Blood. 2006, 107: 4440-4448.
Voswinkel J, Mueller A, Kraemer JA, Lamprecht P, Herlyn K, Holl-Ulrich K, Feller AC, Pitann S, Gause A, Gross WL: B lymphocyte maturation in Wegener's granulomatosis: a comparative analysis of VH genes from endonasal lesions. Ann Rheum Dis. 2006, 65: 859-864.
Segerer S, Heller F, Lindenmeyer MT, Schmid H, Cohen CD, Draganovici D, Mandelbaum J, Nelson PJ, Gröne HJ, Gröne EF, Figel AM, Nössner E, Schlöndorff D: Compartment specific expression of dendritic cell markers in human glomerulonephritis. Kidney Int. 2008, 74: 37-46.
Wilde B, van Paassen P, Damoiseaux J, Heerings-Rewinkel P, van Rie H, Witzke O, Tervaert JW: Dendritic cells in renal biopsies of patients with ANCA-associated vasculitis. Nephrol Dial Transplant. 2009, 24: 2151-2156.
Steinmetz O, Velden J, Kneissler U, Marx M, Klein A, Helmchen U: Analysis and classification of B-cell infiltrates in lupus and ANCA-associated nephritis. Kidney Int. 2008, 74: 448-457.
Scholz J, Lukacs-Kornek V, Engel DR, Specht S, Kiss E, Eitner F, Floege J, Groene HJ, Kurts C: Kidney dendritic cells stimulate IL-10 production in nephrotoxic nephritis and attenuate disease. J Am Soc Nephrol. 2008, 19: 527-537.
Lutz MB, Schuler G: Immature, semi-mature and fully mature dendritic cells: which signals induce tolerance or immunity?. Trends Immunol. 2002, 23: 445-449.
Trzonkowski P, Szarynska M, Mysliwska J, Mysliwski A: Ex vivo expansion of CD4(+)CD25(+) T regulatory cells for immunosuppressive therapy. Cytometry A. 2009, 75: 175-188.
Koenen HJPM, Smeets RL, Vink PM, van Rijssen E, Boots AMH, Joosten I: Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood. 2008, 112: 2340-2352.
Genovese MC, Becker JC, Schiff M, Luggen M, Sherrer Y, Kremer J, Birbara C, Box J, Natarajan K, Nuamah I, Li T, Aranda R, Hagerty DT, Dougados M: Abatacept for rheumatoid arthritis refractory to tumor necrosis factor {alpha} inhibition. N Engl J Med. 2005, 353: 1114-1123.
Vincenti F, Kirkman R, Light S, Bumgardner G, Pescovitz M, Halloran P, Neylan J, Wilkinson A, Ekberg H, Gaston R, Backman L, Burdick J: Interleukin-2- receptor blockade with daclizumab to prevent acute rejection in renal transplantation. N Engl J Med. 1998, 338: 161-165.
Nieuwhof CMG, Damoiseaux J, Tervaert JWC: Successful treatment of cerebral vasculitis in an HIV-positive patient with anti-CD25 treatment. Ann Rheum Dis. 2006, 65: 1677-1678.
Bielekova B, Richert N, Howard T, Blevins G, Markovic-Plese S, McCartin J, Wurfel J, Ohayon J, Waidmann TA, McFarland HF, Martin R: Humanized anti-CD25 (daclizumab) inhibits disease activity in multiple sclerosis patients failing to respond to interferon beta. Proc Natl Acad Sci USA. 2004, 101: 8705-8708.
Hoyer KK, Dooms H, Barron L, Abbas AK: Interleukin-2 in the development and control of inflammatory disease. Immunol Rev. 2008, 226: 19-28.
Stuve O, Gold R, Chan A, Mix E, Zettl U, Kieseier BC: alpha 4-Integrin antagonism with natalizumab effects and adverse effects. J Neurol. 2008, 255: 58-65.
van Paassen P, Tervaert JWC, Heeringa P: Mechanisms of vasculitis: how pauci-immune is ANCA-associated renal vasculitis?. Nephron Exp Nephrol. 2007, 105: e10-16.
Acknowledgements
This work was funded by the European Renal Association-European Dialysis and Transplant Association (ERA-EDTA) and the Dutch Kidney Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Wilde, B., Thewissen, M., Damoiseaux, J. et al. T cells in ANCA-associated vasculitis: what can we learn from lesional versus circulating T cells?. Arthritis Res Ther 12, 204 (2010). https://doi.org/10.1186/ar2923
Published:
DOI: https://doi.org/10.1186/ar2923