
Abstract
Osteoarthritis (OA) affects all articular tissues and finally leads to joint failure. Although articular tissues have long been considered unresponsive to estrogens or their deficiency, there is now increasing evidence that estrogens influence the activity of joint tissues through complex molecular pathways that act at multiple levels. Indeed, we are only just beginning to understand the effects of estrogen deficiency on articular tissues during OA development and progression, as well as on the association between OA and osteoporosis. Estrogen replacement therapy and current selective estrogen receptor modulators have mixed effectiveness in preserving and/or restoring joint tissue in OA. Thus, a better understanding of how estrogen acts on joints and other tissues in OA will aid the development of specific and safe estrogen ligands as novel therapeutic agents targeting the OA joint as a whole organ.
Similar content being viewed by others

Introduction
Osteoarthritis (OA) is a very common chronic disease that affects all joint tissues, causing progressive irreversible damage and, finally, the failure of the joint as an organ [1]. Characteristic pathological changes in OA not only include joint cartilage degeneration but also subchondral bone thickening, osteophyte formation and synovial inflammation, all of which are associated with capsule laxitude and decreased muscle strength [1, 2]. The pathological changes that occur in OA are the result of the action of biomechanical forces coupled with multiple autocrine, paracrine and endocrine cellular events that lead to a breakdown of the normal balance in tissue turnover within the joint [3, 4].
Among the multiple physiopathological mechanisms involved in OA, those related to sex hormone control have been attracting much attention, in particular those involving estrogens [5]. In contrast to other tissues such as the endometrium, breast, brain and non-joint bone, it was traditionally thought that joint tissues were non-responsive to estrogens and estrogen deficit. However, interest in estrogens was stimulated by the large proportion of postmenopausal women with OA and the complexity of their role in this disease. Indeed, considerable efforts have been made to understand the potential role of estrogens in the biology of joint tissues, as well as in the development and progression of OA, which has led to a better understanding of the effects of estrogen on joint tissues and on cartilage in particular [5–7].
There is increasing evidence that estrogens fulfill a relevant role in maintaining the homeostasis of articular tissues and, hence, of the joint itself. The dramatic rise in OA prevalence among postmenopausal women [8, 9], which is associated with the presence of estrogen receptors (ERs) in joint tissues [10–14], suggests a link between OA and loss of ovarian function. This association indicates a potential protective role for estrogens against the development of OA. Indeed, recent in vitro, in vivo, genetic and clinical studies have shed further light on these issues.
This review is based on a literature search of peer-reviewed articles written in English in the Medline and PubMed databases from 1952 to April 2009 carried out using the keywords estrogen, menopause, estrogen replacement therapy (ERT) and selective estrogen receptor modulators (SERMs) alone or in various combinations with joint, cartilage, subchondral bone, synovium, ligaments, muscle, tendons, OA and osteoporosis (OP). Accordingly, it addresses the effect of estrogen deficit on all joint tissues and the dual action of estrogen deficit on the association of OP and cartilage damage. In addition, we emphasize the relevance of these effects in the onset and/or progression of OA as well as summarizing our current knowledge on how estrogen regulates the metabolism of joint tissues. Finally, we examine the effects of ERT and current SERMs in OA, as well as the development of new specific estrogen ligands as potential therapeutic strategies to treat this disease.
The effects of estrogen deficiency on components of the osteoarthritis joint
Different studies have provided compelling information on the relevant effects of estrogen deficiency on joint components in cell culture, animal models or humans. Although much of the attention has focused on the effects of estrogen on articular cartilage, estrogen deficiency also affects other joint tissues during the course of OA, such as the periarticular bone, synovial lining, muscles, ligaments and the capsule (Figure 1).

Estrogen actions on target articular tissues. ACL, anterior cruciate ligament; [Ca2+]i, intracellular calcium concentration; COX-2, cyclooxygenase-2; IGF, insulin-like growth factor; iNOS, inducible nitric oxide synthase; MRI, magnetic resonance imaging; OB, osteoblast; OVX, ovariectomized; PG, proteoglycan.
In vitrostudies
Several experimental studies have shown that estrogens are implicated in the regulation of cartilage metabolism. Indeed, 17β-estradiol (E2) enhances glycosaminoglycan synthesis in cultures of rabbit joint chondrocytes through the up-regulation of the uridine diphosphate glucose dehydrogenase gene [15]. Furthermore, estrogen (1 to 100 M) significantly impairs the release of C-telopeptide of type II collagen from TNF-α and oncostatin M-stimulated bovine cartilage explants ex vivo in a dose-dependent manner [16]. In addition, E2 inhibits cyclooxygenase-2 mRNA expression in bovine articular chondrocytes and protects them from reactive oxygen species-induced damage [17, 18]. However, the effects of high doses of estrogen on chondrocytes are contradictory. High concentrations of E2 lead to deleterious effects such as suppression of DNA synthesis in human chondrocytes [19], as well as the inhibition of proteoglycan synthesis and cell division in both bovine chondrocytes and cartilage explants [20, 21]. A significant difference in ER affinity for its ligand as a function of age was observed. Human chondrocytes from early pubertal individuals display a maximal response to estrogens, while chondrocytes from neonatal children do not respond at all [22]. Similarly, ERs from pubertal rabbit chondrocytes exhibit higher affinity for estrogens than pre-pubertal chondrocytes [23]. Thus, estrogen dose and donor age are the main factors that influence chondrocyte response to estrogen.
These and many other relevant findings in vitro (discussed below) clearly show that estrogen influences the activity of all joint tissues through complex molecular mechanisms acting at multiple levels.
In vivostudies
The effects of estrogen on joint tissues have primarily been studied in ovariectomized (OVX) animal models. Despite these studies, the influence of estrogen deficiency on cartilage remains unclear, even though there is significant evidence of the detrimental effect of estrogen loss in mature female animals [7]. An increase in cartilage turnover and surface erosion was observed in OVX Sprague-Dawley rats [24], as well as in cynomolgus macaques subjected to bilateral OVX [25]. Significantly, intact females had less severe OA than OVX females and although intact male mice showed more severe OA than intact females, orchiectomized mice develop less OA than intact males [26]. By contrast, such associations could not be shown in other earlier studies [7].
Relevant changes have also been described in the subchondral bone of OVX animals. Indeed, OVX cynomolgus monkeys have higher indices of bone turnover in subchondral bone compared to epiphyseal/metaphyseal cancellous bone of the proximal tibia [27]. Moreover, the marginal osteophyte area is positively correlated with subchondral bone thickness in the medial tibial plateau of these animals [28]. Significantly, subchondral bone remodeling has also been described in conjunction with changes in joint cartilage in a guinea pig model of spontaneous OA [29]. We found that rabbit subchondral bone has mixed densitometric characteristics with a marked predominance of cortical bone [30]. In fact, subchondral knee bone mineral density (BMD) is significantly correlated with the BMD of the spine, and trabecular and cortical knee bone in healthy, OA, OP and OP/OA rabbits [31].
Our rabbit model is a valuable tool to study OP because rabbits have much faster bone turnover than rodents or primates, and in contrast to rodents, they reach skeletal maturity soon after their sexual development is complete [32]. Moreover, since OVX itself only causes mild osteopenia, which may be insufficient to provoke OP in these animals, moderate doses of methylprednisolone were administrated to ensure OP development [33]. We evaluated whether estrogen deficiency alone can induce OA alterations in healthy cartilage or, by contrast, whether OP subchondral bone is the origin of the cartilage changes in these animals. Estrogen deficiency leads to mild OA changes 22 weeks after isolated OVX in healthy articular cartilage, while OVX and methylprednisolone-induced OP play an additional role in these osteoarthritic changes (Figure 2). Thus, estrogen deprivation might produce a dual effect: a main direct action upon joint cartilage and a minor indirect effect on subchondral bone.

Osteoarthritic cartilage damage is aggravated by ovariectomy plus glucocorticoid-induced osteoporosis in a rabbit model. Ovariectomy itself induces small disturbances in the cartilage, while no differences were found between articular cartilage from ovariectomized (OVX), osteoporosis (OP) and osteoarthritis (OA) rabbits. Bar graphs showing the total Mankin score from the histological evaluation of joint cartilage at the weight bearing area of the medial femoral chondyle in the different experimental groups. Healthy, controls; OVX, ovariectomized rabbits; OP, osteoporotic rabbits induced by OVX followed by parenteral methyprednisolone injections for 4 weeks; OA, osteoarthritic rabbits induced by partial medial meniscectomy and anterior cruciate ligament section of the knee; OP+OA, rabbits with experimentally induced OP followed by OA induction. Data are expressed as the mean ± standard deviation. #P < 0.05 versus healthy; &P < 0.05 versus OVX; §P < 0.05 versus OP; ¶P < 0.05 versus OA.
The influence of estrogen on the remaining joint tissues has not been studied directly in OA animal models. However, the involvement of these tissues in OA and the changes produced by estrogen in related animal models suggest a potential role of estrogen in OA changes. Indeed, the remodeling of the cruciate ligament is thought to occur early during knee OA in guinea pigs [29] and the potential role of endogenous estrogens in the disproportionate number of anterior cruciate ligament (ACL) injuries seen in female athletes has been studied in different animal models, although to date with negative results [34]. Besides, significant attenuation of histochemical and biochemical indices of muscle damage and inflammatory response were found in female rats after downhill running when compared with their male counterparts. Such an effect may possibly be explained by the higher circulating estrogen levels in these rats [35]. In addition, estrogen deficiency following OVX is often accompanied by an increase in fat mass, which in turn leads to increased adipokine levels, the role of which in OA is also now being investigated.
Human studies
Associations between polymorphisms in the human ERα gene (ESR1) and OA have been studied in different populations with mixed results. Haplotypes of the PvuII and XbaI polymorphisms in the ERα gene have been associated with an increased prevalence of clinical and radiographic knee OA [36–38]. In addition, the exon 8 G/A BtgI polymorphism was also associated with knee OA in Asian populations [38]. However, other studies showed either no or only a modest inverse relationship between ERα gene polymorphisms and OA in Caucasian populations [39, 40].
Numerous clinical studies have also shown that OA is related to estrogen levels [8, 9, 41–47]. Thus, the prevalence of OA is greater in women than men and a clear increase in OA prevalence is associated with the peak age of menopause [8, 9, 41]. Indeed, a nationwide population survey showed that radiographic generalized OA is three times more common in women aged 45 to 64 years compared to their male counterparts [9], and a hospital-based study found a high female to male ratio of 10:1 for OA, with a peak at 50 years of age [42]. In addition, 64% of females with knee OA suffered the onset of symptoms either perimenopausally or within 5 years of natural menopause or hysterectomy. In fact, the onset of symptoms of knee OA occurred before 50 years of age in 58% of females as opposed to only 20% of males [43].
Since the earliest studies of OA, generalized involvement of joints was described in postmenopausal females, and predominant node formation with early signs of inflammation was observed in the proximal and distal interphalangeal joints of the hands [44]. Nodular hand OA is often associated with a polyarticular and symmetric involvement of major joints such as knees and hips [45]. Erosions may occur in the inter-phalangeal joints and are characteristic of erosive OA. This disorder tends to occur in middle-aged women, and it is often an acute condition with features of inflammation that subside over a period of months to years, leaving deformed joints and occasional ankylosis [46]. Lower levels of serum E2 and its metabolite 2-hydroxyestrone in urine were recently reported in postmenopausal women who developed radiographically defined knee OA [47].
Failure of estrogen production at menopause is associated with a relevant loss of muscle mass and, therefore, significant impairment of muscle performance and functional capacity [48]. Diminished strength of the quadriceps in women but not men predict knee OA [49], and peri- and postmenopausal women also seem to have less lean body mass when compared with pre-menopausal women [50]. In addition, varus-valgus laxity has more frequently been described in women than in men [51].
The effect of estrogen deficiency on the association between osteoarthritis and osteoporosis
At this time, a complex and paradoxical relationship seems to exist between OA and OP, although there is increasing evidence supporting a close biomolecular and mechanical association between subchondral bone and cartilage [52]. Indeed, microarray profiles have identified a number of genes differentially expressed in OA bone that are key players in the structure and function of both bone and cartilage, including genes that participate in the Wingless-type mouse mammary tumor virus/β-catenin (Wnt/β-catenin) and transforming growth factor-β/mothers against decapentaplegic (TGF-β/SMAD) signaling pathways and their targets [53]. Wnt5b and other genes involved in osteoclast function are differentially expressed between male and female OA bone [53]. Furthermore, aggrecan production, as well as SOX9, type II collagen and parathyroid hormone-related protein mRNA expression was inhibited in sclerotic but not non-sclerotic osteoblasts (OBs), while expression of matrix metalloproteinases MMP-3 and MMP-13 and osteoblast-specific factor 1 by human OA chondrocytes was augmented in a co-culture system. Thus, sclerotic osteoarthritic subchondral OBs may contribute to cartilage degradation and chondrocyte hypertrophy [54].
Current methodological difficulties in detecting and closely following incipient OA lesions at early stages in humans are a major obstacle to better understanding the relationship between OA and OP. Therefore, animal models provide an alternative to study this relationship. However, some species may not be suitable for such studies since OVX provokes strong subchondral bone remodeling and loss in these animals (for example, rodents), and possibly ensuing indirect cartilage damage. Conversely, there are certain advantages to studying OP in rabbits [32] and, in this context, our group has developed an experimental model in mature rabbits where OP markedly aggravates the severity of OA estimated using the Mankin score (Figure 2). Moreover, the increased cartilage damage is correlated with loss of bone mass, suggesting a direct relationship between OA and OP [31].
Several cross-sectional studies have demonstrated an inverse relationship between OP and OA [55, 56], while others produced opposite results [57]. However, some confounding variables such as race, obesity and physical activity could explain the mutually exclusive relationship between OA and OP. Thus, overweight individuals and/or those that undertake excessive physical activity could have a higher risk of developing OA and of having a higher bone mass. This controversial relationship is also witnessed at the regional level. Indeed, severe hip OA has a protective role against the age-related decrease in structural and mechanical properties of cancellous bone in the principal compressive region of the ipsilateral femoral head [58]. In turn, subchondral tibial BMD was correlated with future joint space narrowing and it has been proposed as a predictor of knee OA progression [59]. However, other studies have shown a decrease in subchondral BMD associated with knee OA. Indeed, in female patients with relatively mild OA of the knee, a significant decrease in periarticular subchondral BMD was evident, whether or not they had a low spine BMD [60].
Mechanisms underlying the effects of estrogen on joint tissues
Estrogen influences the biology of joint tissues by regulating the activity and expression of key signaling molecules in several distinct pathways (Figure 3).

Intracellular signaling pathways used to regulate the activity of estrogens, estrogen receptors, and selective estrogen receptor modulators on articular tissues. Pathway 1: canonical estrogen signaling pathway (estrogen response element (ERE)-dependent) - ligand-activated estrogen receptors (ERs) bind specifically to EREs in the promoter of target genes. Pathway 2: non-ERE estrogen signaling pathway - ligand-bound ERs interact with other transcription factors, such as activator protein (AP)-1, NF-κB and Sp1, forming complexes that mediate the transcription of genes whose promoters do not harbor EREs. Co-regulator molecules regulate the activity of the transcriptional complexes. Pathway 3: non-genomic estrogen signaling pathways - ERs and GP30 localized at or near the cell membrane might elicit the rapid response by activating the phosphatidylinositol-3/Akt (PI3K/Akt) and/or protein kinase C/mitogen activated protein kinase (PKC/MAPK) signal transduction pathways. Pathway 4: ligand-independent pathways - ERs can be stimulated by growth factors such as insulin-like growth factor (IGF)-1, transforming growth factor-β/mothers against decapentaplegic (TGF-β/SMAD), epidermal growth factor (EGF) and the Wnt/β-catenin signaling pathway in the absence of ligands, either by direct interaction or by MAP and PI3/Akt kinase-mediated phosphorylation. Since members of these signaling pathways are transcription factors, some of them, such as SMADs 3/4, can elicit estrogen responses by interacting with ER in the non-ERE-dependent genomic pathway. ERK, extracellular signal regulated kinase; GF, growth factor; GFR, growth factor receptor; MNAR, Modulator of nongenomic action of estrogen receptors; TF, transcription factor.
Canonical estrogen receptor signaling pathway (estrogen response element-dependent)
Estrogen primarily exerts its effects on target tissues by binding to and activating ERs. ERs act as ligand-activated transcription factors in the nucleus that specifically bind to estrogen response elements (EREs) in the promoters of target genes such as the human oxytocin, prolactin, cathepsin D, progesterone receptor, vascular endothelial growth factor, insulin-like growth factor (IGF)-1, or c-fos genes [61], as diagrammatically shown in Figure 3 (pathway 1). The ERE is a 13 base-pair inverted sequence that binds ERs as dimers. Because imperfect palindromic EREs, or even half EREs, are often seen in the regulatory region of estrogen target genes, transcriptional synergism might occur that could include the co-operative recruitment of co-activators, direct interaction between ER dimers, or allosteric modulation of the DNA-ER complexes [62].
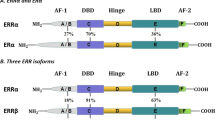
ERs contain four functional domains. The variable amino-terminal A/B domain harbors the constitutive activation function (AF)-1, which modulates transcription in a gene- and cell-specific manner. The central and most conserved C domain contains the DNA binding domain, and it also mediates receptor dimerization. The D domain is a less well understood region. Finally, the carboxy-terminal multifunctional E/F domain holds the ligand-binding domain as well as sites for cofactors, transcriptional activation (AF-2) and nuclear localization (Figure 4) [63]. There are two receptor subtypes, ERα and ERβ, which are different proteins encoded by distinct genes located on chromosomes 6 (q24-q27) and 14 (q21-q22), respectively [64]. These two receptor subtypes have 96% amino acid homology in the DNA binding domain but only 53% identity in the ligand-binding domain. As a result, similar ERE binding properties have been associated with a partially distinct spectrum of ligands for each receptor, although with similar affinities for estrogen. Even weaker amino acid identity is found in the A/B domain of ERα and ERβ (Figure 4). Both receptors also show little conservation in AF-2 and, therefore, several proteins may direct ERα and ERβ to different targets as observed in their contrasting effects at the activator protein (AP)-1 site of the collagenase promoter. Thus, ERα and ERβ have different transcriptional activities that may contribute to their distinct tissue-specific actions [63, 65].

Structural composition of estrogen receptor (ER)α and ERβ. Both receptors have four functional domains that harbor a DNA-binding domain (DBD), a ligand-binding domain (LBD) and two transcriptional activation functions (AF-1 and AF-2), as indicated for ERβ. The percent of homology in these domains between ERα and ERβ is indicated, as well as the location of several phosphorylation sites in ERα whereby this receptor is activated by important kinases that modulate a wide variety of cellular events. aa, amino acids; Akt, serine/threonine specific-protein kinase family encoded by the Akt genes; CDK2, cyclin-dependent kinase 2; MAPK, mitogen activated protein kinase; PKA, protein kinase A; Src: steroid receptor coactivator.
Both ERs are distributed widely throughout the body, displaying distinct but overlapping expression in a variety of tissues. ERα is highly expressed in classical estrogen target tissues such as the uterus, placenta, pituitary and cardiovascular system, whereas ERβ is more abundant in the ventral prostate, urogenital tract, ovarian follicles, lung, and immune system. However, the two ERs are co-expressed in tissues such as the mammary gland, bone, and certain regions of the brain [66]. Although both ER subtypes can be expressed in the same tissue, they may not be expressed in the same cell type. Nonetheless, in cells where the two ER subtypes are co-expressed, ERβ can antagonize ERα-dependent transcription [64]. The generation of human ERα and ERβ mRNA transcripts is a complex process that is controlled by sophisticated regulatory mechanisms leading to the generation of several isoforms/variants for each receptor subtype. Most ERα variants only differ at the 5' untranslated region and they are involved in tissue-specific regulation of ERα gene expression. Several species-specific and common ERβ isoforms have been described, many of which are expressed as proteins in tissues [67].
In articular tissues, both ER types are expressed by the chondrocytes [10], subchondral bone cells [11], synoviocytes [12], ligament fibroblasts [13] and myoblasts [14] in humans and other species. However, ERα is predominant in cortical bone and ERβ predominates in cartilage, cancellous bone and synovium [10, 12, 68]. More mRNA transcripts for both subtypes of ERs were found in male than in female human cartilage, but there were no differences between different joints, or between cartilage from OA patients and the normal population [10]. In bone, ERα and ERβ are expressed by OBs and they are differentially expressed during rat OB maturation [69]. Pre-osteoclasts express ERα, while osteoclast maturation and bone resorption is associated with the loss of ERα expression [70]. ERβ mRNA and protein are predominantly found in the stroma and lining cells of normal human synovium, independent of sex or menopausal status of the tissue donor [12]. Fibroblasts from human ACL, medial cruciate ligament and patellar tendon express functional ER transcripts. Indeed, 4 to 10% of ACL cells express ERs in patients with acute ACL injuries, approximately twice the proportion found in control subjects [13, 71]. In human skeletal muscle, ERα mRNA expression was 180-fold higher than that of ERβ [72]. Remarkably, individuals that undergo high endurance training have more ERα and ERβ mRNA transcripts in skeletal muscles than moderately active individuals [73].
Characterizing the phenotypes of knockout models has advanced our understanding of the role of ER in biological processes. Indeed, ERβ plays a significant role in bone remodeling in female ER knockout mice, whereas ERα does so in both sexes. Thus, male and female ERα-/- mice show decreased bone turnover and greater cancellous bone volume, even though the cortical thickness and BMD was reduced. Female ERβ-/- mice have slightly increased trabecular bone volume, while male animals do not show any change in their bones. Male and female double ER-/- mice showed significant defects in cortical bone and BMD, while female mice alone displayed a profound decrease in trabecular bone volume [74]. A recent study has shown that ERα-/-β-/- double knockout increased osteophytosis and thinning of the lateral subchondral plate, both osteoarthritic characteristics, in the knee of transgenic mice [75]. These results confirm the relevant changes described in subchondral bone of OVX animal models [27–29]. However, no difference in cartilage damage was observed between the ERα-/-, ERβ-/- and ERα-/-β-/- double knockout and wild-type mice at 6 months of age, although the cartilage damage was very mild in all mice [75]. Whether the absence of significant cartilage damage in all ER knockout mice groups reflects some important differences between ER knockout mice, which lack ER expression since birth, and OVX models that show significant OA cartilage changes associated with estrogen depletion at a later age [7, 24–26] remains to be established.
As regards muscle, ERα-/- mice have lower tetanic tension per calculated anatomical cross-sectional and fiber areas in tibialis anterior and gastrocnemius than in wild-type mice. In contrast, ERβ-/-and wild-type mice were comparable in all measures. These results suggest that the effects of estrogen on skeletal muscle are mainly mediated by ERα [76]. With respect to ligaments, no changes in medial cruciate ligament or ACL viscoelastic or tensile mechanical properties were observed in ERβ-/- mice [77].
Non-estrogen response element-mediated genomic ER signaling
The second genomic mechanism involves the interaction of ligand-bound ERs with other transcription factors like Fos/Jun (AP-1-responsive elements), c-Jun/NF-κB and specificity protein 1 (Sp1) recruiting co-regulators to form initiation complexes that regulate the transcription of genes whose promoters do not harbor EREs [64, 78]. In this tethering mechanism, ERs do not bind directly to DNA (Figure 3, mechanism 2) and, thus, ERs can up-regulate the expression of promoters containing AP-1 sites, such as the collagenase and IGF-1 genes. Interestingly, E2 exerts distinct transcriptional activation on the AP-1 site of the collagenase promoter depending on whether ERα or ERβ is involved: it elicits transcriptional activation with ERα, while it represses transcription with ERβ [65, 78]. The interaction of ERs with Sp1 activates uteroglobin, retinoic acid receptor alpha, IGF-binding protein 4 (IGFBP4), TGF-α, bcl2 and the low-density lipoprotein receptor genes [61, 78]. Similarly, suppression of IL-6 expression by E2 occurs through interactions of the ligand bound ER with the NF-κB complex [64].
Ligand-dependent activation of ERs, both ERE and non-ERE-mediated, attracts co-regulator molecules that modify the chromatin state, thereby recruiting or hindering the transcriptional complex and representing another level of control in ER gene regulation [61, 63, 79]. Co-activators stimulate transcription by interacting with helix 12 (H12) of the AF-2 region through their short 'nuclear receptor boxes', transducing ligand signals to the basal transcriptional machinery. The best characterized co-activators include the steroid receptor co-activator (SRC) family (SRC1, SRC2 and SRC3) and members of the mammalian mediator complex (thyroid receptor associated proteins, vitamin-D receptor interacting proteins, activator-recruited cofactor) [63, 79]. Alternatively, co-repressors that impede transcription include the nuclear receptor co-repressor (NCoR) and the silencing mediator for the retinoic acid and thyroid hormone receptor (SMRT), which interact with ligand-free ER through an elongated amino acid sequence called the CoRNR-box. By contrast, if H12 assumes a 'charge clamp' configuration in response to agonist binding, then it could not hold the long NCoR/SMRT helices. Thus, agonist binding reduces the affinity of ERs for co-repressors and increases their affinity for co-activators [63, 79]. In addition, both SMRT and NCoR recruit the protein SIN3 and histone deacetylases to form a large co-repressor complex, implicating histone deacetylation in transcriptional repression [79].
In rabbit articular chondrocytes, ERα activation inhibits NF-κB p65 activity and, subsequently, decreases IL-1β-stimulated inducible nitric oxide synthase expression and nitric oxide production [80]. Moreover, ERα and, particularly, ERβ transfection significantly enhances MMP-13 promoter activity through an AP-1 site, which may be modulated through the sites of the Runt-related (Runx) and PEA-3 Ets transcription factors in a rabbit synovial cell line lacking endogenous ER [81]. A normal balance between classic ERE-mediated and non-ERE-mediated ERα, genomic and non-genomic, pathways in cortical bone have also been described in ERα-/NERKI mice and its disruption can lead to an aberrant response to estrogen [82].
Non-genomic ER signaling pathways
Estrogens may also exert their ligand-dependent effects through non-genomic mechanisms that are responsible for more rapid effects, occurring within seconds or minutes of stimulating cell signal transduction pathways, such as the mitogen activated protein (MAP) kinases, in particular the extracellular signal regulated kinase 1/2 (ERK 1/2), p38 and phosphatidylinositol-3 (PI3) kinase/Akt pathways [64]. A small ER population and/or a G-protein-coupled receptor termed GP30, localized at or close to the cell membrane, may elicit these responses [83, 84]. ER translocation to the cell membrane is nourished by its interaction with membrane proteins such as caveolin 1/2, striatin and the adaptor proteins Shc and p130 Cas [64]. S-palmitoylation and myristoylation of ERα also promote ERα association with the plasma membrane and its interaction with caveolin-1 [64]. Furthermore, interaction between ER, the tyrosine kinase cSrc and an adaptor protein called modulator of nongenomic action of estrogen receptors (MNAR) generates a signaling complex that may be crucial for the important cSrc activation and further kinase phosphorylation [85]. Thus, several molecular processes have been shown to mediate the non-genomic effects of ER (Figure 3, pathway 3). However, the precise mechanisms involved in ER localization in the cell membrane, as well as the interaction between ERs and signaling pathways, are yet to be fully established.
There appears to be sexual dimorphism in the non-genomic pathways described in human articular and rat growth plate chondrocytes. Thus, only female cells respond to estrogens by promoting a rapid protein kinase C (PKC)-α-mediated increase in proteoglycan production and alkaline phospha-tase activity (PKC increase occurred within 9 minutes and was maximal at 90 minutes). Treatment with the PKC inhibitor chelerythrine blocked these effects [86, 87]. PKC activation initiated a signaling cascade involving the ERK1/2 and p38 MAP kinase pathways, which in turn mediate the downstream biological effects of estrogen on alkaline phosphatase activity and [(35)S]-sulfate incorporation in rat growth plate chondrocytes. A membrane receptor has been proposed to elicit this response, although its precise nature remains to be established [88].
Estrogen also regulates intracellular calcium concentrations ([Ca2+]i) in a sex-specific and cell maturation state-dependent manner in rat growth plate chondrocytes. Indeed, E2 more rapidly increased [Ca2+]i in resting zone chondrocytes than in growth-zone chondrocytes from female rats, while no effect was observed in chondrocytes from male rats. This effect is mediated by membrane-associated events, phospholipase C-dependent inositol triphosphate-3 production and Ca2+release from the endoplasmic reticulum [89]. In the light of the higher prevalence of OA in postmenopausal females, it has been proposed that these intrinsic sex-specific differences may contribute to OA development [86]. In addition, inclusion of the gender variable when interpreting experimental data and the functional adaptation of donor cells in transplants between organisms of different sexes should be considered [86].
Both ERK phosphorylation kinetics and the duration of phospho-ERK nuclear retention determine the pro- or anti-apoptotic effects of estrogen in bone cells. In fact, E2-induced transient ERK phosphorylation (lasting 30 minutes) leads to anti-apoptotic effects in OBs and osteocytes, whereas it produces pro-apoptotic signals in osteoclasts through sustained ERK phosphorylation (for at least 24 hours) [90]. Also, the ERK 1/2 and PI3K/Akt/Bad pathways mediate the anti-apoptotic effect of estrogens in C2C12 muscle cells following activation of ERα and ERβ located in diverse cellular compartments such as the mitochondria and perinucleus [91]. Divergent ER-induced gene expression has been found depending on whether the genomic or non-genomic signaling pathways are activated in different cell types. In osteoblastic OB-6 cells, E2 stimulated complement 3 (C3) and IGF-1 expression after 24 hours, which did not occur following estren administration. This discrepancy is explained by the ERE present in the promoter of the C3 gene and by ER regulating IGF-1 through a protein-protein interaction that influences the AP-1 enhancer. Since estren is a non-genotropic ER activator, it did not activate these ERE-or AP-1-containing genes [92].
Ligand-independent signaling pathways
The stimulation of growth factors such as those of the IGF-1, epidermal growth factor, TGF-β/SMAD and Wnt/β-catenin signaling pathways can activate ERs or associated co-regulators via kinase phosphorylation in the absence of ER ligands [64, 93–95]. In turn, ERα may also regulate growth factor signaling [64, 93–95]. Crosstalk between growth factors and ERs occurs in both the nuclear and cytoplasmic compartments, promoting highly active interactions [64, 93–95] (Figure 3, pathway 4).
In OBs, estrogen and TGF-β/SMAD signaling pathways may interact at several levels: activation of the TGF-β pathway by estrogens via TGF-β mRNA induction; increase of estrogen and TGF-β/SMAD signaling due to cytoplasmic MAP kinase activity; direct interaction between ERs and the SMAD proteins in the cytoplasm or nucleus; and interaction between ERs and the TGF-β-inducible early-response gene (TIEG) and Runx-2 transcription factors in the nucleus. Both TIEG and Runx-2 expression are induced by E2 and TGF-β and, furthermore, TIEG appears to be required for the E2 and TGF-β-induced regulation of Runx2 expression [95]. Thus, a relevant inhibition of osteoclastic bone resorption by osteocytes occurs as a result of TGF-β enhancement by estrogen [96].
ERs can interact with members of the Wnt/β-catenin signaling system in both the presence and absence of the ligand [97]. Bone response to mechanical forces can be influenced by interactions between the β-catenin and T-cell factor nuclear complex, and ERα in OBs. Indeed, ER modulators suppressed the accumulation of active β-catenin in the nucleus of OBs in vitro within 3 hours following a single period of dynamic strain of magnitude similar to the estimated strain that OBs regularly experience in vivo. Accordingly, microarray analysis performed with RNA extracted from the tibia of ERα-/- mice demonstrated the abrogation of dynamic axial loading-induced expression of Wnt-responsive genes (compared with RNA from the tibia of wild-type mice) [98]. These results suggest that ERα is required for early Wnt/β-catenin-induced bone cell responses to mechanical strain. Indeed, the reduced effectiveness of the bone cell responses to mechanical load associated with estrogen deficiency may alter the bone mass in postmenopausal OP women.
In cynomolgus monkey joint cartilage, IGFBP2-mediated activation of the IGF system induces IGF-1 production, which in turn leads to increased sulfate incorporation into proteoglycans following estrogen administration [99]. In addition, ERs might interact with the TGF-β and Wnt/β-catenin signaling cascades in articular chondrocytes. Both the Wnt/β-catenin and TGF-β/SMAD signaling pathways play a prominent role in bone and cartilage biology. The TGF-β/SMAD pathway fulfils a beneficial role in bone and cartilage maintenance/repair, although it is also an important protagonist of osteophyte formation [95, 100]. In turn, the Wnt/β-catenin system is essential in many biological aspects of bone, from differentiation, proliferation and cellular apoptosis to bone mass regulation and its ability to respond to mechanical load [101]. Activation of the Wnt/β-catenin pathway has also been implicated in OA cartilage damage, and Wnt inhibitors such as the secreted frizzled related protein 3 and Dickkopf-1 might modulate the susceptibility to, and the progress of, hip OA [102].
Although our understanding of the different molecular mechanisms by which estrogen deficits could act on articular tissues and their contribution to OA development has advanced significantly in recent years, it is still limited and more research will be necessary to identify therapeutic targets for this very prevalent disease.
The effects of estrogen replacement therapy and selective estrogen receptor modulators on articular tissues
ERT has displayed mixed effects on joint tissues in various animal and human studies while SERMS conversely have demonstrated a homogeneous response in these tissues (a general description of the effects of SERMs on different tissues is presented in Table 1).
In vivostudies
Estrogen administration in OVX animals has paradoxical effects on joint cartilage, in contrast to the clear benefits of SERM administration [24]. While intra-articular E2 injections [103] and high supraphysiological estrogen concentrations [104] caused deleterious effects on joint cartilage in a dose-and time-dependent fashion, the beneficial effects of long-term estrogen treatment have been seen in different models [24, 25, 99]. Early estrogen administration maximizes its positive effects on cartilage [16] and, in turn, tamoxifen decreases cartilage damage in a rabbit model of OA, even in males [105]. Furthermore, tamoxifen antagonized the chondro-destructive effects of high dose intra-articular E2 during early knee OA in rabbits [106]. Also, NNC 45-0781 and levormeloxifen both inhibited the OVX-induced acceleration of cartilage and bone turnover, and they significantly suppressed cartilage damage in female Sprague-Dawley rats [24, 107].
In subchondral bone, the effects of long-term ERT have only recently begun to be studied. ERT limits bone formation in both subchondral bone and epiphyseal/metaphyseal cancellous bone of the proximal tibia in OVX cynomolgus monkeys [27]. ERT also reduces the prevalence of marginal osteophytes, particularly in the lateral tibial plateau, while the presence of axial osteophytes is not affected. However, neither the cross-sectional area in osteophytes nor its static and dynamic histomorphometric parameters are significantly influenced by ERT [28, 108]. In addition, a significant effect of ERT has been described on several components of the IGF system in the synovial fluid of OVX female adult cynomolgus monkeys, suggesting a potential stimulatory effect of estrogen on joint tissues in vivo [109]. In turn, estrogen administration reversed OVX-induced contractile muscle and myosin dysfunction, as well as the OVX-induced increase of muscle wet mass in mature female mice caused by fluid accumulation [110].
Clinical studies
The effect of ERT on the risk of developing OA and on its progression in postmenopausal women remains unclear. Unlike observational clinical studies, some radiographic studies have suggested a protective effect of ERT on the radiographic detection of OA or its progression [111–115]. In a cross-sectional study, ERT significantly reduced the risk of radiographic hip OA, particularly in long-term users [111]. Similarly, an initial cross-sectional analysis of two of the largest studies found an inverse association between ERT use and radiological knee OA, suggesting that ERT may have a chondroprotective effect. However, a subsequent follow-up analysis failed to show significant ERT protection against either the development or progression of radiographic knee OA [112–115]. Additionally, contradictory results were described regarding the association between ERT and the requirement for arthroplasty [116]. Nevertheless, in the largest study, females that received estrogen alone had significantly fewer arthroplasties, particularly in the hip. Thus, unopposed estrogen administration might have a protective effect against the risk of joint replacement, an effect that may be particularly relevant in hip compared to knee OA [117].
Magnetic resonance imaging-estimated subchondral bone attrition and bone-marrow abnormalities associated with cartilage degradation in knee OA was delayed or prevented by ERT or alendronate in postmenopausal women [118]. In turn, ERT may preserve muscle performance. A 12-month trial showed that ERT protects against the detrimental effects of estrogen deficiency on skeletal muscle in early postmenopausal women, thereby positively influencing muscle performance and structure. Moreover, high-impact physical training provided additional benefits [119].
Development of novel estrogen ligands
Recently, novel ER ligands, both pathway-selective and ERβ-selective, have been developed due to the potent anti-inflammatory activity they have been attributed [120, 121] (Table 1). Indeed, the pathway-selective ER ligands WAY-169916 and WAY-204688 inhibit NF-κB transcriptional activity in the absence of conventional estrogenic activity in different animal models of inflammatory diseases [122, 123].
The suppressive effects of estrogen on inflammatory mediators, including NF-κB, inducible nitric oxide synthase, cyclooxygenase-2, and reactive oxygen species in articular chondrocytes [17, 18, 80], in association with other selective estrogenic benefits on joint tissues might reflect their potential utility in OA treatment.
Conclusion
Progressive structural and functional changes on articular structures commence at early menopause and persist post-menopause, leading to an increase in the prevalence of OA in the latter population and representing a big impact on health costs worldwide. Both experimental and observational evidence support a relevant role for estrogens in the homeostasis of joint tissues and, hence, in the health status of joints. Indeed, estrogens influence their metabolism at many crucial levels and through several complex molecular mechanisms. These effects of estrogens at joints are either significantly dampened or lost as a result of postmenopausal ovary insufficiency.
A better understanding of the role that estrogen and its deficiency plays in the molecular mechanisms of menopause-induced osteoarthritic changes that affect the different joint structures will help further development of new and precise therapeutic strategies to prevent and/or restore damaged articular tissues in OA. These improved therapeutic approaches must be devoid of the widely known undesirable effects of estrogens in other target tissues. Thus, in OA, which represents a particularly challenging disease due to its effects upon different joint structures, these therapeutic options should target the joint as a whole organ rather than focusing only on cartilage damage.
Abbreviations
- ACL:
-
anterior cruciate ligament
- AF:
-
activation function
- AP:
-
activator protein
- BMD:
-
bone mineral density
- E2:
-
17β-estradiol
- ER:
-
estrogen receptor
- ERE:
-
estrogen response element
- ERK:
-
extracellular signal regulated kinase
- ERT:
-
estrogen replacement therapy
- IGF:
-
insulin-like growth factor
- IGFBP:
-
IGF-binding protein
- IL:
-
interleukin
- MAP:
-
mitogen activated protein
- MMP:
-
matrix metalloproteinase
- NCoR:
-
nuclear receptor co-repressor
- NF:
-
nuclear factor
- OA:
-
osteoarthritis
- OB:
-
osteoblast
- OP:
-
osteoporosis
- OVX:
-
ovariectomized
- PI3:
-
phosphatidylinositol-3
- PKC:
-
protein kinase C
- SERM:
-
selective estrogen receptor modulators
- SMAD:
-
mothers against decapentaplegic
- SMRT:
-
silencing mediator for the retinoic acid and thyroid hormone receptor
- Sp:
-
specificity protein
- TGF:
-
transforming growth factor
- TNF:
-
tumor necrosis factor.
References
Brandt KD, Radin EL, Dieppe PA, Putte van de L: Yet more evidence that osteoarthritis is not a cartilage disease. Ann Rheum Dis. 2006, 65: 1261-1264. 10.1136/ard.2006.058347.
Sellam J, Herrero-Beaumont G, Berenbaum F: Osteoarthritis: pathogenesis, clinical aspects and diagnosis. EULAR Compendium on Rheumatic Diseases. Edited by: Hans Bijlsma. 2009, London, UK: BMJ Publishing Group Ltd, 444-463.
Brandt KD, Dieppe P, Radin EL: Etiopathogenesis of osteoarthritis. Rheum Dis Clin North Am. 2008, 34: 531-559. 10.1016/j.rdc.2008.05.011.
Herrero-Beaumont G, Roman-Blas JA, Castañeda S, Jimenez SA: Primary osteoarthritis: three subsets with etiological, clinical and therapeutic characteristics. Semin Arthritis Rheum. 2009 [Epub ahead of print].
Gokhale JA, Frenkel SR, Dicesare PE: Estrogen and osteoarthritis. Am J Orthop. 2004, 33: 71-80.
Tankó LB, Søndergaard BC, Oestergaard S, Karsdal MA, Christiansen C: An update review of cellular mechanisms conferring the indirect and direct effects of estrogen on articular cartilage. Climacteric. 2008, 11: 4-16. 10.1080/13697130701857639.
Sniekers YH, Weinans H, Bierma-Zeinstra SM, van Leeuwen JP, van Osch GJ: Animal models for osteoarthritis: the effect of ovariectomy and estrogen treatment - a systematic approach. Osteoarthritis Cartilage. 2008, 16: 533-541. 10.1016/j.joca.2008.01.002.
Srikanth VK, Fryer JL, Zhai G, Winzenberg TM, Hosmer D, Jones G: A meta-analysis of sex differences prevalence, incidence and severity of osteoarthritis. Osteoarthritis Cartilage. 2005, 13: 769-781. 10.1016/j.joca.2005.04.014.
Lawrence RC, Helmick CG, Arnett FC, Deyo RA, Felson DT, Giannini EH, Heyse SP, Hirsch R, Hochberg MC, Hunder GG, Liang MH, Pillemer SR, Steen VD, Wolfe F: Estimates of the prevalence of arthritis and selected musculoskeletal disorders in the United States. Arthritis Rheum. 1998, 41: 778-799. 10.1002/1529-0131(199805)41:5<778::AID-ART4>3.0.CO;2-V.
Ushiyama T, Ueyama H, Inoue K, Ohkubo I, Hukuda S: Expression of genes for estrogen receptors alpha and beta in human articular chondrocytes. Osteoarthritis Cartilage. 1999, 7: 560-566. 10.1053/joca.1999.0260.
Braidman IP, Hainey L, Batra G, Selby PL, Saunders PT, Hoyland JA: Localization of estrogen receptor beta protein expression in adult human bone. J Bone Miner Res. 2001, 16: 214-220. 10.1359/jbmr.2001.16.2.214.
Dietrich W, Haitel A, Holzer G, Huber JC, Kolbus A, Tschugguel W: Estrogen receptor-beta is the predominant estrogen receptor subtype in normal human synovia. J Soc Gynecol Investig. 2006, 13: 512-517.
Sciore P, Frank CB, Hart DA: Identification of sex hormone receptors in human and rabbit ligaments of the knee by reverse transcription polymerase chain reaction: evidence that receptors are present in tissue from both male and female subjects. J Orthop Res. 1998, 16: 604-610. 10.1002/jor.1100160513.
Kahlert S, Grohé C, Karas RH, Löbbert K, Neyses L, Vetter H: Effects of estrogen on skeletal myoblast growth. Biochem Biophys Res Commun. 1997, 232: 373-378. 10.1006/bbrc.1997.6223.
Maneix L, Beauchef G, Servent A, Wegrowski Y, Maquart FX, Boujrad N, Flouriot G, Pujol JP, Boumediene K, Galéra P, Moslemi S: 17Beta-oestradiol up-regulates the expression of a functional UDP-glucose dehydrogenase in articular chondrocytes: comparison with effects of cytokines and growth factors. Rheumatology. 2008, 47: 281-288. 10.1093/rheumatology/kem323.
Oestergaard S, Sondergaard BC, Hoegh-Andersen P, Henriksen K, Qvist P, Christiansen C, Tankó LB, Karsdal MA: Effects of ovariectomy and estrogen therapy on type II collagen degradation and structural integrity of articular cartilage in rats: implications of the time of initiation. Arthritis Rheum. 2006, 54: 2441-2451. 10.1002/art.22009.
Morisset S, Patry C, Lora M, de Brum-Fernandes AJ: Regulation of cyclooxygenase-2 expression in bovine chondrocytes in culture by interleukin 1β, tumor necrosis factor-α, glucocorticoids, and 17β-estradiol. J Rheumatol. 1998, 25: 1146-1153.
Claassen H, Schünke M, Kurz B: Estradiol protects cultured articular chondrocytes from oxygen-radical-induced damage. Cell Tissue Res. 2005, 319: 439-445. 10.1007/s00441-004-1029-9.
Scranton PE, McMaster JH, Diamond PE: Hormone suppression of DNA synthesis in cultured Hormone suppression of DNA synthesis in cultured chondrocyte and osteosarcoma cell line. Clin Orthop Relat Res. 1975, 112: 340-348. 10.1097/00003086-197510000-00036.
Mackintosh D, Mason RM: Pharmacological actions of 17 beta-oestradiol on articular cartilage chondrocytes and chondrosarcoma chondrocytes in the absence of oestrogen receptors. Biochim Biophys Acta. 1988, 964: 295-302.
Talwar RM, Wong BS, Svoboda K, Harper RP: Effects of estrogen on chondrocyte proliferation and collagen synthesis in skeletally mature articular cartilage. J Oral Maxillofac Surg. 2006, 64: 600-609. 10.1016/j.joms.2005.12.006.
Blanchard O, Tsagris L, Rappaport R, Duval-Beaupere G, Corvol M: Age-dependent responsiveness of rabbit and human cartilage cells to sex steroids in vitro. J Steroid Biochem Mol Biol. 1991, 40: 711-716. 10.1016/0960-0760(91)90295-G.
Dayani N, Corvol MT, Robel P, Eychenne B, Moncharmont B, Tsagris L, Rappaport R: Estrogen receptors in cultured rabbit articular chondrocytes: influence of age. J Steroid Biochem. 1988, 31: 351-356. 10.1016/0022-4731(88)90361-5.
Høegh-Andersen P, Tankó LB, Andersen TL, Lundberg CV, Mo JA, Heegaard AM, Delaissé JM, Christgau S: Ovariectomized rats as a model of postmenopausal osteoarthritis: validation and application. Arthritis Res Ther. 2004, 6: R169-180. 10.1186/ar1152.
Ham KD, Loeser RF, Lindgren BR, Carlson CS: Effects of long-term estrogen replacement therapy on osteoarthritis severity in cynomolgus monkeys. Arthritis Rheum. 2002, 46: 1956-1964. 10.1002/art.10406.
Ma HL, Blanchet TJ, Peluso D, Hopkins B, Morris EA, Glasson SS: Osteoarthritis severity is sex dependent in a surgical mouse model. Osteoarthritis Cartilage. 2007, 15: 695-700. 10.1016/j.joca.2006.11.005.
Ham KD, Carlson CS: Effects of estrogen replacement therapy on bone turnover in subchondral bone and epiphyseal metaphyseal cancellous bone of ovariectomized cynomolgus monkeys. J Bone Miner Res. 2004, 19: 823-829. 10.1359/JBMR.040309.
Olson EJ, Lindgren BR, Carlson CS: Effects of long-term estrogen replacement therapy on the prevalence and area of periarticular tibial osteophytes in surgically postmenopausal cynomolgus monkeys. Bone. 2007, 41: 282-289. 10.1016/j.bone.2007.04.175.
Quasnichka HL, Anderson-MacKenzie JM, Bailey AJ: Subchondral bone and ligament changes precede cartilage degradation in guinea pig osteoarthritis. Biorheology. 2006, 43: 389-397.
Castañeda S, Largo R, Calvo E, Rodríguez-Salvanés F, Marcos ME, Díaz-Curiel M, Herrero-Beaumont G: Bone mineral measurements of subchondral and trabecular bone in healthy and osteoporotic rabbits. Skeletal Radiol. 2006, 35: 34-41. 10.1007/s00256-005-0022-z.
Calvo E, Castañeda S, Largo R, Fernández-Valle ME, Rodríguez-Salvanés F, Herrero-Beaumont G: Osteoporosis increases the severity of cartilage damage in an experimental model of osteoarthritis in rabbits. Osteoarthritis Cartilage. 2007, 15: 69-77. 10.1016/j.joca.2006.06.006.
Gilsanz V, Roe TF, Gibbens DT, Schulz EE, Carlson ME, Gonzalez O, Boechat MI: Effect of sex steroids on peak bone density of growing rabbits. Am J Physiol Endocrinol Metab. 1988, 255: E416-421.
Castañeda S, Calvo E, Largo R, González-González R, de la Piedra C, Díaz-Curiel M, Herrero-Beaumont G: Characterization of a new experimental model of osteoporosis in rabbits. J Bone Miner Metab. 2008, 26: 53-59. 10.1007/s00774-007-0797-1.
Strickland SM, Belknap TW, Turner SA, Wright TM, Hannafin JA: Lack of hormonal influences on mechanical properties of sheep knee ligaments. Am J Sports Med. 2003, 31: 210-215.
Komulainen J, Koskinen SO, Kalliokoski R, Takala TE, Vihko V: Gender differences in skeletal muscle fibre damage after eccentrically biased downhill running in rats. Acta Physiol Scand. 1999, 165: 57-63. 10.1046/j.1365-201x.1999.00481.x.
Bergink AP, van Meurs JB, Loughlin J, Arp PP, Fang Y, Hofman A, van Leeuwen JP, van Duijn CM, Uitterlinden AG, Pols HA: Estrogen receptor alpha gene haplotype is associated with radiographic osteoarthritis of the knee in elderly men and women. Arthritis Rheum. 2003, 48: 1913-1922. 10.1002/art.11046.
Valdes AM, Van Oene M, Hart DJ, Surdulescu GL, Loughlin J, Doherty M, Spector TD: Reproducible genetic associations between candidate genes and clinical knee osteoarthritis in men and women. Arthritis Rheum. 2006, 54: 533-539. 10.1002/art.21621.
Jin SY, Hong SJ, Yang HI, Park SD, Yoo MC, Lee HJ, Hong MS, Park HJ, Yoon SH, Kim BS, Yim SV, Park HK, Chung JH: Estrogen receptor-alpha gene haplotype is associated with primary knee osteoarthritis in Korean population. Arthritis Res Ther. 2004, 6: R415-421. 10.1186/ar1207.
Loughlin J, Sinsheimer JS, Mustafa Z, Carr AJ, Clipsham K, Bloomfield VA, Chitnavis J, Bailey A, Sykes B, Chapman K: Association analysis of the vitamin D receptor gene, the type I collagen gene COL1A1, and the estrogen receptor gene in idiopathic osteoarthritis. J Rheumatol. 2000, 27: 779-784.
Lian K, Lui L, Zmuda JM, Nevitt MC, Hochberg MC, Lee JM, Li J, Lane NE: Estrogen receptor alpha genotype is associated with a reduced prevalence of radiographic hip osteoarthritis in elderly Caucasian women. Osteoarthritis Cartilage. 2007, 15: 972-978. 10.1016/j.joca.2007.02.020.
Felson DT, Zhang Y, Hannan MT, Naimark A, Weissman BN, Aliabadi P, Levy D: The incidence and natural history of knee osteoarthritis in the elderly. The Framingham Osteoarthritis Study. Arthritis Rheum. 1995, 38: 1500-1505. 10.1002/art.1780381017.
Wood P: Age and the rheumatic diseases. Population Studies of the Rheumatic Diseases. Edited by: Bennett PH, Wood PH. 1982, Amsterdam: Excerpta Medica, 4: 26-37.
Nadkar MY, Samant RS, Vaidya SS, Borges NE: Relationship between osteoarthritis of knee and menopause. J Assoc Phys India. 1999, 47: 1161-1163.
Kellgren JH, Moore R: Generalized osteoarthritis and Heberdens nodes. BMJ. 1952, 1: 181-187. 10.1136/bmj.1.4751.181.
Felson DT, Nevitt MC: The effects of estrogen on osteoarthritis. Curr Opin Rheumatol. 1998, 10: 269-272. 10.1097/00002281-199805000-00019.
Punzi L, Ramonda R, Sfriso P: Erosive osteoarthritis. Best Pract Res Clin Rheumatol. 2004, 18: 739-758. 10.1016/j.berh.2004.05.010.
Sowers MR, McConnell D, Jannausch M, Buyuktur AG, Hochberg M, Jamadar DA: Estradiol and its metabolites and their association with knee osteoarthritis. Arthritis Rheum. 2006, 54: 2481-2487. 10.1002/art.22005.
Sipilä S: Body composition and muscle performance during menopause and hormone replacement therapy. J Endocrinol Invest. 2003, 26: 893-901.
Slemenda C, Heilman DK, Brandt KD, Katz BP, Mazzuca SA, Braunstein EM, Byrd D: Reduced quadriceps strength relative to body weight: a risk factor for knee osteoarthritis in women?. Arthritis Rheum. 1998, 41: 1951-1959. 10.1002/1529-0131(199811)41:11<1951::AID-ART9>3.0.CO;2-9.
Sipilä S, Poutamo J: Muscle performance, sex hormones and training in peri-menopausal and post-menopausal women. Scand J Med Sci Sports. 2003, 13: 19-25. 10.1034/j.1600-0838.2003.20210.x.
Sharma L, Lou C, Felson DT, Dunlop DD, Kirwan-Mellis G, Hayes KW, Weinrach D, Buchanan TS: Laxity in healthy and osteoarthritic knees. Arthritis Rheum. 1999, 42: 861-870. 10.1002/1529-0131(199905)42:5<861::AID-ANR4>3.0.CO;2-N.
Karsdal MA, Leeming DJ, Dam EB, Henriksen K, Alexandersen P, Pastoureau P, Altman RD, Christiansen C: Should subchondral bone turnover be targeted when treating osteoarthritis?. Osteoarthritis Cartilage. 2008, 16: 638-646. 10.1016/j.joca.2008.01.014.
Hopwood B, Tsykin A, Findlay DM, Fazzalari NL: Microarray gene expression profiling of osteoarthritic bone suggests altered bone remodelling, WNT and transforming growth factor-beta/bone morphogenic protein signalling. Arthritis Res Ther. 2007, 9: R100-10.1186/ar2301.
Sanchez C, Deberg MA, Piccardi N, Msika P, Reginster JY, Henrotin YE: Osteoblasts from the sclerotic subchondral bone downregulate aggrecan but upregulate metalloproteinases expression by chondrocytes. This effect is mimicked by inter-leukin-6, -1beta and oncostatin M pre-treated non-sclerotic osteoblasts. Osteoarthritis Cartilage. 2005, 13: 979-987. 10.1016/j.joca.2005.03.008.
Stewart A, Black A: Bone mineral density in osteoarthritis. Curr Opin Rheumatol. 2000, 12: 464-467. 10.1097/00002281-200009000-00021.
Dequeker J, Aerssens J, Luyten FP: Osteoarthritis and osteoporosis: clinical and research evidence of inverse relationship. Aging Clin Exp Res. 2003, 15: 426-439.
Ng MC, Revell PA, Beer M, Boucher BJ, Cohen RD, Currey LF: Incidence of metabolic bone disease in rheumatoid arthritis and osteoarthritis. Ann Rheum Dis. 1984, 43: 370-377. 10.1136/ard.43.3.370.
Perilli E, Baleani M, Ohman C, Baruffaldi F, Viceconti M: Structural parameters and mechanical strength of cancellous bone in the femoral head in osteoarthritis do not depend on age. Bone. 2007, 41: 760-768. 10.1016/j.bone.2007.07.014.
Bruyere O, Dardenne C, Lejeune E, Zegels B, Pahaut A, Richy F, Seidel L, Ethgen O, Henrotin Y, Reginster JY: Subchondral tibial bone mineral density predicts future joint space narrowing at the medial femorotibial compartment in patients with knee osteoarthritis. Bone. 2003, 32: 541-545. 10.1016/S8756-3282(03)00059-0.
Karvonen RL, Miller PR, Nelson DA, Granda JL, Fernandez-Madrid F: Periarticular osteoporosis in osteoarthritis of the knee. J Rheumatol. 1998, 25: 2187-2194.
Klinge CM: Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29: 2905-2919. 10.1093/nar/29.14.2905.
Gruber CJ, Gruber DM, Gruber IM, Wieser F, Huber JC: Anatomy of the estrogen response element. Trends Endocrinol Metab. 2004, 15: 73-78. 10.1016/j.tem.2004.01.008.
Ellmann S, Sticht H, Thiel F, Beckmann MW, Strick R, Strissel PL: Estrogen and progesterone receptors: from molecular structures to clinical targets. Cell Mol Life Sci. 2009, 66: 2405-2426. 10.1007/s00018-009-0017-3.
Cheskis BJ, Greger JG, Nagpal S, Freedman LP: Signaling by estrogens. J Cell Physiol. 2007, 213: 610-617. 10.1002/jcp.21253.
Weihua Z, Andersson S, Cheng G, Simpson ER, Warner M, Gustafsson JA: Update on estrogen signaling. FEBS Lett. 2003, 546: 17-24. 10.1016/S0014-5793(03)00436-8.
Matthews J, Gustafsson JA: Estrogen signaling: a subtle balance between ER alpha and ER beta. Mol Interv. 2003, 3: 281-292. 10.1124/mi.3.5.281.
Hirata S, Shoda T, Kato J, Hoshi K: Isoform/variant mRNAs for sex steroid hormone receptors in humans. Trends Endocrinol Metab. 2003, 14: 124-129. 10.1016/S1043-2760(03)00028-6.
Bord S, Horner A, Beavan S, Compston J: Estrogen receptors alpha and beta are differentially expressed in developing human bone. J Clin Endocrinol Metab. 2001, 86: 2309-2314. 10.1210/jc.86.5.2309.
Onoe Y, Miyaura C, Ohta H, Nozawa S, Suda T: Expression of estrogen receptor beta in rat bone. Endocrinology. 1997, 138: 4509-4512. 10.1210/en.138.10.4509.
Oreffo RO, Kusec V, Virdi AS, Flanagan AM, Grano M, Zambonin-Zallone A, Triffitt JT: Expression of estrogen receptor-alpha in cells of the osteoclastic lineage. Histochem Cell Biol. 1999, 111: 125-133. 10.1007/s004180050342.
Faryniarz DA, Bhargava M, Lajam C, Attia ET, Hannafin JA: Quantitation of estrogen receptors and relaxin binding in human anterior cruciate ligament fibroblasts. In Vitro Cell Dev Biol Anim. 2006, 42: 176-181. 10.1290/0512089.1.
Wiik A, Glenmark B, Ekman M, Esbjörnsson-Liljedahl M, Johansson O, Bodin K: Oestrogen receptor beta is expressed in adult human skeletal muscle both at the mRNA and protein level. Acta Physiol Scand. 2003, 179: 381-387. 10.1046/j.0001-6772.2003.01186.x.
Wiik A, Gustafsson T, Esbjörnsson M, Johansson O, Ekman M, Sundberg CJ, Jansson E: Expression of oestrogen receptor alpha and beta is higher in skeletal muscle of highly endurance-trained than of moderately active men. Acta Physiol Scand. 2005, 184: 105-112. 10.1111/j.1365-201X.2005.01433.x.
Sims NA, Dupont S, Krust A, Clement-Lacroix P, Minet D, Resche-Rigon M, Gaillard-Kelly M, Baron R: Deletion of estrogen receptors reveals a regulatory role for estrogen receptors-beta in bone remodeling in females but not in males. Bone. 2002, 30: 18-25. 10.1016/S8756-3282(01)00643-3.
Sniekers YH, van Osch GJ, Ederveen AG, Inzunza J, Gustafsson JA, van Leeuwen JP, Weinans H: Development of osteoarthritic features in estrogen receptor knockout mice. Osteoarthritis Cartilage. 2009, Apr 17 [Epub ahead of print].
Brown M, Ning J, Ferreira JA, Bogener JL, Lubahn DB: Estrogen receptor-{alpha} and -{beta} and aromatase knockout effects on lower limb muscle mass and contractile function in female mice. Am J Physiol Endocrinol Metab. 2009, 296: E854-861. 10.1152/ajpendo.90696.2008.
Warden SJ, Saxon LK, Castillo AB, Turner CH: Knee ligament mechanical properties are not influenced by estrogen or its receptors. Am J Physiol Endocrinol Metab. 2006, 290: E1034-1040. 10.1152/ajpendo.00367.2005.
Safe S, Kim K: Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J Mol Endocrinol. 2008, 41: 263-275. 10.1677/JME-08-0103.
Lonard DM, O'malley BW: Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Mol Cell. 2007, 27: 691-700. 10.1016/j.molcel.2007.08.012.
Richette P, Dumontier MF, Tahiri K, Widerak M, Torre A, Benallaoua M, Rannou F, Corvol MT, Savouret JF: Oestrogens inhibit interleukin 1beta-mediated nitric oxide synthase expression in articular chondrocytes through nuclear factor-kappa B impairment. Ann Rheum Dis. 2007, 66: 345-350. 10.1136/ard.2006.059550. Erratum in: Ann Rheum Dis 2007, 66:708
Lu T, Achari Y, Rattner JB, Hart DA: Evidence that estrogen receptor beta enhances MMP-13 promoter activity in HIG-82 cells and that this enhancement can be influenced by ligands and involves specific promoter sites. Biochem Cell Biol. 2007, 85: 326-336. 10.1139/O07-016.
Syed FA, Mödder UI, Fraser DG, Spelsberg TC, Rosen CJ, Krust A, Chambon P, Jameson JL, Khosla S: Skeletal effects of estrogen are mediated by opposing actions of classical and non-classical estrogen receptor pathways. J Bone Miner Res. 2005, 20: 1992-2001. 10.1359/JBMR.050713.
Levin ER: Cellular functions of plasma membrane estrogen receptors. Steroids. 2002, 67: 471-475. 10.1016/S0039-128X(01)00179-9.
Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER: A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005, 307: 1625-1630. 10.1126/science.1106943.
Wong CW, McNally C, Nickbarg E, Komm BS, Cheskis BJ: Estrogen receptor interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. Proc Natl Acad Sci USA. 2002, 99: 14783-14788. 10.1073/pnas.192569699.
Kinney RC, Schwartz Z, Week K, Lotz MK, Boyan BD: Human articular chondrocytes exhibit sexual dimorphism in their responses to 17b-estradiol. Osteoarthritis Cartilage. 2005, 13: 330-337. 10.1016/j.joca.2004.12.003.
Boyan BD, Schwartz Z: Rapid vitamin D-dependent PKC signaling shares features with estrogen-dependent PKC signaling in cartilage and bone. Steroids. 2004, 69: 591-597. 10.1016/j.steroids.2004.05.008.
McMillan J, Fatehi-Sedeh S, Sylvia VL, Bingham V, Zhong M, Boyan BD, Schwartz Z: Sex-specific regulation of growth plate chondrocytes by estrogen is via multiple MAP kinase signaling pathways. Biochem Biophys Acta. 2006, 1763: 381-392. 10.1016/j.bbamcr.2006.02.010.
Eckstein Ekstein J, Nasatzky E, Boyan BD, Ornoy A, Schwartz Z: Growth-plate chondrocytes respond to 17beta-estradiol with sex-specific increases in IP3 and intracellular calcium ion signalling via a capacitative entry mechanism. Steroids. 2005, 70: 775-786. 10.1016/j.steroids.2005.04.007.
Chen JR, Plotkin LI, Aguirre JI, Han L, Jilka RL, Kousteni S, Bellido T, Manolagas SC: Transient versus sustained phosphorylation and nuclear accumulation of ERKs underlie anti-versus pro-apoptotic effects of estrogens. J Biol Chem. 2005, 280: 4632-4638. 10.1074/jbc.M411530200.
Boland R, Vasconsuelo A, Milanesi L, Ronda AC, de Boland AR: 17beta-Estradiol signaling in skeletal muscle cells and its relationship to apoptosis. Steroids. 2008, 73: 859-863. 10.1016/j.steroids.2007.12.027.
Almeida M, Han L, O'brien CA, Kousteni S, Manolagas SC: Classical genotropic versus kinase-initiated regulation of gene transcription by the estrogen receptor alpha. Endocrinology. 2006, 147: 1986-1996. 10.1210/en.2005-1314.
Osborne CK, Shou J, Massarweh S, Schiff R: Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin Cancer Res. 2005, 11: 865s-870s.
Fagan DH, Yee D: Crosstalk between IGF1R and estrogen receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia. 2008, 13: 423-429. 10.1007/s10911-008-9098-0.
Hawse JR, Subramaniam M, Ingle JN, Oursler MJ, Rajamannan NM, Spelsberg TC: Estrogen-TGFbeta cross-talk in bone and other cell types: role of TIEG, Runx2, and other transcription factors. J Cell Biochem. 2008, 103: 383-392. 10.1002/jcb.21425.
Heino TJ, Hentunen TA, Väänänen HK: Osteocytes inhibit osteoclastic bone resorption through transforming growth factor-beta: enhancement by estrogen. J Cell Biochem. 2002, 85: 185-197. 10.1002/jcb.10109.
Kouzmenko AP, Takeyama K, Ito S, Furutani T, Sawatsubashi S, Maki A, Suzuki E, Kawasaki Y, Akiyama T, Tabata T, Kato S: Wnt/beta-catenin and estrogen signaling converge in vivo. J Biol Chem. 2004, 279: 40255-40258. 10.1074/jbc.C400331200.
Armstrong VJ, Muzylak M, Sunters A, Zaman G, Saxon LK, Price JS, Lanyon LE: Wnt/beta-catenin signaling is a component of osteoblastic bone cell early responses to load-bearing and requires estrogen receptor alpha. J Biol Chem. 2007, 282: 20715-20727. 10.1074/jbc.M703224200.
Richmond RS, Carlson CS, Register TC, Shanker G, Loeser RF: Functional estrogen receptors in adult articular cartilage. Estrogen replacement therapy increases chondrocyte synthesis of proteoglycans and insulin-like growth factor binding protein 2. Arthritis Rheum. 2000, 43: 2081-2090. 10.1002/1529-0131(200009)43:9<2081::AID-ANR20>3.0.CO;2-I.
Blaney Davidson EN, Kraan van der PM, Berg van den WB: TGF-beta and osteoarthritis. Osteoarthritis Cartilage. 2007, 15: 597-604. 10.1016/j.joca.2007.02.005.
Johnson ML, Kamel MA: The Wnt signaling pathway and bone metabolism. Curr Opin Rheumatol. 2007, 19: 376-382. 10.1097/BOR.0b013e32816e06f9.
Corr M: Wnt-beta-catenin signaling in the pathogenesis of osteoarthritis. Nat Clin Pract Rheumatol. 2008, 4: 550-556. 10.1038/ncprheum0904.
Tsai CL, Liu TK: Estradiol-induced knee osteoarthrosis in ovariectomized rabbits. Clin Orthop Relat Res. 1993, 291: 295-302.
Ng MC, Harper RP, Le CT, Wong BS: Effects of estrogen on the condylar cartilage of the rat mandible in organ culture. J Oral Maxillofac Surg. 1999, 57: 818-823. 10.1016/S0278-2391(99)90823-6.
Rosner IA, Boja BA, Goldberg VM, Moskowitz RW: Tamoxifen therapy in experimental osteoarthritis. Curr Ther Res. 1983, 34: 409-414.
Tsai CL, Liu TK: Inhibition of estradiol-induced early osteoarthritic changes by tamoxifen. Life Sci. 1992, 50: 1943-1951. 10.1016/0024-3205(92)90523-R.
Christgau S, Tankó LB, Cloos PA, Mouritzen U, Christiansen C, Delaissé JM, Høegh-Andersen P: Suppression of elevated cartilage turnover in postmenopausal women and in ovariectomized rats by estrogen and a selective estrogen-receptor modulator (SERM). Menopause. 2004, 11: 508-518. 10.1097/01.WCB.0000121484.18437.98.
Olson EJ, Lindgren BR, Carlson CS: Effects of long-term estrogen replacement therapy on bone turnover in periarticular tibial osteophytes in surgically postmenopausal cynomolgus monkeys. Bone. 2008, 42: 907-913. 10.1016/j.bone.2007.12.007.
Fernihough JK, Richmond RS, Carlson CS, Cherpes T, Holly JM, Loeser RF: Estrogen replacement therapy modulation of the insulin-like growth factor system in monkey knee joints. Arthritis Rheum. 1999, 42: 2103-2111. 10.1002/1529-0131(199910)42:10<2103::AID-ANR10>3.0.CO;2-S.
Moran AL, Nelson SA, Landisch RM, Warren GL, Lowe DA: Estradiol replacement reverses ovariectomy-induced muscle contractile and myosin dysfunction in mature female mice. J Appl Physiol. 2007, 102: 1387-1393. 10.1152/japplphysiol.01305.2006.
Nevitt MC, Cummings SR, Lane NE, Hochberg MC, Scott JC, Pressman AR, Genant HK, Cauley JA: Association of estrogen replacement therapy with the risk of osteoarthritis of the hip in elderly white women. Study of Osteoporotic Fractures Research Group. Arch Intern Med. 1996, 156: 2073-2080. 10.1001/archinte.156.18.2073.
Spector TD, Nandra D, Hart DJ, Doyle DV: Is hormone replacement therapy protective for hand and knee osteoarthritis in women? The Chingford Study. Ann Rheum Dis. 1997, 56: 432-444. 10.1136/ard.56.7.432.
Hart DJ, Doyle DV, Spector TD: Incidence and risk factors for radiographic knee osteoarthritis in middle-aged women: the Chingford Study. Arthritis Rheum. 1999, 42: 17-24. 10.1002/1529-0131(199901)42:1<17::AID-ANR2>3.0.CO;2-E.
Hannan MT, Felson DT, Anderson JJ, Naimark A, Kannel WB: Estrogen use and radiographic osteoarthritis of the knee in women: the Framingham Osteoarthritis Study. Arthritis Rheum. 1990, 33: 525-532. 10.1002/art.1780330410.
Zhang Y, McAlindon TE, Hannan MT, Chaisson CE, Klein R, Wilson PW, Felson DT: Estrogen replacement therapy and worsening of radiographic knee osteoarthritis: the Framingham Study. Arthritis Rheum. 1998, 41: 1867-1873. 10.1002/1529-0131(199810)41:10<1867::AID-ART20>3.0.CO;2-W.
de Klerk BM, Schiphof D, Groeneveld FP, Koes BW, van Osch GJ, van Meurs JB, Bierma-Zeinstra SM: Limited evidence for a protective effect of unopposed oestrogen therapy for osteoarthritis of the hip: a systematic review. Rheumatology. 2009, 48: 104-12. 10.1093/rheumatology/ken390.
Cirillo DJ, Wallace RB, Wu L, Yood RA: Effect of hormone therapy on risk of hip and knee joint replacement in the Women's Health Initiative. Arthritis Rheum. 2006, 54: 3194-3204. 10.1002/art.22138.
Carbone LD, Nevitt MC, Wildy K, Barrow KD, Harris F, Felson D, Peterfy C, Visser M, Harris TB, Wang BW, Kritchevsky SB, Health, Aging and Body Composition Study: The relationship of antiresorptive drug use to structural findings and symptoms of knee osteoarthritis. Arthritis Rheum. 2004, 50: 3516-3525. 10.1002/art.20627.
Taaffe DR, Sipilä S, Cheng S, Puolakka J, Toivanen J, Suominen H: The effect of hormone replacement therapy and/or exercise on skeletal muscle attenuation in postmenopausal women: a yearlong intervention. Clin Physiol Funct Imaging. 2005, 25: 297-304. 10.1111/j.1475-097X.2005.00628.x.
Harnish DC: Estrogen receptor ligands in the control of pathogenic inflammation. Curr Opin Investig Drugs. 2006, 7: 997-1001.
Shelly W, Draper MW, Krishnan V, Wong M, Jaffe RB: Selective estrogen receptor modulators: an update on recent clinical findings. Obstet Gynecol Surv. 2008, 63: 163-181.
Harris HA, Albert LM, Leathurby Y, Malamas MS, Mewshaw RE, Miller CP, Kharode YP, Marzolf J, Komm BS, Winneker RC, Frail DE, Henderson RA, Zhu Y, Keith JC: Evaluation of an estrogen receptor-β agonist in animal models of human disease. Endocrinology. 2003, 144: 4241-4249. 10.1210/en.2003-0550.
Keith JC, Albert LM, Leathurby Y, Follettie M, Wang L, Borges-Marcucci L, Chadwick CC, Steffan RJ, Harnish DC: The utility of pathway selective estrogen receptor ligands that inhibit nuclear factor-kappa B transcriptional activity in models of rheumatoid arthritis. Arthritis Res Ther. 2005, 7: R427-438. 10.1186/ar1692.
Acknowledgements
The authors would like to thank Dr Mark Sefton for his assistance in editing the manuscript, and Dr Lenny A Mendoza Torres for her help in preparing the figures. Gabriel Herrero-Beaumont was supported by the SAF2006-02704 research grant from the Spanish Science and Education Ministry, 2006-2008.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Roman-Blas, J.A., Castañeda, S., Largo, R. et al. Osteoarthritis associated with estrogen deficiency. Arthritis Res Ther 11, 241 (2009). https://doi.org/10.1186/ar2791
Published:
DOI: https://doi.org/10.1186/ar2791