
Abstract
The past 10 years have seen the description of families of receptors that drive proinflammatory cytokine production in infection and tissue injury. Two major classes have been examined in the context of inflammatory joint disease - the Toll-like receptors (TLRs) and NOD-like receptors (NLRs). TLRs such as TLR2 and TLR4 are being implicated in the pathology of rheumatoid arthritis, ankylosing spondylitis, lyme arthritis and osteoarthritis. Nalp3 has been identified as a key NLR for IL-1β production and has been shown to have a particular role in gout. These findings present new therapeutic opportunities, possibly allowing for the replacement of biologics with small molecule inhibitors.
Similar content being viewed by others
Introduction
Proinflammatory cytokines such as TNF, IL-6 and IL-1 have proven to be excellent therapeutic targets for diseases such as rheumatoid arthritis (RA). More recently, however, attention has focused on the mechanisms whereby these cytokines are induced. In this regard there has been remarkable progress in the elucidation of receptors that drive their production as well as other inflammatory mediators. This progress has led to a renaissance of interest in innate immunity among immunologists, since these receptors also sense microbial products to drive host defense.
Two particular classes - the Toll-like receptors (TLRs) and NOD-like receptors (NLRs), which are pattern recognition receptors (PRRs) - have been most extensively studied. Certain TLRs (for example, TLR2, TLR4 and TLR9) and certain NLRs (for example, Nalp3) have been implicated in various inflammatory arthopathies. More recently evidence has been presented that these TLRs and NLRs might also be activated by noninfectious endogenous signals, making them even more attractive as important drivers of cytokines in diseases with no obvious infection.
In the present review we will summarise the current state of knowledge in TLRs and NLRs, and also speculate on their roles in the pathogenesis of autoinflammatory joint diseases.
Toll-like receptors
The past 10 years have seen over 11,000 papers published on TLRs, which is a testament to the importance placed upon them by inflammation biologists and immunologists. Ten TLRs occur in humans, and the roles of nine of them (TLR1 to TLR9) have been determined [1].
TLR2 senses lipopeptides from bacteria, with TLR1/2 dimers sensing triacylated lipopeptides and TLR2/6 dimers sensing diacylated lipopeptides. In addition, TLR2 also senses zymosan from fungi. The structure of the TLR1/2 dimer has been solved [2], as has the structure of TLR4 in complex with its ligand lipopolysacharide from Gram-negative bacteria that are presented to TLR4 by MD2 [3]. TLR4 can also sense F protein from respiratory syncytial virus and glycerophosphatidylinositol anchors from parasites [4, 5]. This provides a receptor repertoire to respond to all pathogens that infect humans.
The signaling pathways activated by TLRs have also been worked out in great detail and involve the selective recruitment of adapter proteins (MyD88, Mal, Trif and Tram) [6]. These lead to activation of NF-κB, which is a major response to TLRs. Certain TLRs (TLR4 and nucleic acid-sensing TLRs) can also engage with a pathway leading to the activation of the transcription factor interferon regulatory factor-3. Both NF-κB and interferon regulatory factor-3 are required for the induction of a wide range of cytokines.
NOD-like receptors
NLRs are intracellular sensors of pathogen-associated or endogenous danger-associated molecular patterns. The NLR family consists of 22 cytoplasmic proteins including the NOD and NALP subfamilies, with the 14 NALPs representing the largest subfamily. NLR family members share common structural features, including a nucleotide binding domain (nucleotide binding site or NACHT domain) central to the molecule, flanked by a leucine rich-repeat domain at the C-terminus and a caspase-recruitment domain and a pyrin domain at the N-terminus.
The best characterised NLR is NALP3, which when activated forms a large oligomer able to interact with intermediate proteins ASC and Cardinal, creating a complex able to recruit procaspase-1. Through an autocatalytic process, procaspase-1 is then activated - resulting in a multimeric structure termed the inflammasome, which is able to induce maturation and secretion of proinflammatory cytokines IL-1β and IL-18 [7].
Gain of function mutations in the NALP3 gene leading to elevated levels of processed IL-1β cause hereditary periodic fever syndromes in humans, including Mucke-Wells syndrome, chronic infantile cutaneous neurologic articular syndrome and familial cold-induced autoinflammatory syndrome [8]. Fever, joint pain and systemic inflammation are common features of these disorders and provided the first clue that the inflammasome has a potential role in rheumatic diseases [9]. The effectiveness of IL-1β blockade (Anakinra) in treating inherited periodic fever syndromes has transformed the understanding and management of these disorders and has implications for future therapies in rheumatic diseases.
Important links and synergies are evident between TLRs and NLRs. TLRs are required to induce pro-IL1β, and the Nalps then activate caspase-1 to process it, so both act in concert for IL-1 production [10]. Another important aspect is the link between these receptors and adaptive immunity. Nalp3 has been shown to be a target for the adjuvant Alum, although whether it is required for antibody production is less clear. TLRs, however, are important for inducing the T-cell co-stimulatory molecules CD80 and CD86. This is particularly the case with TLR4, which achieves this via induction of IFNβ [11]. B cells and T cells have also been shown to express certain TLRs - TLR9 has been shown to induce B-cell proliferation [12], whilst TLR2 has been shown to be present on regulatory T cells and to activate them [13]. These kinds of studies highlight the role of innate immunity in the adaptive response, and the two responses are increasingly seen as inter-linked.
Rheumatoid arthritis
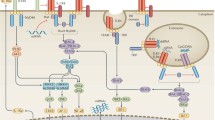
There has been a longstanding hypothesis that infection plays a role in the initiation of RA (Figure 1). Molecules of microbial origin have been found in the joints of patients with RA [14, 15], where they can trigger inflammatory reactions through PRRs. These inflammatory reactions damage the host tissue, releasing molecules (danger signals) that can activate the PRRs resulting in vicious cycles of inflammation.

Signaling through pathogen-associated and damage-associated molecular patterns drives chronic inflammation in diseases like rheumatoid arthritis. Bacterial DNA, peptidoglycans, muramyl dipeptide and viral molecules have been found in arthritic joints. These microbial pathogen-associated molecular patterns (PAMPs) can drive inflammation through the membrane-bound (Toll-like receptor (TLR)) and cytosolic (NOD-like receptor (NLR)) pattern recognition receptors (PRRs). The resulting release in inflammatory cytokines can drive the damage of host tissue releasing damage-associated molecular patterns (DAMPs), such as high-mobility group box protein 1, GP96, heat shock proteins and ATP, which also activate both types of PRR resulting in a vicious cycle of inflammation.
This sterile inflammation induced by endogenous danger signals released from the inflamed host tissue is thought to lead to the pathological joint destruction associated with RA. There is increasing evidence that TLRs, and more recently NLRs, have a role in RA pathology.
Ospelt and colleagues comparatively analysed the expression of TLRs in synovial tissues during the early and late stages of RA, and found that TLR3 and TLR4 were elevated in both early and late RA samples compared with samples from osteoarthritis (OA) synovium [16]. These results concur with studies from Brentano and colleagues, who also detected elevated levels of TLR3 expression in RA synovial fibroblasts over OA synovial fibroblasts [17]. Similarly, elevated levels of TLR7 have also been detected in synovium from RA patients compared with OA patients or healthy volunteers [18]. In addition to synovial fibroblasts, differences in TLR expression/activity have also been detected in macrophages isolated from synovium of RA patients. Huang and colleagues discovered elevated levels of TLR2 and TLR4 activity in macrophages isolated from RA synovium compared with control synovium [19]. Spontaneous production of proinflammatory cytokines and matrix metalloproteinases from RA synovial membrane cultures has been shown to be inhibited by overexpressing dominant negative constructs of Mal and MyD88, essential adaptors molecules for TLR2 and TLR4 signaling [20].
A later study investigating the use of a novel TLR4 antagonist has shown the most convincing evidence for TLR involvement in RA, as shown in Figure 2[21]. In this study, two mouse models of RA were used to test a TLR4 antagonist for efficacy. An IL1-receptor antagonist knockout model, where the mice develop arthritis spontaneously, was run alongside a collagen-induced arthritis model that requires the use of an adjuvant containing TLR ligands. In both models the TLR4 antagonist showed impressive therapeutic effects. Another study by the same group crossed TLR2, TLR4 and TLR9 knockout mice with the IL1-receptor antagonist knockout mice that spontaneously develop arthritis [22]. Agreeing with the results from their TLR4 antagonist study, Abdollahi-Roodsaz and colleagues found that IL1rn-/-TLR4-/- animals are protected against arthritis whereas IL1rn-/-TLR2-/-animals develop a more severe arthritis - suggesting an anti-inflammatory role for TLR2 in this model. A lack of TLR9 did not affect the progression of arthritis. The anti-inflammatory nature of TLR2 in the IL1-receptor antagonist knockout models is in contrast to results obtained in a streptococcal cell wall induced model of arthritis, where mice deficient for TLR2 were shown to have a reduced severity of arthritis [23]. TLR4 has been shown to be involved in the chronic erosive stage of arthritis in this model of disease [24].

Treating spontaneous arthritis with a TLR4 antagonist suppresses the clinical and histological characteristics of arthritis. Abdollahi-Roodsaz and colleagues have recently shown that treating collagen-induced arthritis (left-hand side) with a TLR4 antagonist suppresses the clinical and histological characteristics of arthritis (right-hand side). Histological images of knee joints are shown, stained with hematoxylin and eosin. Arrow indicates inflammatory cell influx and chondrocyte cell death. Image taken from [21]. Reproduced with permission of John Wiley and Sons.
As already mentioned, the role of TLRs in RA is believed to be driven by inflammation in response to danger signals (endogenous host cell molecules released from stressed cells) as well as TLR ligands of microbial origin. Similar to the microbial TLR ligands, endogenous TLR ligands have been found in the joints or serum of RA patients and their levels have been correlated with disease activity scores [25]. These ligands - including heat shock proteins, fibronectin, high-mobility group box chromosomal protein-1 (HMGB1) and breakdown products of heparan sulfate and hyaluronic acid - activate TLR2, TLR4, or both. The most recent addition to the growing list of endogenous TLR ligands is GP96 [26]. GP96 is a heat shock glycoprotein detected at high levels in RA synovial tissues that is capable of activating TLRs. Like HMGB1, this endogenous ligand has been shown to drive inflammation by signaling through both TLR2 and TLR4. Considering the extensive evidence linking TLR signaling and RA pathology, it is surprising that no TLR polymorphisms have been identified involved in the susceptibility and severity of RA [16, 27, 28].
While TLRs appear to be the principal PRRs implicated in RA pathology, evidence is emerging that NLRs may also have a role in RA. NOD1 and NOD2 have been shown to be expressed in RA synovial tissue samples, and the microbial ligand for NOD2, muramyl dipeptide, has been detected in RA synovium [29, 30]. Using NOD1 and NOD2 knockout mice, Joosten and colleagues have shown a proinflammatory role for NOD2 and an anti-inflammatory role for NOD1 in a streptococcal cell wall induced model of arthritis [30].
Lyme arthritis and TLR2
Lyme arthritis is caused by infection with the tick-borne spirochete Borrelia burgdorferi. A subacute inflammatory arthritis develops in 60% of individuals not treated at the time of the tick bite, and is associated with invasion of the joint tissue by spirochetes. Immune responses of the host toward B. burgdorferi are predominantly mediated by the recognition of proteins modified with tripalmitoyl-S-glyceryl-cysteine by TLR2 [31]. TLR2 knockout mice have been shown to be hyporesponsive to vaccination with lipopeptides, and hyporesponsiveness in humans is linked with low levels of TLR1 expression [32]. In contrast to the studies in the TLR2 knockout mice, a polymorphism resulting in a nonfunctional TLR2 receptor (Arg753Gln) in vitro has been shown to be protective from the clinical symptoms of late-stage infection with B. burgdorferi [33].
Systemic lupus erythematosus, Toll-like receptors and the AIM2 inflammasome
Systemic lupus erythematosus (SLE) is a prototypic systemic autoimmune disease, the cause of which has not yet been fully elucidated. Immune complexes of autoantibodies to chromatin and RNA protein particles (snRNP) are characteristic of SLE and play an important role in the pathogenesis of the disease. Increased levels of serum IFNα have been found in many patients with SLE, and these levels correlate with disease severity and disease markers such as the DNA autoantibodies. Evidence for the crucial role of type 1 interferon in the pathology of lupus comes indirectly from findings that patients with nonautoimmune disorders treated with recombinant IFNα produce autoantibodies to DNA and develop clinical syndromes that resemble SLE [34, 35].
There is good evidence that TLRs are involved in SLE. TLR9-expressing B cells are expanded in SLE patients with active disease, and this is correlated with levels of autoantibodies against DNA [36]. Activation of endosomal TLRs is believed to drive the elevated levels of IFNα that promote and maintain SLE disease progression. Nephritis is a condition associated with SLE, and in a murine model of the disease (MRLlpr/lpr) immunisation with unmethylated CpG, an exogenous TLR9 ligand, aggravates the condition [37]. This is consistent with observed association of lupus flares with viral infection. Using TLR7 and TLR9 oligonucleotide-based inhibitors, mammalian DNA and RNA in the form of immune complexes from SLE patient serum have been shown to act as endogenous ligands for TLR7 and TLR9, respectively [38]. In lupus-prone (NZB × NZW)F1 mice that spontaneously develop symptoms similar to human lupus, administration of a TLR7/TLR9 dual oligonucleotide inhibitor showed efficacy at suppressing the production of autoantibodies, reducing kidney damage and increasing survival of treated mice [39]. In the MRLlpr/lpr lupus model, mice deficient for MyD88 failed to produce DNA autoantibodies [40]. In the same lupus animal model, TLR7 deficiency has shown reduced autoimmune disease as expected, while TLR9 deficiency resulted in exacerbated autoimmune disease [41].
The pathogenic rather than protective effect observed in the TLR9 knockout in the MRLlpr/lpr lupus mouse model does not correlate with the earlier in vitro studies linking TLR9 activation to disease progression. It has been suggested that human-mouse differences in the expression, distribution and functional response of TLR7 and TLR9, as well as drawbacks in the animal model used, may explain the pathogenic effect observed in the TLR9 knockout MRLlpr/lpr mouse model [42]. Three studies have failed to correlate a certain set of polymorphisms in TLR9 with SLE [43–45]; however, a Japanese group recently identified two alleles that downregulated TLR9 expression in a reporter assay but are associated with increased SLE susceptibility [46]. This linkage would indicate that the TLR9 knockout data from the MRLlpr/lpr mice may be correct and that TLR9 has an anti-inflammatory function in SLE.
It remains to be seen whether endosomal TLR agonist or antagonists will be beneficial for the treatment of SLE; however, endosomal TLR signaling certainly appears to be involved in SLE pathology. Interestingly a polymorphism in Mal, the signaling adaptor used by TLR2 and TLR4, has been shown to be protective against SLE [47]. This polymorphism attenuates Mal signal transduction, which would diminish signaling through TLR2 and TLR4 [48]. Interestingly, HMGB1-containing DNA immune complexes that have been shown to bind RAGE on plasmactytoid dendritic cells and B cells [49] have recently been shown to induce proinflammatory cytokine production in macrophages in a TLR2-dependent manner [50]. These results indicate that there may be a more complex interplay between cell surface TLRs, their adaptors and endosomal TLRs in the pathology of SLE.
A cytoplasmic DNA-sensing inflammasome has been described more recently that is NALP3 independent. Absent in melanoma-2 (AIM2) is an interferon-inducible HIN200 family member that binds DNA through the HIN domain and has a pyrin domain that interacts with ASC to activate NF-κB and caspase-1. Knockdown of AIM2 using shRNA blocks recognition of cytoplasmic dsDNA in human macrophages [51–53]. SLE is characterised by elevated levels of interferon and by the presence of DNA:antibody complexes. In addition, genetic mapping studies have identified a susceptibility locus for SLE that contains the AIM2 gene, raising the possibility that AIM2 has a role to play in the pathology of SLE. Further studies are required to fully elucidate any link between AIM2 and SLE. The identification of AIM2 may in addition help to explain the results observed by Kawane and colleagues, who observed a TLR-independent polyarthritic phenotype in mice deficient for DNaseII and IFNIR as a consequence of the inability of macrophages to efficiently degrade cytosolic DNA [54].
Ankylosing spondylitis, TLR2 and TLR4
Ankylosing spondylitis is a multifactorial and polygenic inflammatory rheumatic disease with a poorly understood pathophysiology. Apart from HLA, other genes are likely to play a role in disease susceptibility and indigenous bacteria also appear to be involved in the pathology. This suggests that both adaptive and innate immune responses are required for disease progression. Expression studies looking at the CD4+CD28nullT-cell populations from ankylosing spondylitis patients have shown that TLR2 and TLR4 levels are increased and that this effect can be reduced by therapeutic blockade of TNFα [55]. Polymorphisms in TLR4 have been described and there are several studies that have looked at the association between these polymorphisms and susceptibility to ankylosing spondylitis. There is good evidence for a link between both Asp299Gly and Thr399Ile polymorphisms and ankylosing spondylitis [56], but no link with the Asp896Gly polymorphism [57]. The functional consequences of these polymorphisms and the mechanistic link to ankylosing spondylitis remain to be established. The S180L polymorphism in TIRAP/Mal that has been shown to be protective against SLE [47] has no association with axial spondyloarthritis [58].
Psoriatic arthritis
Psoriatic arthritis is an inflammatory arthritis associated with psoriasis in which the CD8+ T cell plays a pivotal role. The data on TLRs in psoriatic arthritis are restricted to a few studies of expression levels of TLR2 and TLR4. Candia and colleagues have shown that TLR2 expression was increased in immature dendritic cells from patients with psoriatic arthritis, although mature dendritic cells did not show statistically significant differences [59]. No effect was seen on TLR4 expression. Conversely, Raffeiner and colleagues looked at CD4+CD28null T cells and showed an increase in surface levels of TLR4 but no effects on TLR2 [55]. Further detailed analysis of TLRs in psoriatic arthritis is required to better understand whether there is a role in the pathogenesis of the disease.
Gout, pseudogout, TLR2 and Nalp3
Gout and pseudogout are crystal-induced arthropathies, gout being the most common autoinflammatory arthritis with increasing incidence over the past decade [60]. Gout is characterised by elevated serum urate and recurrent attacks of intra-articular crystal deposition of monosodium urate, whereas pseudogout is associated with calcium pyrophosphate dihydrate crystals and has a poorly understood pathophysiology.
Uric acid crystals stimulate dendritic cell maturation, enhance antigen-specific immune responses and directly activate T cells leading to elevated levels of CD70 [61]. The role of the innate immune system in gout has now been firmly established with the realisation that the uptake of monosodium urate crystals by monocytes involves interactions with TLR2 and CD14 [62] and that intracellularly monosodium urate crystal-induced inflammation is mediated by the NALP3 inflammasome [63]. The role of the NALP3 inflammasome was confirmed in a monosodium-urate-induced peritonitis mouse model that mimics an acute gout attack. Intraperitoneal injection of monosodium urate induces recruitment of neutrophils, and this effect was abrogated when either Anakinra or an anti-IL-1R antibody was co-administered with monosodium urate [63]. This monosodium-urate-induced mouse gout model clearly establishes the role of IL-1 in gout and led to an open-label study of Anakinra in 10 patients with gout that could not tolerate or had failed standard anti-inflammatory therapies. All patients received Anakinra daily for 3 days and all showed rapid positive responses with no adverse effects observed [64]. In addition, there is one report of Anakinra delivering a positive effect in a steroid-resistant pseudogout patient [65].
Osteoarthritis and Toll-like receptors
Synovial inflammation is increasingly recognised as an important pathophysiological process in OA, and endogenous ligands released as a consequence of synovial and cartilage catabolism (for example, fibronectin and hyaluronan fragments) are likely to be recognised by PRRs [66].
Histology and expression studies using isolated chondrocytes and cartilage have shown that human articular chondrocytes predominantly express TLR1, TLR2, TLR3, TLR4 and TLR5 [67–69]. Expression of TLR2 and TLR4 is elevated in OA particularly at sites of lesions in cartilage [67, 69]. Treatment of isolated cells with inflammatory cytokines and fibronectin proteolytic fragments results in increased expression of TLR2, and culture in the presence of TLR1/2 or TLR2/6 ligands but not TLR3 ligands results in elevated levels of matrix metalloproteinases and significantly increased collagenolysis and aggrecanolysis [67, 69].
OA is also associated with crystal deposition in synovial fluid - in particular, calcium pyrophosphate dihydrate and basic calcium phosphate [70], as well as hydroxyapatite [71] and silicon dioxide [72]. The physiological relevance of crystals to disease pathology is keenly debated but it seems probable that recognition of these crystals by the inflammasome will contribute to local inflammation in the joint [73].
Conclusions and future therapeutics opportunities
The roles of TLRs and Nalp3 in arthropathies are becoming clearer and they remain exciting therapeutic options. One interesting example is in aseptic loosening that occurs in 10% of joint replacements, resulting in revision surgery. Evidence is emerging to suggest that aseptic loosening of total joint replacements is driven through implant debris activation of the inflammasome leading to locally elevated levels of inflammatory cytokines [74]. More obviously Nalp3, TLR2 and TLR4 are attractive targets for RA and OA, whilst TLR7 and/or TLR9 and AIM2 represent therapeutic potentials for joint inflammation in SLE.
There has been considerable focus on identification of small molecule agonists and antagonists of TLRs over the past 5 years, with several successful examples now undergoing clinical evaluation. If the preclinical observations described in the present review [21, 39] translate to the clinic, then inhibition of TLRs and NLRs using small molecules may provide viable replacements for current biologic agents. In any event, the hope is that these new insights into innate immunity will ultimately translate into better therapies for inflammatory arthropathies that continue to represent a major burden on humanity.
Note
The Scientific Basis of Rheumatology: A Decade of Progress
This article is part of a special collection of reviews, The Scientific Basis of Rheumatology: A Decade of Progress, published to mark Arthritis Research & Therapy's 10th anniversary.
Other articles in this series can be found at: http://arthritis-research.com/sbr
Abbreviations
- AIM2:
-
absent in melanoma-2
- CpG:
-
cytosine phosphate guanine
- dsDNA:
-
double-stranded DNA
- HMGB1:
-
high-mobility group box protein 1
- IFN:
-
interferon
- IL:
-
interleukin
- NALP:
-
Nacht domain-containing, leucine-rich repeat-containing, and pyrin domain-containing protein
- NF:
-
nuclear factor
- NLR:
-
nucleotide-binding oligomerisation domain-like and leucine-rich repeat receptors
- NOD:
-
nucleotide-binding oligomerisation domains
- OA:
-
osteoarthritis
- PRR:
-
pattern recognition receptor
- RA:
-
rheumatoid arthritis
- RAGE:
-
receptor for advanced glycation end products
- shRNA:
-
short hairpin RNA
- SLE:
-
systemic lupus erythematosus
- snRNP:
-
small nuclear ribonucleoproteins
- TLR:
-
Toll-like receptor
- TNF:
-
tumor necrosis factor.
References
O'Neill LA: The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev. 2008, 226: 10-18. 10.1111/j.1600-065X.2008.00701.x.
Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, Lee H, Lee JO: Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007, 130: 1071-1082. 10.1016/j.cell.2007.09.008.
Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, Lee JO: Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007, 130: 906-917. 10.1016/j.cell.2007.08.002.
Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, Walsh EE, Freeman MW, Golenbock DT, Anderson LJ, Finberg RW: Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000, 1: 398-401. 10.1038/80833.
Awomoyi AA, Rallabhandi P, Pollin TI, Lorenz E, Sztein MB, Boukhvalova MS, Hemming VG, Blanco JC, Vogel SN: Association of TLR4 polymorphisms with symptomatic respiratory syncytial virus infection in high-risk infants and young children. J Immunol. 2007, 179: 3171-3177.
Kenny EF, O'Neill LA: Signalling adaptors used by Toll-like receptors: an update. Cytokine. 2008, 43: 342-349. 10.1016/j.cyto.2008.07.010.
Martinon F, Mayor A, Tschopp J: The inflammasomes: guardians of the body. Annu Rev Immunol. 2009, 27: 229-265. 10.1146/annurev.immunol.021908.132715.
Verma D, Lerm M, Blomgran Julinder R, Eriksson P, Soderkvist P, Sarndahl E: Gene polymorphisms in the NALP3 inflammasome are associated with interleukin-1 production and severe inflammation: relation to common inflammatory diseases?. Arthritis Rheum. 2008, 58: 888-894. 10.1002/art.23286.
Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J: NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004, 20: 319-325. 10.1016/S1074-7613(04)00046-9.
Church LD, Cook GP, McDermott MF: Primer: inflammasomes and interleukin 1β in inflammatory disorders. Nat Clin Pract Rheumatol. 2008, 4: 34-42. 10.1038/ncprheum0681.
Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, Freudenberg M, Beutler B: CD14 is required for MyD88-independent LPS signaling. Nat Immunol. 2005, 6: 565-570. 10.1038/ni1207.
Herlands RA, Christensen SR, Sweet RA, Hershberg U, Shlomchik MJ: T cell-independent and toll-like receptor-dependent antigen-driven activation of autoreactive B cells. Immunity. 2008, 29: 249-260. 10.1016/j.immuni.2008.06.009.
Ganley-Leal LM, Liu X, Wetzler LM: Toll-like receptor 2-mediated human B cell differentiation. Clin Immunol. 2006, 120: 272-284. 10.1016/j.clim.2006.04.571.
Heijden van der IM, Wilbrink B, Tchetverikov I, Schrijver IA, Schouls LM, Hazenberg MP, Breedveld FC, Tak PP: Presence of bacterial DNA and bacterial peptidoglycans in joints of patients with rheumatoid arthritis and other arthritides. Arthritis Rheum. 2000, 43: 593-598. 10.1002/1529-0131(200003)43:3<593::AID-ANR16>3.0.CO;2-1.
Blaschke S, Schwarz G, Moneke D, Binder L, Muller G, Reuss-Borst M: Epstein-Barr virus infection in peripheral blood mononuclear cells, synovial fluid cells, and synovial membranes of patients with rheumatoid arthritis. J Rheumatol. 2000, 27: 866-873.
Ospelt C, Brentano F, Rengel Y, Stanczyk J, Kolling C, Tak PP, Gay RE, Gay S, Kyburz D: Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: toll-like receptor expression in early and longstanding arthritis. Arthritis Rheum. 2008, 58: 3684-3692. 10.1002/art.24140.
Brentano F, Schorr O, Gay RE, Gay S, Kyburz D: RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via Toll-like receptor 3. Arthritis Rheum. 2005, 52: 2656-2665. 10.1002/art.21273.
Roelofs MF, Joosten LA, Abdollahi-Roodsaz S, van Lieshout AW, Sprong T, Hoogen van den FH, Berg van den WB, Radstake TR: The expression of toll-like receptors 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of toll-like receptors 3, 4, and 7/8 results in synergistic cytokine production by dendritic cells. Arthritis Rheum. 2005, 52: 2313-2322. 10.1002/art.21278.
Huang Q, Ma Y, Adebayo A, Pope RM: Increased macrophage activation mediated through toll-like receptors in rheumatoid arthritis. Arthritis Rheum. 2007, 56: 2192-2201. 10.1002/art.22707.
Sacre SM, Andreakos E, Kiriakidis S, Amjadi P, Lundberg A, Giddins G, Feldmann M, Brennan F, Foxwell BM: The Toll-like receptor adaptor proteins MyD88 and Mal/TIRAP contribute to the inflammatory and destructive processes in a human model of rheumatoid arthritis. Am J Pathol. 2007, 170: 518-525. 10.2353/ajpath.2007.060657.
Abdollahi-Roodsaz S, Joosten LA, Roelofs MF, Radstake TR, Matera G, Popa C, Meer van der JW, Netea MG, Berg van den WB: Inhibition of Toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum. 2007, 56: 2957-2967. 10.1002/art.22848.
Abdollahi-Roodsaz S, Joosten LA, Koenders MI, Devesa I, Roelofs MF, Radstake TR, Heuvelmans-Jacobs M, Akira S, Nicklin MJ, Ribeiro-Dias F, Berg van den WB: Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest. 2008, 118: 205-216. 10.1172/JCI32639.
Joosten LA, Koenders MI, Smeets RL, Heuvelmans-Jacobs M, Helsen MM, Takeda K, Akira S, Lubberts E, Loo van de FA, Berg van den WB: Toll-like receptor 2 pathway drives streptococcal cell wall-induced joint inflammation: critical role of myeloid differentiation factor 88. J Immunol. 2003, 171: 6145-6153.
Abdollahi-Roodsaz S, Joosten LA, Helsen MM, Walgreen B, van Lent PL, Bersselaar van den LA, Koenders MI, Berg van den WB: from toll-like receptor 2 (TLR-2) toward TLR-4 dependency in the erosive stage of chronic streptococcal cell wall arthritis coincident with TLR-4-mediated interleukin-17 production. Arthritis Rheum. 2008, 58: 3753-3764. 10.1002/art.24127.
Goldstein RS, Bruchfeld A, Yang L, Qureshi AR, Gallowitsch-Puerta M, Patel NB, Huston BJ, Chavan S, Rosas-Ballina M, Gregersen PK, Czura CJ, Sloan RP, Sama AE, Tracey KJ: Cholinergic anti-inflammatory pathway activity and high mobility group box-1 (HMGB1) serum levels in patients with rheumatoid arthritis. Mol Med. 2007, 13: 210-215. 10.2119/2006-00108.Goldstein.
Huang QQ, Sobkoviak R, Jockheck-Clark AR, Shi B, Mandelin AM, Tak PP, Haines GK, Nicchitta CV, Pope RM: Heat shock protein 96 is elevated in rheumatoid arthritis and activates macrophages primarily via TLR2 signaling. J Immunol. 2009, 182: 4965-4973. 10.4049/jimmunol.0801563.
Kilding R, Akil M, Till S, Amos R, Winfield J, Iles MM, Wilson AG: A biologically important single nucleotide polymorphism within the toll-like receptor-4 gene is not associated with rheumatoid arthritis. Clin Exp Rheumatol. 2003, 21: 340-342.
Sanchez E, Orozco G, Lopez-Nevot MA, Jimenez-Alonso J, Martin J: Polymorphisms of toll-like receptor 2 and 4 genes in rheumatoid arthritis and systemic lupus erythematosus. Tissue Antigens. 2004, 63: 54-57. 10.1111/j.1399-0039.2004.00162.x.
Ospelt C, Brentano F, Jungel A, Rengel Y, Kolling C, Michel BA, Gay RE, Gay S: Expression, regulation, and signaling of the pattern-recognition receptor nucleotide-binding oligomerizastion domain 2 in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2009, 60: 355-363. 10.1002/art.24226.
Joosten LA, Heinhuis B, Abdollahi-Roodsaz S, Ferwerda G, Lebourhis L, Philpott DJ, Nahori MA, Popa C, Morre SA, Meer van der JW, Girardin SE, Netea MG, Berg van den WB: Differential function of the NACHT-LRR (NLR) members Nod1 and Nod2 in arthritis. Proc Natl Acad Sci USA. 2008, 105: 9017-9022. 10.1073/pnas.0710445105.
Hirschfeld M, Kirschning CJ, Schwandner R, Wesche H, Weis JH, Wooten RM, Weis JJ: Cutting edge: inflammatory signaling by Borrelia burgdorferi lipoproteins is mediated by toll-like receptor 2. J Immunol. 1999, 163: 2382-2386.
Alexopoulou L, Thomas V, Schnare M, Lobet Y, Anguita J, Schoen RT, Medzhitov R, Fikrig E, Flavell RA: Hyporesponsiveness to vaccination with Borrelia burgdorferi OspA in humans and in TLR1- and TLR2-deficient mice. Nat Med. 2002, 8: 878-884.
Schroder NW, Diterich I, Zinke A, Eckert J, Draing C, von Baehr V, Hassler D, Priem S, Hahn K, Michelsen KS, Hartung T, Burmester GR, Gobel UB, Hermann C, Schumann RR: Heterozygous Arg753Gln polymorphism of human TLR-2 impairs immune activation by Borrelia burgdorferi and protects from late stage Lyme disease. J Immunol. 2005, 175: 2534-2540.
Gota C, Calabrese L: Induction of clinical autoimmune disease by therapeutic interferon-alpha. Autoimmunity. 2003, 36: 511-518. 10.1080/08916930310001605873.
Ronnblom LE, Alm GV, Oberg KE: Possible induction of systemic lupus erythematosus by interferon-alpha treatment in a patient with a malignant carcinoid tumour. J Intern Med. 1990, 227: 207-210. 10.1111/j.1365-2796.1990.tb00144.x.
Anders HJ, Vielhauer V, Eis V, Linde Y, Kretzler M, Perez de Lema G, Strutz F, Bauer S, Rutz M, Wagner H, Grone HJ, Schlondorff D: Activation of toll-like receptor-9 induces progression of renal disease in MRL-Fas(lpr) mice. FASEB J. 2004, 18: 534-536.
Ramos-Casals M, Cuadrado MJ, Alba P, Sanna G, Brito-Zeron P, Bertolaccini L, Babini A, Moreno A, D'Cruz D, Khamashta MA: Acute viral infections in patients with systemic lupus erythematosus: description of 23 cases and review of the literature. Medicine (Baltimore). 2008, 87: 311-318. 10.1097/MD.0b013e31818ec711.
Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, Chang B, Duramad O, Coffman RL: Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005, 202: 1131-1139. 10.1084/jem.20050914.
Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL: Treatment of lupusprone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol. 2007, 37: 3582-3586. 10.1002/eji.200737815.
Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A: RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005, 202: 1171-1177. 10.1084/jem.20050630.
Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ: Toll-like receptor 7 and TLR9 dictate autoanti-body specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006, 25: 417-428. 10.1016/j.immuni.2006.07.013.
Barrat FJ, Coffman RL: Development of TLR inhibitors for the treatment of autoimmune diseases. Immunol Rev. 2008, 223: 271-283. 10.1111/j.1600-065X.2008.00630.x.
De Jager PL, Richardson A, Vyse TJ, Rioux JD: Genetic variation in toll-like receptor 9 and susceptibility to systemic lupus erythematosus. Arthritis Rheum. 2006, 54: 1279-1282. 10.1002/art.21755.
Hur JW, Shin HD, Park BL, Kim LH, Kim SY, Bae SC: Association study of Toll-like receptor 9 gene polymorphism in Korean patients with systemic lupus erythematosus. Tissue Antigens. 2005, 65: 266-270. 10.1111/j.1399-0039.2005.00374.x.
Ng MW, Lau CS, Chan TM, Wong WH, Lau YL: Polymorphisms of the toll-like receptor 9 (TLR9) gene with systemic lupus erythematosus in Chinese. Rheumatology (Oxford). 2005, 44: 1456-1457. 10.1093/rheumatology/kei120.
Tao K, Fujii M, Tsukumo S, Maekawa Y, Kishihara K, Kimoto Y, Horiuchi T, Hisaeda H, Akira S, Kagami S, Yasutomo K: Genetic variations of Toll-like receptor 9 predispose to systemic lupus erythematosus in Japanese population. Ann Rheum Dis. 2007, 66: 905-909. 10.1136/ard.2006.065961.
Castiblanco J, Varela DC, Castano-Rodriguez N, Rojas-Villarraga A, Hincapie ME, Anaya JM: TIRAP (MAL) S180L polymorphism is a common protective factor against developing tuberculosis and systemic lupus erythematosus. Infect Genet Evol. 2008, 8: 541-544. 10.1016/j.meegid.2008.03.001.
Khor CC, Chapman SJ, Vannberg FO, Dunne A, Murphy C, Ling EY, Frodsham AJ, Walley AJ, Kyrieleis O, Khan A, Aucan C, Segal S, Moore CE, Knox K, Campbell SJ, Lienhardt C, Scott A, Aaby P, Sow OY, Grignani RT, Sillah J, Sirugo G, Peshu N, Williams TN, Maitland K, Davies RJ, Kwiatkowski DP, Day NP, Yala D, Crook DW, Marsh K, Berkley JA, O'Neill LA, Hill AV: A Mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat Genet. 2007, 39: 523-528. 10.1038/ng1976.
Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ: Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007, 8: 487-496. 10.1038/ni1457.
Urbonaviciute V, Furnrohr BG, Meister S, Munoz L, Heyder P, De Marchis F, Bianchi ME, Kirschning C, Wagner H, Manfredi AA, Kalden JR, Schett G, Rovere-Querini P, Herrmann M, Voll RE: Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008, 205: 3007-3018. 10.1084/jem.20081165.
Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES: AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009, 458: 509-513. 10.1038/nature07710.
Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA: AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009, 458: 514-518. 10.1038/nature07725.
Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, Superti-Furga G: An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009, 10: 266-272. 10.1038/ni.1702.
Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, Yoshikawa H, Nagata S: Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. 2006, 443: 998-1002. 10.1038/nature05245.
Raffeiner B, Dejaco C, Duftner C, Kullich W, Goldberger C, Vega SC, Keller M, Grubeck-Loebenstein B, Schirmer M: Between adaptive and innate immunity: TLR4-mediated perforin production by CD28null T-helper cells in ankylosing spondylitis. Arthritis Res Ther. 2005, 7: R1412-R1420. 10.1186/ar1840.
Snelgrove T, Lim S, Greenwood C, Peddle L, Hamilton S, Inman R, Rahman P: Association of toll-like receptor 4 variants and ankylosing spondylitis: a case-control study. J Rheumatol. 2007, 34: 368-370.
Paardt van der M, Crusius JB, de Koning MH, Morre SA, Stadt van de RJ, Dijkmans BA, Pena AS, Horst-Bruinsma van der IE: No evidence for involvement of the Toll-like receptor 4 (TLR4) A896G and CD14-C260T polymorphisms in susceptibility to ankylosing spondylitis. Ann Rheum Dis. 2005, 64: 235-238. 10.1136/ard.2004.021105.
Cantaert T, Stone MA, ter Borg M, Mogg R, De Vries N, Wilson AG, Tak PP, Baeten D: A functional polymorphism of TIR-domain-containing adaptor protein is not associated with axial spondyloarthritis. Ann Rheum Dis. 2008, 67: 720-722. 10.1136/ard.2007.082784.
Candia L, Marquez J, Hernandez C, Zea AH, Espinoza LR: Toll-like receptor-2 expression is upregulated in antigen-presenting cells from patients with psoriatic arthritis: a pathogenic role for innate immunity?. J Rheumatol. 2007, 34: 374-379.
So A: Developments in the scientific and clinical understanding of gout. Arthritis Res Ther. 2008, 10: 221-10.1186/ar2509.
Webb R, Jeffries M, Sawalha AH: Uric acid directly promotes human T-cell activation. Am J Med Sci. 2009, 337: 23-27. 10.1097/MAJ.0b013e31817727af.
Scott P, Ma H, Viriyakosol S, Terkeltaub R, Liu-Bryan R: Engagement of CD14 mediates the inflammatory potential of monosodium urate crystals. J Immunol. 2006, 177: 6370-6378.
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J: Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006, 440: 237-241. 10.1038/nature04516.
So A, De Smedt T, Revaz S, Tschopp J: A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 2007, 9: R28-10.1186/ar2143.
McGonagle D, Tan AL, Madden J, Emery P, McDermott MF: Successful treatment of resistant pseudogout with anakinra. Arthritis Rheum. 2008, 58: 631-633. 10.1002/art.23119.
Scanzello CR, Plaas A, Crow MK: Innate immune system activation in osteoarthritis: is osteoarthritis a chronic wound?. Curr Opin Rheumatol. 2008, 20: 565-572. 10.1097/BOR.0b013e32830aba34.
Zhang Q, Hui W, Litherland GJ, Barter MJ, Davidson R, Darrah C, Donell ST, Clark IM, Cawston TE, Robinson JH, Rowan AD, Young DA: Differential Toll-like receptor-dependent collagenase expression in chondrocytes. Ann Rheum Dis. 2008, 67: 1633-1641. 10.1136/ard.2007.079574.
Su SL, Tsai CD, Lee CH, Salter DM, Lee HS: Expression and regulation of Toll-like receptor 2 by IL-1β and fibronectin fragments in human articular chondrocytes. Osteoarthritis Cartilage. 2005, 13: 879-886. 10.1016/j.joca.2005.04.017.
Kim HA, Cho ML, Choi HY, Yoon CS, Jhun JY, Oh HJ, Kim HY: The catabolic pathway mediated by Toll-like receptors in human osteoarthritic chondrocytes. Arthritis Rheum. 2006, 54: 2152-2163. 10.1002/art.21951.
Whelan LC, Morgan MP, McCarthy GM: Basic calcium phosphate crystals as a unique therapeutic target in osteoarthritis. Front Biosci. 2005, 10: 530-541. 10.2741/1549.
Pay S, Terkeltaub R: Calcium pyrophosphate dihydrate and hydroxyapatite crystal deposition in the joint: new developments relevant to the clinician. Curr Rheumatol Rep. 2003, 5: 235-243. 10.1007/s11926-003-0073-x.
Oliviero F, Frallonardo P, Peruzzo L, Ramonda R, Sfriso P, Scanu A, Tauro L, Liote F, Punzi L: Evidence of silicon dioxide crystals in synovial fluid of patients with osteoarthritis. J Rheumatol. 2008, 35: 1092-1095.
Ea HK, Liote F: Advances in understanding calcium-containing crystal disease. Curr Opin Rheumatol. 2009, 21: 150-157. 10.1097/BOR.0b013e3283257ba9.
Caicedo MS, Desai R, McAllister K, Reddy A, Jacobs JJ, Hallab NJ: Soluble and particulate Co-Cr-Mo alloy implant metals activate the inflammasome danger signaling pathway in human macrophages: a novel mechanism for implant debris reactivity. J Orthop Res. 2008, 27: 847-854. 10.1002/jor.20826.
Acknowledgements
LAO'N acknowledges Science Foundation Ireland for research funding.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
WJM and AEP are employees of Opsona Therapeutics, a drug discovery and development company focused on the role of TLRs and inflammasome signaling in human immunology. LAO'N is a founder of Opsona therapeutics and is a member of its scientific advisory board.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
McCormack, W.J., Parker, A.E. & O'Neill, L.A. Toll-like receptors and NOD-like receptors in rheumatic diseases. Arthritis Res Ther 11, 243 (2009). https://doi.org/10.1186/ar2729
Published:
DOI: https://doi.org/10.1186/ar2729