
Abstract
The neuroendocrine immune (NEI) system is regarded as a fundamental network for the maintenance of health status (homeostasis), and it plays an important role in several systemic diseases, including autoimmune disorders. Among the major players of NEI pathways are steroid hormones of the adrenal (cortisol) and gonadal glands (sex hormones), neurohormones such as melatonin, and more recently the vitamin D endocrine system. Estrogens, melatonin and chronic stress (inducing decreased adrenal glucocorticoid release over a long time) strongly modulate the NEI system and stimulate the immune response. The vitamin D endocrine system is regarded as a potential immunosuppressive factor. Consequently, estrogens (especially in patients affected by B-cell-driven immunity) and melatonin should be avoided, and glucocorticoids (as replacement therapy) and vitamin D are allowed in the treatment of autoimmunity.
Similar content being viewed by others

Introduction
The neuroendocrine immune (NEI) system is generally considered a fundamental network and its integrity is essential for the maintenance of health status in humans [1]. As a consequence, several systemic diseases, including autoimmune disorders, originate from the altered balance/activation of the NEI system.
Modulators of the immune system include different hormones, and major players of NEI pathways are steroid hormones of the adrenal and gonadal glands, as well as neurohormones such as melatonin (MLT) [1]. Steroid hormones are not stored in endocrine glands in the form of the final bioactive hormones, but their precursor cholesterol is metabolized by different enzyme steps leading to cortisol, the bioactive highly antiinflammatory endogenous glucocorticoid. The enzyme steps are regulated by microenvironmental factors such as cytokines. In the tissue, cortisol is degraded to cortisone, which can be reactivated by the 11β-hydroxysteroid de-hydrogenase type 1 [2]. In addition, the adrenal glands produce the major androgen precursors dehydroepiandrosterone and androstenedione, which can be converted into active sex hormones such as testosterone and estrogens in peripheral tissues.
Gonadal glands mainly synthesize sex hormones such as testosterone (in the testicles) and estrogens (in the ovaries), again from the precursor cholesterol. Testosterone can also be viewed as a precursor of estrogens in tissue with high aromatase activity (that is, rheumatoid synovial tissue).
Interestingly, another hormone arising from cholesterol, namely vitamin D and its endocrine system, is involved in various biological processes that modulate immune responses (mainly immunosuppressive), and has an important role in autoimmune diseases [3].
Role of glucocorticoids in autoimmunity and inflammation
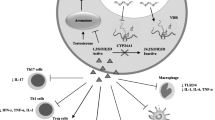
Several factors are involved in the pathogenesis of auto-immune rheumatic diseases, including genetic aspects, chronic infections, sex hormones (estrogens) and stress (Figure 1).

Factors involved in the pathogenesis of autoimmune rheumatic diseases. The major risk factors in autoimmunity include specific genetic background, chronic infections as triggers of the immune response, estrogens as general enhancers of the immune response (at least B-cell-driven), and chronic stress over a long time inducing a decreased adrenal glucocorticoid release.
In particular, stress (that is, interpersonal, severe surgery, chronic infections) is now recognized as an important risk factor [4]. Patients with insufficient stress response axes demonstrate paradoxically decreased stress responses and, consequently, proinflammatory side effects [5]. The loss of adequate stress responses is reflected by low serum levels of cortisol and also low concentrations of norepinephrine in the tissue (nerve fiber loss) [1, 5]. Minor stress and probably also major stress are therefore not accompanied by an adequate stress response, especially over a long time, which leads to inadequately low concentrations of stress axes mediators (that is, endogenous glucocorticoids) in relation to the immune/inflammatory situation [5].
Therapeutically, a reduction in stress episodes or a change of stress management must be implemented.
Based on their decreased concentrations in chronic diseases, glucocorticoids are now considered important disease-modifying antirheumatic drugs at least in rheumatoid arthritis (RA) [1, 6]. In addition, mild exercise and training and a decrease in the proinflammatory load – as obtained also by the use of anticytokine therapy – can normalize stress axes, leading to favorable responses and decreased appeal of exogenous glucocorticoids [7].
Suppression of the hypothalamic–pituitary–adrenal axis, however – especially in polymyalgia rheumatica, an inflammatory syndrome affecting older people – plays an important pathogenetic role [8]. In fact, older people per se represent a condition of endocrine senescence including adrenal hypofunction, and the presence of chronic stress represents a harmful stimulus to seriously compromised endogenous glucocorticoid production. Interestingly, serum cytokine (mainly IL-6) and steroidal hormone patterns suggest that patients with polymyalgia rheumatica have an intensive inflammatory reaction (much higher then in RA), and glucocorticoid administration represents the most effective replacement treatment [9].
Insights into therapeutic optimization of neuroendocrine immune system modulation in autoimmunity
It has been known for many decades that disease symptoms in immune-mediated diseases such as RA follow obvious circadian rhythms, with an increase of activity in the early morning hours, abatement during the day, and a smaller new increase in the early evening [10].
A number of articles have reported a temporal relationship between elevated levels of proinflammatory cytokines and symptoms of RA, such as morning stiffness [11, 12]. Several of these cytokines are highly elevated in patients with active RA in the early hours of the day, but after noon their levels are almost zero. Their release pattern and serum concentrations, respectively, are modulated by hormones and neuronal pathways coordinated by a subordinate neuroendocrine centre in the hypothalamus, which follows a strict 24-hour daily cycle.
The closest similarity with the daily pattern of RA symptoms, such as morning stiffness, joint pain and functional disability, seems to exist for serum concentrations of IL-6 (Figure 2).

Daily pattern of rheumatoid arthritis symptoms for serum concentrations of IL-6. Proinflammatory hormones (that is, melatonin) start to rise during the early night before the onset of rheumatoid arthritis symptoms and before the endogenous cortisol rise in these patients. Grey shading, trigger time for the IL-6 rise.
Proinflammatory hormones (that is, MLT) start to rise during the night before the onset of RA symptoms and before an endogenous cortisol rise in these patients [11] (Figure 2). The role of IL-6 in the regulation of inflammatory and immune responses, particularly in RA, is well established, but an increased production of other proinflammatory cytokines such as TNF, IL-1, IL-8, IL-12 and IL-17 has also been reported [13].
RA has also been considered characterized by an inadequate antiinflammatory response that may contribute to morning stiffness. This lack of anti-inflammatory response is not yet totally understood. Cortisol secretion and glucocorticoid receptor density, however, have been reported to be altered in patients with RA. Furthermore, changes of peripheral metabolism of endogenous glucocorticoids may also contribute to the early morning manifestation of the disease symptoms in RA [13].
Based on these considerations, the usual administration of glucocorticoids between 06:00 and 08:00 hours has been suggested as not optimal [14]. This could simply be too late since the night-time pathophysiological processes have already led to inflammation, pain and subjective symptoms. Consequently, it has been hypothesized that it could be easier to prevent the circadian increase of proinflammatory cytokine levels, and therefore the consequently observed clinical signs and symptoms of the disease, than to treat these signs and symptoms once they are established in the morning [10].
A new modified-release delivery system has been developed that adapts the release of the administered glucocorticoids to the circadian rhythms of endogenous cortisol and disease symptoms to improve glucocorticoid therapy [15]. The efficacy and safety of a new modified-release prednisone tablet (intake at 22:00 hours, release at 02:00 hours) compared with immediate-release prednisone (intake at 07:00 hours) in RA patients was recently tested. The mean relative change in duration of morning stiffness of the joints from baseline to end of treatment (12 weeks) was significantly higher with modified-release prednisone than with immediate-release prednisone (-22.7% vs. -0.4%; difference = 22.4% (95% confidence interval = 0.49 to 44.30); P = 0.045). Patients in the modified-release prednisone group achieved a mean reduction of morning stiffness of 44.0 minutes (standard deviation = 136.6 minutes) compared with baseline.
The absolute difference between the treatment groups was 29.2 minutes (95% confidence interval = -2.59 to 61.9) in favor of modified-release prednisone (P = 0.072). The safety profile did not differ between treatments [15].
Optimization of the timing (night) and dosage (low) of glucocorticoids has been recently recommended to improve the tolerance and efficacy in autoimmune rheumatic diseases [16].
Insights into neurohormonal effects of melatonin in autoimmunity
MLT is an important neurohormone mainly synthesized by the pineal gland with a circadian rhythm that peaks between 01:00 and 03:00 hours.
At physiological concentrations in human peripheral blood mononuclear cells, MLT has been reported to stimulate the production of IFNγ, IL-1, IL-2, IL-6 and IL-12, but not production of IL-4 [17]. In addition, MLT was found to enhance production of inflammatory cytokines from cultured human monocytes/macrophages, including IL-12, and to turn the MLT/IL-2 connection towards the enhancement of T-cell immunity [18].
MLT serum levels at 20:00 and 08:00 hours have been found to be significantly higher in patients with RA than in healthy control individuals (P < 0.05). MLT was found to be detectable at high concentration in synovial fluids from patients who had RA, and binding sites for MLT were present in synovial macrophages [18]. Interestingly, IFNγ, IL-1, IL-6, IL-2, IL-12 and TNF production reach their peak during the night and early morning, shortly after MLT serum levels are highest and plasma cortisol is lowest [19] (Figure 2). This is in line with the hypothesis that MLT upregulates cytokine production and immune system activity [18].
Since until recently there was still a need to obtain clinically-based evidence about the possible role of MLT as a disease-promoting or a disease-protecting hormone in RA, a double-blind placebo-controlled study investigating the effects of MLT administration in patients with RA was initiated [20]. The results obtained were somewhat disappointing and surprising, as the authors stated in the discussion by considering MLT an in vitro potent antioxidant [20]. MLT was therefore expected to decrease oxidative processes such as lipid peroxidation that decreased in RA patients, but the erythrocyte sedimentation rate and neopterin levels increased compared with patients treated with placebo. This observation is consistent with an antioxidant effect, but also suggests some proinflammatory activity [20]. In addition, no reduction of RA disease activity was observed, and the disease appeared to be worse in some MLT-treated RA patients.
Serum TNF was recently found to be higher in Northern European patients with RA than in their controls and was found to be significantly correlated with the early increased serum MLT concentrations, at least during the winter [21]. The increased serum concentrations and circadian rhythm of MLT and a relative adrenal insufficiency in chronic RA (low cortisol) therefore allow at least T-helper type 1 cytokines to be produced in higher amounts during the late night under the enhancing effect of the pineal hormone (Figure 3).

Altered balance between nocturnal hormones in rheumatoid arthritis. The altered balance between nocturnal hormone production in chronic diseases such as rheumatoid arthritis (RA) is characterized by increased levels and steady-state duration of melatonin (enhancer of the immune/inflammatory reaction) and by decreased adrenal cortisol availability (downregulator of the immune/inflammatory reaction).
In conclusion, the translation from basic research to clinical medicine clearly showed that MLT treatment does not improve RA and must be avoided [22].
Insights into estrogen involvement in neuroendocrine immune disturbances and autoimmunity
Generally, based on epidemiological and immunological evaluations, estrogens enhance the humoral response (antibody production, T-helper type 2 immune response, B-cell immunity); but, at the same time, estrogens might inhibit T cells and macrophages at normal to high estrogen concentrations [23]. The translation of the study results on modulatory effects of estrogens obtained from animal and in vitro investigations to the human condition, however, is always difficult and complex [24]. In addition, in most animal studies only 17β-estradiol was used as the investigated estrogen and in most human studies a crude mixture of conjugated estrogens was used, which can have proinflammatory effects, as recently reviewed [25].
Different concentrations used in in vitro or in vivo testing might also render estrogen friend or foe in immune/inflammatory conditions. Moreover, different cells involved in the immune/inflammatory response react in an opposite manner to different estrogen concentrations. In addition, the expression of estrogen receptors (estrogen receptor alpha or estrogen receptor beta) might be quite different under inflammatory conditions depending on the microenvironment and the type of disease. Generally, estrogens enhance cell proliferation and reduce cell apoptosis [26]. Finally, the role of local estrogen concentrations and the type of peripheral estrogen metabolites at the level of inflammatory foci is of great importance in order to explain the sometime opposite modulatory effects exerted by these hormones on the immune/inflammatory reaction [27].
Immunological evidence suggests that female gonadal hormones exert an important role in the etiology and course of chronic autoimmune diseases since the menstrual cycle, pregnancy and menopausal status are recognized as significant influencing factors [28].
Generally, the immune supportive role exerted by estrogens is evident in trauma/sepsis and some chronic autoimmune disorders such as systemic lupus erythematosus (SLE) or Sjögren syndrome [28]. Interestingly, studies in women using oral contraceptives versus those not using oral contraceptives demonstrated no significant increased risk of developing SLE, but hormone replacement therapy in postmenopausal women seems to increase the risk of developing SLE [29]. Oral contraceptive use was not associated with changes in the disease course in premenopausal women with SLE, at least in the nonactive phase, but hormone replacement therapy increased the risk of mild flares in postmenopausal patients [23]. This information indicates that the positive effect of estrogens on B cells does not play a role in premenopausal women with normal menstrual cycles and low disease activity, but estrogens mildly stimulate SLE in women with postmenopausal levels of estrogens [23].
Nevertheless, mainly for strictly B-cell-dependent diseases, the female to male preponderance can be explained by the propagating effects of estrogens [30]. On the other hand, because men never experience high estrogen (or proges-terone) levels like women during pregnancy, the apparent gender dimorphism of chronic inflammatory diseases during the reproductive period of women can be explained. In addition, higher androgen levels in men most often exert inhibitory effects on many immune phenomena (the opposite for low androgen levels; that is, in Klinefelter syndrome), which is an other important argument why women with low androgen levels are protected from infectious diseases but are more prone to B-cell-dependent autoimmunity [31, 32].
In conclusion, in the presence of active immune-mediated diseases such as SLE (or antiphospholipid syndrome and others), the administration of estrogens should be avoided.
Vitamin D endocrine system in autoimmunity
The discovery of the vitamin D receptor (VDR), a member of the nuclear hormone receptor superfamily, in the cells of the immune system suggested that vitamin D could have immunoregulatory properties [3].
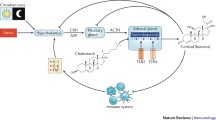
The vitamin D endocrine system is involved in various biological processes that modulate immune responses, and plays an important role in autoimmune diseases [33] (Figure 3). In addition to exerting direct modulatory effects on T-cell and B-cell function, VDR agonists influence the phenotype and function of dendritic cells, thereby promoting tolerogenic properties that favor the induction of regulatory, rather than effector, T cells [34]. VDR agonistic effects have been demonstrated in several experimental models and could be utilized to treat several autoimmune diseases and other immune-mediated pathologies that are characterized by chronic inflammatory responses [3].
1,25-Dihydroxyvitamin D3 (1,25(OH)2D3) – which is produced by macrophages, dendritic cells, T cells and B cells – seems to contribute physiologically, via the VDR expressed in these cell types, to the autocrine and paracrine regulation of both innate and adaptive immune responses [35–37] (Figure 4). This tight control of bioactive hormone production by cells of the immune system itself further supports the relevance of the vitamin D endocrine system in the modulation of immune responses in health and disease. A physiological role for vitamin D in the immune system is also supported by the presence of the VDR in primary lymphoid organs. The primary lymphoid organs (bone marrow and thymus) are the centers where the immune system develops and differentiates [38].

Downregulatory and upregulatory influences exerted by vitamin D on the immune response. Synthetic presentation of the major downregulatory (yellow) and upregulatory (with) influences exerted by vitamin D on the immune response. Effects on T lymphocytes and B lymphocytes and related cytokines, as well as on antigen-presenting cells (APC) and dendritic cells (DC). NKT, natural killer T cells; TGFβ1, transforming growth factor beta 1; Th, T-helper type cells; Treg, regulatory T cells.
T cells have been shown to play fundamental roles in autoimmune diseases. Quiescent CD4+ T cells express VDRs at low numbers, which increase fivefold after activation [39]. The effects of 1,25(OH)2D3 on the acquired, antigen-specific immune response are characterized by inhibition of T-lymphocyte activation, particularly of the T-helper type 1 arm. Treatment of CD4 T cells with 1,25(OH)2D3 inhibits T-helper type 1 cell proliferation and cytokine production [40]. Other observations demonstrated inhibition of both T-helper type 1 and T-helper type 2 cell cytokine production, including inhibition of IL-4 [41, 42].
Interestingly, 1,25(OH)2D3 has been found to regulate the proliferation of activated B cells and their subsequent differentiation, as well as enhancing IL-10 expression, further suggesting its role in the maintenance of B-cell homeostasis; this regulation also shows that the correction of vitamin D deficiency may be useful in the treatment of B-cell-mediated autoimmune disorders as well as allergic immune responses [37, 43]. Addition of 1,25(OH)2D3 was also shown to inhibit the expression of IL-6, an important factor that stimulates T-helper type 17 cells, which seem considered a critical component of the autoimmune reaction [44].
Interestingly, vitamin D has been shown to inhibit antibody secretion and autoantibody production in B cells [45]. In vitro, 1,25(OH)2D3 stimulates phagocytosis and killing of bacteria by macrophages but suppresses the antigen-presenting capacity of these cells and of dendritic cells [46].
Vitamin D has been found to promote the induction of monocytic differentiation to macrophages and to modulate macrophage responses, preventing them from releasing proinflam-matory cytokines and chemokines [47]. In synthesis, the most evident effects of the vitamin D hormone on the immune system seem to be in the control of T-helper type-1-driven autoimmunity and partially the T-helper type-2-driven immune response.
Decreased vitamin D plasma levels have been linked to autoimmune diseases in humans [41]. A large population-based study (Nurses Health Study I and II) showed recently that women in the highest quintile of vitamin D intake had a 40% reduced rate of developing multiple sclerosis [48]. Experimentally it has been shown that vitamin D deficiency exacerbates both inflammatory bowel disease and multiple sclerosis in animals, and vitamin D hormone has been shown to suppress experimental encephalitis and colitis in mice [41].
Low serum levels of vitamin D might be related, among other factors, to prolonged daily darkness (reduced activation of the provitamin D by ultraviolet B sunlight), to different genetic background (that is, vitamin D receptor polymorphism) and to nutritional factors. This might also explain the latitude-related prevalence of autoimmune diseases such SLE and RA by considering the potential immunosuppressive roles of vitamin D [49].
The 25(OH)D3 plasma levels have recently been found to be inversely correlated with RA disease activity, showing a circannual rhythm (more severe in winter) [50]. In addition, greater intake of vitamin D was associated with a lower risk of RA, as well as a significant clinical improvement being strongly correlated with the immunomodulating potential in vitamin-D-treated RA patients [50].
Patients with SLE have multiple risk factors for vitamin D deficiency, and the disease complexity seems to correlate with lower 25-OH vitamin D plasma levels [51]. Consideration of the possibility of vitamin D deficiency and its treatment should therefore be mandatory in SLE patients, but also in patients with undifferentiated connective tissue disease [51–54].
Conclusion
Presently, by considering some of the major players of the NEI system, it is evident that estrogens, MLT and chronic stress (chronic stress inducing a decreased adrenal glucocorticoid release over a long time) can support the immune response. In contrast, the integrity of the vitamin D endocrine system is regarded as a potential immunosuppressive hormonal system.
Estrogens (especially in patients affected by B-cell-driven immunity) and MLT should consequently be avoided, and glucocorticoids (as replacement therapy) and vitamin D are allowed in the treatment of autoimmunity.
Abbreviations
- IFN:
-
interferon
- IL:
-
interleukin
- MLT:
-
melatonin
- NEI:
-
neuroendocrine immune
- 1:
-
25(OH)2D3: 1,25-dihydroxyvitamin D3
- RA:
-
rheumatoid arthritis
- SLE:
-
systemic lupus erythematosus
- TNF:
-
tumor necrosis factor
- VDR:
-
vitamin D receptor.
References
Cutolo M, Straub RH, Bijlsma JW: Neuroendocrine-immune interactions in synovitis. Nat Clin Pract Rheumatol. 2007, 3: 627-634.
Buttgereit F, Zhou H, Seibel MJ: Arthritis and endogenous glucocorticoids: the emerging role of the 11β-HSD enzymes. Ann Rheum Dis. 2008, 67: 1201-1203.
Adorini L, Penna G: Control of autoimmune diseases by the vitamin D endocrine system. Nat Clin Pract Rheumatol. 2008, 4: 404-412.
Elenkov IJ, Chrousos GP: Stress system – organization, physiology and immunoregulation. Neuroimmunomodulation. 2006, 13: 257-267.
Cutolo M, Straub RH: Stress as a risk factor in the pathogenesis of rheumatoid arthritis. Neuroimmunomodulation. 2006, 13: 277-282.
Bijlsma JW, Hoes JN, Van Everdingen AA, Verstappen SM, Jacobs JW: Are glucocorticoids DMARDs?. Ann NY Acad Sci. 2006, 1069: 268-274.
Straub RH, Pongratz G, Cutolo M, Wijbrandts CA, Baeten D, Fleck M, Atzeni F, Grunke M, Kalden JR, Schölmerich J, Lorenz HM, Tak PP, Sarzi-Puttini P: Increased cortisol relative to adrenocorticotropic hormone predicts improvement during anti-tumor necrosis factor therapy in rheumatoid arthritis. Arthritis Rheum. 2008, 58: 976-978.
Cutolo M, Cimmino MA, Sulli A: Polymyalgia rheumatica vs late-onset rheumatoid arthritis. Rheumatology (Oxford). 2009, 48: 93-95.
Straub RH, Cutolo M: Further evidence for insufficient hypothalamic–pituitary–glandular axes in polymyalgia rheumatica. J Rheumatol. 2006, 33: 1219-1223.
Straub RH, Cutolo M: Circadian rhythms in rheumatoid arthritis: implications for pathophysiology and therapeutic management. Arthritis Rheum. 2007, 56: 399-408.
Cutolo M, Seriolo B, Craviotto C, Pizzorni C, Sulli A: Circadian rhythms in RA. Ann Rheum Dis. 2003, 62: 593-596.
Arvidson NG, Gudbjornsson B, Elfman L, Ryden AC, Totterman TH, Hallgren R: Circadian rhythm of serum interleukin-6 in rheumatoid arthritis. Ann Rheum Dis. 1994, 53: 521-524.
Cutolo M, Straub RH, Buttgereit F: Circadian rhythms of nocturnal hormones in rheumatoid arthritis: translation from bench to bedside. Ann Rheum Dis. 2008, 67: 905-908.
Bijlsma JW, Jacobs J: Innovative use of glucocorticoids in patients with rheumatoid arthritis. Lancet. 2008, 371: 183-184.
Buttgereit F, Doering G, Schaeffler A, Witte S, Sierakowski S, GromnicaIhle E, Jeka S, Krueger K, Szechinski J, Alten R: Efficacy of modified-release versus standard prednisone to reduce duration of morning stiffness of the joints in rheumatoid arthritis (CAPRA-1): a double-blind, randomised controlled trial. Lancet. 2008, 371: 183-184.
Hoes JN, Jacobs JW, Boers M, Boumpas D, Buttgereit F, Caeyers N, Choy EH, Cutolo M, Da Silva JA, Esselens G, Guillevin L, Hafstrom I, Kirwan JR, Rovensky J, Russell A, Saag KG, Svensson B, Westhovens R, Zeidler H, Bijlsma JW: EULAR evidence-based recommendations on the management of systemic glucocorticoid therapy in rheumatic diseases. Ann Rheum Dis. 2007, 66: 1560-1567.
Garcia-Maurino S, Gonzalez-Haba MG, Calvo JR, Rafii-El-Idrissi M, Sanchez-Margalet V, Goberna R, Guerrero JM: Melatonin enhances IL-2, IL-6, and IFN-γ production by human circulating CD4+ cells. J Immunol. 1997, 159: 574-581.
Cutolo M, Villaggio B, Candido F, Valenti S, Giusti M, Felli L, Sulli A, Accardo S: Melatonin influences interleukin-12 and nitric oxide production by primary cultures of rheumatoid synovial macrophages and THP-1 cells. Ann NY Acad Sci. 1999, 876: 246-254.
Petrovsky N, McNair P, Harrison LC: Diurnal rhythms of pro-inflammatory cytokines: regulation by plasma cortisol and therapeutic implications. Cytokine. 1998, 10: 307-312.
Forrest CM, Mackay GM, Stoy N, Stone TW, Darlington LG: Inflammatory status and kynurenine metabolism in rheumatoid arthritis treated with melatonin. Br J Clin Pharmacol. 2007, 64: 517-526.
Cutolo M, Maestroni GJ, Otsa K, Aakre O, Villaggio B, Capellino S, Montagna P, Fazzuoli L, Veldi T, Peets T, Hertens E, Sulli A: Circadian melatonin and cortisol levels in rheumatoid arthritis patients in winter time: a north and south Europe comparison. Ann Rheum Dis. 2005, 64: 212-216.
Maestroni G, Otsa K, Cutolo M: Melatonin treatment does not improve rheumatoid arthritis. Br J Clin Pharmacol. 2008, 65: 797-798.
Straub RH: The complex role of estrogens in inflammation. Endocr Rev. 2007, 28: 521-574.
Cutolo M: Sex and rheumatoid arthritis: mouse model versus human disease. Arthritis Rheum. 2007, 56: 1-3.
Cutolo M, Capellino S, Straub RH: Oestrogens in rheumatic diseases: friend or foe?. Rheumatology (Oxford). 2008, 47 (Suppl 3): iii2-iii5.
Castagnetta LA, Carruba G, Granata OM, Stefano R, Miele M, Schmidt M, Cutolo M, Straub RH: Increased estrogen formation and estrogen to androgen ratio in the synovial fluid of patients with rheumatoid arthritis. J Rheumatol. 2003, 30: 2597-2605.
Cutolo M, Sulli A, Capellino S, Villaggio B, Montagna P, Seriolo B, Straub RH: Sex hormones influence on the immune system: basic and clinical aspects in autoimmunity. Lupus. 2004, 13: 635-638.
Hughes GC, Clark EA: Regulation of dendritic cells by female sex steroids: relevance to immunity and autoimmunity. Autoimmunity. 2007, 40: 470-481.
Cohen-Solal JF, Jeganathan V, Grimaldi CM, Peeva E, Diamond B: Sex hormones and SLE: influencing the fate of autoreactive B cells. Curr Top Microbiol Immunol. 2006, 305: 67-88.
Cutolo M, Capellino S, Sulli A, Serioli B, Secchi ME, Villaggio B, Straub RH: Estrogens and autoimmune diseases. Ann NY Acad Sci. 2006, 1089: 538-547.
Cutolo M, Sulli A, Capellino S, Villaggio B, Montagna P, Seriolo B, Straub RH: Sex hormones influence on the immune system: basic and clinical aspects in autoimmunity. Lupus. 2004, 13: 635-638.
Ganesan K, Selvam R, Abhirami R, Raju KV, Manohar BM, Puvanakrishnan R: Gender differences and protective effects of testosterone in collagen induced arthritis in rats. Rheumatol Int. 2008, 28: 345-353.
Adorini L: Intervention in autoimmunity: the potential of vitamin D receptor agonists. Cell Immunol. 2007, 233: 115-124.
van Etten E, Mathieu C: Immunoregulation by 1,25-dihydroxyvitamin D3: basic concepts. J Steroid Biochem Mol Biol. 2005, 97: 93-101.
Hewison M, Freeman L, Hughes SV, Evans KN, Bland R, Eliopoulos AG, Kilby MD, Moss PA, Chakraverty R: Differential regulation of vitamin D receptor and its ligand in human monocyte-derived dendritic cells. J Immunol. 2003, 170: 5382-5390.
Cadranel J, Garabedian M, Milleron B, Guillozo H, Akoun G, Hance AJ: 1,25(OH)2 D3 production by T lymphocytes and alveolar macrophages recovered by lavage from normocalcemic patients with tuberculosis. J Clin Invest. 1990, 85: 1588-1593.
Chen S, Sims GP, Chen XX, Gu YY, Chen S, Lipsky PE: Modulatory effects of 1,25-dihydroxyvitamin D3 on human B cell differentiation. J Immunol. 2007, 179: 1634-1647.
Deluca HF, Cantorna MT: Vitamin D: its role and uses in immunology. FASEB J. 2001, 15: 2579-2585.
Mahon BD, Wittke A, Weaver V, Cantorna MT: The targets of vitamin D depend on the differentiation and activation status of CD4 positive T cells. J Cell Biochem. 2003, 89: 922-932.
Boonstra A, Barrat FJ, Crain C, Heath VL, Savelkoul HF, O'Garra A: 1α,25-Dihydroxyvitamin d3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J Immunol. 2001, 167: 4974-4980.
Cantorna MT, Mahon BD: Mounting evidence for vitamin D as an environmental factor affecting autoimmune disease prevalence. Exp Biol Med (Maywood). 2004, 229: 1136-1142.
Staeva-Vieira TP, Freedman LP: 1,25-dihydroxyvitamin D3 inhibits IFNγ and IL-4 levels during in vitro polarization of primary murine CD4+ T cells. J Immunol. 2002, 168: 1181-1189.
Heine G, Niesner U, Chang HD, Steinmeyer A, Zügel U, Zuberbier T, Radbruch A, Worm M: 1,25-dihydroxyvitamin D(3) promotes IL-10 production in human B cells. Eur J Immunol. 2008, 38: 2210-2218.
Stockinger B: Th17 cells: an orphan with influence 4. Immunol Cell Biol. 2007, 85: 83-84.
Linker-Israeli M, Elstner E, Klinenberg JR, Wallace DJ, Koeffler HP: Vitamin D(3) and its synthetic analogs inhibit the spontaneous in vitro immunoglobulin production by SLE-derived PBMC. Clin Immunol. 2001, 99: 82-93.
Griffin MD, Lutz WH, Phan VA, Bachman LA, McKean DJ, Kumar R: Potent inhibition of dendritic cell differentiation and maturation by vitamin D analogs. Biochem Biophys Res Commun. 2000, 270: 701-708.
Helming L, Bose J, Ehrchen J, Chiebe S, Frahm T, Geffers R, Probst-Kepper M, Balling R, Lengeling A: 1α,25-Dihydroxyvitamin D3 is a potent suppressor of interferon gamma-mediated macrophage activation. Blood. 2005, 106: 4351-4358.
Munger KL, Zhang SM, O'Reilly E, Hernán MA, Olek MJ, Willett WC, Ascherio A: Vitamin D intake and incidence of multiple sclerosis. Neurology. 2004, 62: 60-65.
Cutolo M, Otsa K, Uprus M, Paolino S, Seriolo B: Vitamin D in rheumatoid arthritis. Autoimmun Rev. 2007, 7: 59-64.
Cutolo M, Otsa K, Laas K, Yprus M, Lehtme R, Secchi ME, Sulli A, Paolino S, Seriolo B: Circannual vitamin D serum levels and disease activity in rheumatoid arthritis: Northern versus Southern Europe. Clin Exp Rheumatol. 2006, 24: 702-704.
Ruiz-Irastorza G, Egurbide MV, Olivares N, Martinez-Berriotxoa A, Aguirre C: Vitamin D deficiency in systemic lupus erythematosus: prevalence, predictors and clinical consequences. Rheumatology (Oxford). 2008, 47: 920-923.
Zold E, Szodoray P, Gaal J, Kappelmayer J, Csathy L, Gyimesi E, Zeher M, Szegedi G, Bodolay E: Vitamin D deficiency in undifferentiated connective tissue disease. Arthritis Res Ther. 2008, 10 (5): R123-
Cutolo M: Vitamin D or hormone D deficiency in autoimmune rheumatic diseases, including undifferentiated connective tissue disease. Arthritis Res Ther. 2008, 6: 123-
Cutolo M, Otsa K, Paolino S, Yprus M, Veldi T, Seriolo B: Vitamin D involvement in rheumatoid arthritis and systemic lupus erythaematosus. Ann Rheum Dis. 2009, 68: 446-447.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Cutolo, M., Straub, R.H. Insights into endocrine-immunological disturbances in autoimmunity and their impact on treatment. Arthritis Res Ther 11, 218 (2009). https://doi.org/10.1186/ar2630
Published:
DOI: https://doi.org/10.1186/ar2630