
Abstract
The nucleotide-binding and oligomerization domain, leucine-rich repeat (also known as NOD-like receptors, both abbreviated to NLR) family of intracellular pathogen recognition receptors are increasingly being recognized to play a pivotal role in the pathogenesis of a number of rare monogenic diseases, as well as some more common polygenic conditions. Bacterial wall constituents and other cellular stressor molecules are recognized by a range of NLRs, which leads to activation of the innate immune response and upregulation of key proinflammatory pathways, such as IL-1β production and translocation of nuclear factor-κB to the nucleus. These signalling pathways are increasingly being targeted as potential sites for new therapies. This review discusses the role played by NLRs in a variety of inflammatory diseases and describes the remarkable success to date of these therapeutic agents in treating some of the disorders associated with aberrant NLR function.
Similar content being viewed by others

Introduction
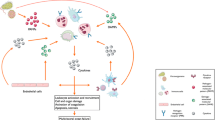
Innate immunity plays a critical role in host protection and employs an array of receptor molecules, including Toll-like receptors (TLRs), NOD-like receptors (nucleotide-binding and oligomerization domain, leucine-rich repeat; both abbreviated to NLR), retinoic acid-inducible gene-like receptors and C-type lectin receptors (CLRs). Pathogen recognition receptors (PRRs), which serve to alert and activate the defence system, are highly conserved at the molecular level between yeast 'stress' proteins, plants (the resistance [R] proteins), invertebrates (the Drosophilia Toll molecules) and vertebrates (Figure 1).

Species homology between the Toll, TLRs, NLRs and plant resistance (R) proteins. Central to innate immunity are the highly conserved core domains that are found in drosophilae, mammals and plants. The Toll family of proteins share homology in their signalling domains with IL-1RI; this family includes Drosophila Toll, plant R proteins, and mammalian TLRs and NLRs. CARD, caspase activation and recruitment; IL-1RI, type I IL-1 receptor; LRR, leucine rich repeat; NALP1, NACHT, leucine rich repeat and pyrin domain containing 1; NLR, NOD-like receptor; PYD, pyrin domain; TLR, Toll-like receptor.
The unexpected finding that the Toll family of proteins share homology in their signalling domains with the type 1 IL-1 (IL-1β) receptor has considerably improved our understanding of IL-1 signalling pathways. This discovery was drawn from many sources, including Drosophilia developmental genetics, yeast genetics and studies of disease in plants. The IL-1 family plays an important role in the genesis of inflammation and host defence, and up to 11 members of this family have been identified to date [1, 2]. Functional roles have been attributed to five members of this family (IL-1α, IL-1β, IL-18, IL-1 receptor antagonist and the more recently reported IL-33). Both IL-1α and IL-1β are proinflammatory cytokines that are synthesized as precursor molecules, but the IL-1α precursor, unlike IL-1β, is biologically active. Pro-IL-1β requires enzymatic cleavage by caspase-1 to be activated [3, 4], which is also true of IL-18 and possibly IL-33 -the more recently discovered member of IL-1 family.
A series of coordinated interactions between the two major groups of receptor molecules in the mammalian innate immune system, the TLRs and NLRs, lead to comprehensive detection of toxins and 'stress' signals at both intracellular and extra-cellular levels, resulting in a specific response being mounted against a range of pathogens. The mammalian family of TLRs is composed primarily of cell-surface receptors, characterized by the presence of an extracellular leucine-rich repeat (LRR) motif. The NLRs, which also contain LRR domains, are part of an intracellular detection system for microbial and danger-associated molecules from both the extracellular and intracellular microenvironments. The range of patterns that is recognized by these molecules is collectively referred to as pathogen-associated molecular patterns (PAMPs) [5], and these in turn promote upregulation of costimulatory molecules, with subsequent priming of T cells, and secretion of inflammatory cytokines by innate immune cells [6–9]. Thus, the PRRs provide an effective recognition system for both PAMPs and damage-associated molecular patterns (DAMPs), which are a second variety of molecules released as a result of tissue injury [10].
At this point it is worth noting that an agreed standard nomenclature for the NLR family is still lacking; in this review we follow the historic precedent of NLR being defined as 'NOD-like receptor', with acknowledgement that the Human Genome Organization Gene Nomenclature Committee has proposed the term 'nucleotide-binding domain, leucine rich repeat containing family' as an alternative description for the NLR abbreviation [11]. However, there remains considerable inconsistency concerning nomenclature of the NLR group found in various publications. For instance, NOD1 (nucleotide-binding oligomerization domain containing 1) may also be termed NLRC1 (NLR family, CARD domain containing 1), and NALP1 (NACHT, leucine rich repeat and PYD [pyrin domain] containing 1) termed NLRP1 (NLR family, pyrin domain containing 1), and so on. We again refer to the historic terminology of NOD and NALP throughout, rather than the proposed Human Genome Organization terminology.
Members of the NLR family share common structural and functional similarities with the TLRs, which include a carboxyl-terminal LRR; a central nucleotide binding domain (NACHT) domain, which has intrinsic ATPase activity; and an amino-terminal protein-protein interaction domain, which contains either a caspase activation and recruitment (CARD) domain or a baculovirus inhibitory repeat domain [12]. The carboxyl-terminal LRR of the NLRs is responsible for sensing PAMPs, thereby performing a similar role to that of TLRs. For a comprehensive description of the tripartite structures of the NLR family members, agonists and the adaptor molecules, the reader is referred to the review by Sirad and coworkers [13].
There are two broad functional divisions within the NLRs, both of which are associated with the presence of large intracytoplasmic protein complexes; these are the inflammasomes, which include the NALP and IL-1β converting enzyme protease activating factor (Ipaf) inflammasomes, involved in proinflammatory cytokine production [14], and the Nodosomes, which induce antimicrobial effectors such as peptides and nitric oxide as well as stimulating proinflammatory signalling and cytokine networks [15]. The inflammasomes all essentially contain either a NALP or an Ipaf central protein, plus an adaptor protein, and a caspase recruitment domain (CARD), which facilitates the activation of caspase-1 or caspase-5 (Figure 2). The NALP1 inflammasome was the first such multimeric complex to be described, in 2002 by Martinon and coworkers [16], when it was found to assemble as a result of bacterial intracellular stress signals or toxins, with subsequent caspase-1 and caspase-5 activation. Previous studies had found an association of the adaptor protein PYCARD (also termed apoptosis-associated speck-like protein [ASC], which we use in this review) with IL-1β; this conversion of pro-IL-1β to its active form required the activation of caspase-1 [17], but a second stimulus, such as ATP, nigericin or bacterial toxins, was also required to induce the formation of the inflammasome, and to enhance the proteolytic maturation and secretion of IL-1β [18].

The NALP1 and NALP3 inflammasome complexes. Both NALP1 and NALP3 associate through homotypic interactions between CARD, ASC and the PYD domains. NALP3 requires a secondary adaptor protein Cardinal to facilitate the activation of caspase-1 and the subsequent cleavage of pro-IL-1, in addition to the adaptor protein ASC. This is not required for the NALP1 inflammasome, which has additional FIIND and CARD domains attached to the core NALP1 protein. ASC, apoptosis-associated speck-like protein; CARD, caspase activation and recruitment; FIIND, domain with a function to find; IL, interleukin; LRR, leucine rich repeat; NALP, NACHT, leucine rich repeat and pyrin domain containing 1; PYD, pyrin domain.
IL-1β is involved in the pathogenesis of numerous diseases with an inflammatory component [19], which is best demonstrated by the therapeutic benefits of treating these conditions with IL-1 agonists, such as IL-1 receptor antagonist. These diseases include hereditary periodic fevers, the prototypic autoinflammatory syndromes [20, 21], which are discussed in greater detail below.
The inflammasomes
NALP1 inflammasome
To date, 14 NALP proteins have been identified in the mammalian host [22], some with undetermined functions. Those NALPs that have been demonstrated to form inflammasome complexes (NALP1 and NALP3) play a major role in the initiation of the innate immune system, as well as priming adaptive immunity, and are essential for cytosolic detection of multiple DAMPs and PAMPs (Figure 2).
NALP1 (NLRP1, CARD7, DEFCAP, CLR17.1) was the first NALP protein to be identified [16, 23, 24], and after discovery of the NALP1 inflammasome other proteins with homology to NALP1 were also found to form similar large intracellular complexes. NALP1 recruits the ASC adaptor protein, as well as caspase-1 and caspase-5, to form its inflammasome [25], thereby activating IL-1β from its inactive pro form. In vitro studies suggest that the bacterial cell wall product muramyl dipeptide (MDP) binds directly and activates NALP1, although some in vivo studies have been inconclusive on this point [26]. The involvement of ASC in the assembly of the NALP1 inflammasome is also somewhat controversial, because in vitro reconstitution experiments have demonstrated that ASC enhances but is not an absolute requirement for NALP1-mediated caspase-1 activation [18], although it may be required in vivo [17].
NALP1 is widely expressed at low levels in many cell types, but it is highly expressed in immune cells, particularly T cells and Langerhans' cells [27]. There are two splice variants of NALP1, one of which does not contain an LRR. Deletion of this domain renders the protein active and able to bind ATP, without need for MDP binding to prime the complex [18]. Variants of NALP1 confer susceptibility to vitiligo, a condition in which white patches appear on the skin due to a loss of pigment-producing cells [28]. Absence of the LRR domain leads to constitutive activation of the NALP1 inflammasome, suggesting that there is no requirement for ligand binding to facilitate cleavage of IL-1β, with associated elevated IL-1β serum levels found in patients with vitiligo. NALP1 can also induce apoptosis in a variety of cell types, and over-expression stimulates caspase-mediated apoptosis [23, 24, 29].
NALP3 inflammasome
NALP3 (cryopyrin, PYPAF1, CIAS1, CLR1.1, NLRP3) also forms an inflammasome complex, similar to NALP1 [16], which mediates intracellular processing of proinflammatory caspases and cytokine production [30]. This inflammasome has largely been studied in the human acute monocytic leukemia cell line THP-1, and its precise physiological role in primary cells is yet to be fully elucidated. This inflammasome is comprised of NALP3; ASC; and pyrin protein, which contains a pyrin domain (PYD), caspase-1 and Cardinal. The function of NALP3 is better characterized than those of other NALP proteins, and its inflammasome assembles in response to both exogenous and endogenous PAMPs and DAMPs. Activators of the NALP3 inflammasome include bacterial peptidoglycan; extracellular ATP, which activates the purigenic P2X7 receptor [31]; low intracellular potassium [32]; nigericin [33]; changes in ionic composition and uric acid crystals within the cyoplasm [32]; and the presence of DNA/RNA [34] and silica [35–37], which have both recently been described.
Mutations in the NALP3 (NLRP3, CIAS1) gene, which encodes the NALP3 protein, have been associated with a group of autoinflammatory diseases termed the cryopyrin-associated periodic syndromes (CAPS; cyropyrinopathies) [38, 39]. These rare monogenic conditions include familial cold autoinflammatory syndrome; Muckle-Wells syndrome; and chronic infantile neurologic, cutaneous and articular syndrome (CINCA)/neonatal onset multisystem inflammatory disease (NOMID). CAPS are caused by gain of function mutations [40] and are thought to share a common mechanism, whereby the closed and inactive structure of NALP3 is disrupted by the various mutations, leading to activation of the inflammasome complex and IL-1β release [41].
The CAPS disorders are classified individually, but they have overlapping symptoms that include fevers, urticarial skin rashes, varying degrees of arthragias/arthritis, neutrophil-mediated inflammation and an acute-phase response [42]. CINCA/NOMID is the most severe clinical phenotype, with signs of central nervous system inflammation and skeletal malformations. Functional studies of macrophages from patients with CINCA/NOMID and Muckle-Wells syndrome have revealed constitutive increases in the secretion of IL-1β and IL-18 [43–45], suggesting that mutations in NALP3 (NLRP3, CIAS1) increase production of these proinflammatory cytokines. Preliminary data reported by Takada and colleagues [46] indicate that a mutation in exon 3 of NALP3 (NLRP3, CIAS1) enhanced monocytic cell death in peripheral blood mononuclear cells of a patient with a mild phenotype of CINCA/NOMID, in response to lipopolysaccharide stimulation.
Mutations in other components of the NALP3 inflammasome platform have also been shown to perpetuate excessive IL-1β production. Pyrin (the protein encoded by the MEFV gene) is mutated in familial Mediterranean fever, an autosomal recessive autoinflammatory disorder in which mutated pyrin is thought to lead to a reduced ability to moderate IL-1β activity [47]. Pyrin interacts with the NALP3 and ASC proteins through homotypic PYD-PYD domains, and it has been proposed by some workers that pyrin negatively regulates caspase-1 by competing for binding with ASC. In patients with familial Mediterranean fever the mutated MEFV results in altered conformation of the B30.2 (SPRY) domain at the carboxyl-terminus, leading to impaired ligand binding and thereby affecting inflammasome activity and IL-1β production [48]. Impaired pyrin-mediated IL-1β regulation is also implicated in the pathogenesis of an autosomal dominant autoinflammatory condition termed pyogenic sterile arthritis, pyoderma gangrenosum and acne (PAPA) syndrome. In these patients a mutation in the PSTPIP1 (proline serine threonine phosphatase-interacting protein 1) gene leads to an increased interaction between PSTPIP and pyrin, resulting in reduced modulation of the NALP3 inflammasome by pyrin [49]. This, in turn, causes a proinflammatory clinical phenotype; thus, there is a biochemical pathway that is common to both familial Mediterranean fever and PAPA, although the precise mechanisms have not been fully elucidated [50].
Both the NALP3 (NLRP3, CIAS1) and MEFV genes were also associated with psoriatic juvenile idiopathic arthritis [51], suggesting the potential for shared disease mechanisms between various autoinflammatory syndromes, involving abnormal production of IL-1β. The MEFV gene is also mutated in a significant proportion of patients with ulcerative colitis, with a number of these having an associated inflammatory arthritis [52, 53]. NALP3 expression may also be increased in complex conditions such as hypertension [54], rheumatoid arthritis [55] and osteoarthritis [56], although the precise roles in these conditions are yet to be elucidated.
NALP3 and biological therapy
Activation of the NALP3 inflammasome leads to production of active cleaved forms of IL-1β and IL-18. Biological therapies that target IL-1β, and the proinflammatory effects of this cytokine, include receptor antagonists (IL-1 receptor antagonist) and biological molecules such as monoclonal antibodies and soluble receptors that block IL-1β (see below). Martinon and coworkers [57] demonstrated that the NALP3 inflammasome was activated by monosodium urate crystals, which are deposited in joints and periarticular tissues in gout, and by crystals of calcium pyrophosphate dihydrate, which is the causative agent in pseudogout, leading to the maturation of IL-1β and IL-18. The mouse model of monosodium urate crystal induced inflammation has successfully been treated with anakinra [57], a recombinant the IL-1 receptor antagonist, and this work has led to successful human trials and a pilot study of 10 patients with gout. All of these patients responded to treatment with anakinra [58], demonstrating the potential to treat gout and pseudogout patients with this agent [59, 60].
Anakinra has also been used therapeutically in a number of diseases that are associated with excessive IL-1β production, including Muckle-Wells syndrome [61–68], familial cold auto-inflammatory syndrome [65, 69–73], NOMID/CINCA [74–76] and Schnitzler's syndrome [77] (Table 1).
The NALP3 inflammasome may also be associated with common autoimmune diseases with IL-1β involvement, including rheumatoid arthritis. The human IL-1β monoclonal antibody ACZ855 (produced by Novartis, basel, Switzerland) has been used in a small clinical study of patients with rheumatoid arthritis, and initial findings indicate greater efficiency of ACZ855 in rheumatoid arthritis compared with anakinra, and that the half-life is extended [78].
IL-1β Trap (rilonacept), a fusion protein consisting of human cytokine receptor extracellular domains and the Fc portion of human IgG1, incorporates the extracellular signalling domain of both IL-1 receptors, namely the type I IL-1 receptor and the IL-1 accessory protein. Rilonacept has been used in pilot studies for the treatment of systemic-onset juvenile idiopathic arthritis, atherosclerosis and CAPS [79].
In patients with rheumatoid arthritis receiving the biological response modifier (biologic) infliximab, a monoclonal antibody to tumour necrosis factor (TNF), there were significantly lower NALP3 transcript levels in those patients who later were classified as responders (according to the EULAR [European League Against Rheumatism] DAS28 [Disease Activity Score using 28 joint counts] criteria) before starting treatment (baseline) with this therapy [80]. NALP3 mRNA levels were reduced further after treatment, suggesting that the NALP3 inflammasome plays a specific role in the pathogenesis of rheumatoid arthritis and in the response of these patients to treatment.
These preliminary data contrast with the findings of Karababa and coworkers [81] in the experimental in vivo antigen-induced arthritis model, in which it was recently demonstrated that NALP3 and Ipaf were not necessary for the development of arthritis, but that the ASC adaptor protein was essential. It was suggested that there is involvement of an inflammasome complex containing ASC in this model, with possible interactions with other members of the NALP family.
Inflammasomes and inflammatory skin disease
There has been considerable recent interest in the pathogenesis of other autoinflammatory skin diseases such as psoriasis and contact hypersensitivity. The latter is a common T lymphocyte mediated allergic disease that is characterized by local inflammatory skin reactions, following contact with small reactive compounds called haptens, in which the inflammatory skin lesions are associated with inflammasome activation. In psoriasis, there is activation of caspase-1 and IL-18 secretion, which is regulated in a p38 mitogen-activated protein kinase/caspase-1 dependent manner [82].
Ipaf inflammasome
The Ipaf (NLRC4, CARD12, CLAN, CLR2.1) protein, which is homologous to NALP1 and NALP3, also forms an inflammasome in response to the detection of flagellin within the cytoplasm, and this also causes activation of caspase-1 [83, 84]. The Ipaf inflammasome contains an amino-terminal CARD, a central NACHT domain and a carboxyl-terminal LRR, and activation of this complex induces the combined activation of the TLR and NLR pathways. The extracellular portion of flagellin is detected by TLR5, and the intracellular portion of flagellin promotes formation of this inflammasome [85]. The appearance of flagellin within the cytoplasm, which announces the arrival of a virulent form of bacteria, prompts the development of both an adaptive response (initiated by TLR5) and an innate immune response. This combined intracellular and extracellular recognition of microbial components mediates rapid pathogen clearance [14].
Mutations in other NALP family members
Mutations in genes encoding other NALP family members also have pathogenic consequences: the NALP1 locus is associated with vitiligo-associated autoimmune disease [28, 86]; NALP7 (NOD12, NLRP7, PYPAF3, CLR19.4) mutations may result in hydatidiform mole [87]; CIITA mutations are associated with bare lymphocyte syndrome [88] and multiple sclerosis [89]; and NOD2 mutations are associated with Crohn's disease and Behçet's syndrome [90, 91]. All of these disease associations emphasize the role played by the NALP family in the pathogenesis of the autoinflammatory-autoimmune disease continuum [92].
Although the NALP1, NALP3 and Ipaf inflammasomes were originally regarded as separate complexes that assemble upon the detection of different stimuli, it is possible that the central component may induce activation of various complexes in a different manner, depending on the nature of the stimuli. Thus, ASC and Ipaf were originally described as being part of different complexes, and Ipaf and caspase-1 (but not ASC) are implicated in Legionella flagellin recognition [85]. Shigella induces caspase-1 activation and IL-1β production by a mechanism involving both ASC and Ipaf [93], which are regarded as components of separate inflammasomes.
The Nodosomes
NOD1 and NOD2
NOD1 and NOD2 are two further NLRs that recognize PAMPs and are implicated in innate immune responses. NOD1 recognizes γ-D-glutamyl-meso-diaminopimelic acid (DAP), a dipeptide derived from peptidoglycans of most Gram-negative bacteria; NOD2 senses MDP, which is a constituent of most Gram-negative and Gram-positive bacterial peptidoglycans [94]. In the basal state, the LRR region of NOD2 represses activation of the nucleotide-binding domain, preventing spontaneous oligomerization [18]; however, upon DAP and MDP sensing, a conformational change in the LRR region allows for oligomerization of the NACHT domain and subsequent activation of CARD, thereby allowing for downstream activation of effector molecules [95].
NOD signalling
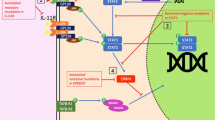
In response to muropeptides, both NOD1 and NOD2 recruit an adaptor protein containing a CARD domain, namely the serine threonine kinase receptor-interacting protein (RIP)2 (also known as RICK and CARDIAK), which assembles via CARD-CARD homotypic binding. This, in turn, allows for oligomerization of RIP2 and interaction with the IκB kinase (IKK) complex (IKKα, IKKβ, and nuclear factor-κB [NF-κB] essential modifier, abbreviated to NEMO). Ubiquitination of this inhibitory complex results in the release and nuclear translocation of the NF-κB transcription factor and subsequent transcription of NF-κB-dependent proinflammatory genes [96, 97] (Figure 3). RIP2 is crucial in this signalling pathway, as demonstrated in RIP2-/- mice [98], in which MDP-induced NOD activation of NF-κB is abolished. RIP2 has also recently been shown to signal specifically for NOD but not TLRs [99], and indeed NOD signalling is independent of Myd88, which is a key adaptor molecule in the TLR signalling pathway [100]. In addition to NF-κB activation, NOD signalling also leads to activation of mitogen-activated protein kinases, further enhancing the proinflammatory state [99, 101].

Nodosome signalling. Ligand binding to the LRR region regulates oligomerization of the NACHT domain and homotypic interactions between CARD domains and RIP2. Ubiquitination of the IKK complex following oligomerization of RIP2 allows for nuclear translocation of NF-κB and subsequent upregulation of proinflammatory cytokines. CARD, caspase activation and recruitment; IKK, IκB kinase; LRR, leucine rich repeat; NEMO, NF-κB essential modifier; NF-κ, nuclear factor-κB; RIP, receptor-interacting protein.
NOD1
NOD1 has been extensively implicated in the handling of a variety of bacteria, and the intracellular nature of such sensing has also been confirmed. An invasive strain of the Gram-negative bacterium Shigella flexneri can also activate NF-κB and IL-8 expression in colonic epithelial cells, but the noninvasive strain does not have this effect. This process is driven by lipopolysaccharide but does not involve sensing by TLRs [102, 103]; indeed, colonic epithelium is refractory to extracellular lipopolysaccharide stimulation, thereby preventing aberrant cellular responses to commensal bacteria. Subsequent to this work, it was demonstrated that oligomerization of NOD1 was responsible for the intra-cellular pathogenicity of S. flexneri and consequent activation of NF-κB [101, 103]. Helicobacter pylori, another Gram-negative noninvasive bacterium, is recognized by NOD1 in epithelial cells in cag pathogenicity island positive bacteria [104]. More severe pathological consequences of H. pylori infection are determined by the cag pathogenicity island, and only strains containing cag pathogenicity island activate NF-κB proinflammatory cytokines [105]. The delivery of muropeptide from this noninvasive bacterium appears to be via a type IV secretion system, directly into the host cell [106], again suggesting pathogen sensing independent of TLRs. NOD2 is also implicated in H. pylori sensing, and the NOD2 mutant R720W increases risk for gastric lymphoma [107], which is a recognized consequence of chronic H. pylori infection.
NOD1 has also been demonstrated to be the PRR for many other bacteria, including the common pathogens Campylobacter jejuni [108], Pseudomonas aeruginosa [109], Escherichia coli [103], and Chlamydia trachomatis and Chlamydia muridarum, with a dominant negative NOD1, or NOD1 depletion, being less effective in activating NF-κB in the case of Chlamydia spp. [110].
NOD1 mutants are implicated in disease to a lesser extent than NOD2 mutants. The NOD1 gene is found on chromosome 7p14, a region that has already been linked to atopy [111]. Weidinger and coworkers [112] analyzed 11 polymorphisms in the NOD1 gene for associations with atopic phenotypes, with some polymorphisms exhibiting association with atopic eczema and asthma. With respect to Crohn's disease (a chronic granulomatous inflammatory disorder of the bowel found in patients carrying mutations in NOD2 in up to 40% of cases [90]), NOD1 mutants have not been reported to confer disease susceptibility to this disorder [113–115].
NOD2
NOD2 mutations has been implicated in several inflammatory disorders, including Crohn's disease [90], Blau syndrome [91], which is a rare autosomal dominant disorder that causes granulomatous inflammation of the skin, arthritis, uveitis and lymphadenopathy, as well as early onset sarcoidosis [116, 117]. NOD2 has been most extensively investigated in inflammatory bowel disease (IBD). It was described as the first susceptibility locus for Crohn's disease in 2001, within the IBD1 region on chromosome 16 [90, 118]. This was the first evidence of a link between the innate immune system and inflammatory processes in Crohn's disease, a disease that was widely accepted to be T-helper-1 driven until that point, and therefore assumed to be a disease of the adaptive immune system [119].
Much work since then has addressed whether mutations in the NOD2 gene lead to a gain or loss of function of the NOD protein. There are three major NOD2 single nucleotide polymorphisms (SNPs), two missense mutations (Arg702Trp and Gly908Arg) and one frameshift mutation (3020insC → 1007fs) [118, 120]. All of these SNPs affect the LRR region of the NOD2 protein, resulting in defective sensing of MDP [121]. The inability of mutant NOD2 to detect microbial constituents translates into a lack of activation of NF-κB and subsequent decreased IL-1β release [122]. However, this does not concur with the clinical picture of active Crohn's disease, and indeed it has been known for many years that IL-1β levels are significantly increased in patients with active Crohn's disease [123], as are other cytokines that are NF-κB dependent, such as IL-6 and IL-12 [124, 125].
This apparent dichotomy may be explained by appreciating the cellular function of NOD2 and its interaction with other PRRs, such as TLRs. Peptidoglycan, from which MDP is derived, is also the PAMP recognized by TLR2; on stimulating NOD-/- cells, which are incapable of sensing MDP, with peptidoglycan, there is an enhanced TLR2 response. Levels of the c-rel subunit of NF-κB increase, thereby regulating an increase in IL-12 and hence increased inflammation. These data suggest an inhibitory regulatory function of NOD2 with respect to TLR2 signalling, because in cells with wild-type NOD2, which sense MDP, the TLR2 peptidoglycan response is inhibited [126, 127]. In human monocytes, Borm and coworkers [128] have demonstrated that low levels of MDP stimulate a synergistic response between TLR2 and NOD2, but this synergism is lost at higher doses of MDP, with decreased inflammatory responses. Mutant NOD2 cannot sense MDP, and so this inhibitory effect is lost and the TLR2 response is heightened [126–128]. The dose-dependent inhibition of TLR2 may help to explain why functioning NOD2 is able to handle commensal bacteria in the gastrointestinal tract, without the aberrant inflammation seen in NOD2 mutants. However, other studies have failed to corroborate the TLR2 story, with Kobayashi and colleagues [129] reporting similar responses to TLR ligands in wild-type and NOD2-/-cells, and indeed an increase in IL-6 in wild-type NOD2 cells on stimulation with Pam3CSK4, a TLR2 ligand [129].
A further consideration is the role that NOD2 plays in mucosal defence. It is widely postulated that the pathogenesis of Crohn's disease is, at least in part, due to defective intestinal barrier function. Paneth cells, found in the crypts of Lieberkuhn in the small intestine, are specialized intestinal innate immune cells, which are responsible for the production and secretion of antimicrobial peptides, such as defensins, in response to luminal bacterial products, such as MDP [130, 131].
NOD2 is most abundantly distributed in Paneth cells in the terminal ileum of patients with Crohn's disease and healthy control individuals, particularly in ileal crypts [132]. The mutated NOD2 phenotype is most frequently associated with ileal disease [133, 134], which therefore may implicate defective Paneth cell function as a disease mechanism. Indeed, decreased expression of human α-defensins HD5 and HD6 is reported in Crohn's disease patients who have mutated NOD2, leading to a subsequent increase in microbial flora in transgenic mice models [135]. The increase in luminal bacteria, and the decreased clearance, may therefore perpetuate bacterial stasis in the intestinal crypts and further exacerbate the inflammatory response [129].
Finally, NOD2 mutations may decrease the expression of the regulatory cytokine IL-10 in dendritic cells at least, which may implicate NOD2 in disordered regulation of inflammatory cytokines, such as TNF, IL-12 and suppressor T cells, and allow for an aberrant inflammatory response [136]. However, despite the NOD2 story in Crohn's disease being quite compelling, it does not explain the whole picture in this polygenic disorder. Recent genome studies have implicated several other new genes that confer susceptibility to Crohn's disease, including two autophagy genes, namely ATG16L1 [137] and IRGM [138], as well as IL-23 receptor polymorphisms [139], suggesting a role for aberrant T-helper-17 responses. Also, various studies have shown that the NOD2 gene does not confer susceptibility to Crohn's disease in certain populations, such as Japanese cohorts [140, 141].
An association of NOD2 with the other major IBD, ulcerative colitis, is less clear, with initial studies showing no association of ulcerative colitis and NOD2 [90]. Subsequent work has demonstrated that NOD2 may modify the risk for developing ulcerative colitis in patients who have the IBD susceptibility locus IBD5 [142]. However, as previously discussed, associations in ulcerative colitis patients with the pyrin protein indicate that these may modify susceptibility to ulcerative colitis particularly with inflammatory arthritis [52, 53].
NOD2 has other disease associations also, such as Blau syndrome [143]. A total of four missense mutations (R334Q, R334W, L469F and E383) have been identified as conferring disease susceptibility [91, 144], all of which are located in the central NACHT domain, which is in contrast to the LRR variants seen in Crohn's disease. These variants lead to NF-κB upregulation on MDP stimulation [145, 146].
Early onset sarcoidosis shares considerable phenotypic overlap with Blau syndrome and has also been associated with the R334W mutation in NOD2 [116, 146]. However, other granulomatous disorders, such as adult-onset sarcoidosis and Wegener's granulomatosis, have not been associated with NOD2 [147, 148].
NOD2 has also been studied in sepsis. Brenmoehl and coworkers [149] showed that mortality from sepsis in the intensive care unit setting is higher in patients carrying the frameshift variant in NOD2 (57% versus 31%), in cohorts of patients who were broadly matched for clinical indices of severity of disease. This may represent the consequences of decreased intracellular sensing of bacterial products and decreased bacterial clearance, leading to a potentiation of infection and proinflammatory cascades, ultimately leading to cardiovascular collapse and shock. In transplant medicine, donor and recipient NOD2 status appears important in graft versus host disease and transplant mortality in allogeneic stem cell transplantation, with an increased likelihood of both of these conditions occurring in the presence of an increasing number of NOD2 mutations in donor and recipient cohorts [150].
In relation to inflammatory arthritis, there is relatively little in the literature suggesting a role for the nodosome. Joosten and colleagues recently demonstrated that NOD2 deficiency in mice is protective against acute joint inflammation and early cartilage destruction induced by bacteria [151]. NOD1 deficiency leads to increased inflammation and cytokine production. This pattern was replicated in human peripheral blood mononuclear cells with NOD1/2 mutants [151]. However, it was previously shown in several studies that NOD2 mutant alleles do not confer susceptibility to rheumatoid arthritis [152].
Until recently, evidence for overlap or crosstalk between individual NLRs had not been identified. However, it appears that NOD2 and NALP3 SNPs may have a synergistic contribution toward susceptibility to Crohn's disease. Cummings and coworkers [153] showed that the rs1539019 SNP in the NALP3 (NLRP3, CIAS1) gene conferred susceptibility to Crohn's disease in the presence of a NOD2 mutation (P = 0.0006). With high levels of IL-1 seen in Crohn's disease patients, mutations within NALP3 make this an attractive candidate gene for further study in Crohn's disease. Of further interest, recent genome-wide association studies are consistently uncovering new genes that are associated with Crohn's disease, with around 30 genes now implicated in susceptibility to this disease [154].
NOD2 and biological therapy
Infliximab was the first anti-TNF therapy to be used in the treatment of Crohn's disease, with response rates of around 70% and remission rates of around 30% [155]. However, two large studies [156, 157] have not suggested a correlation between NOD2 mutations and response or predictors of response or nonresponse to infliximab. Anakinra, however, makes Crohn's disease worse [158]. In Blau syndrome there are case reports of two patients, with the R334W change in the NACHT domain, responding to infliximab, with almost entire resolution of symptoms, but not to etanercept [159]. Whether this effect is mutation specific or a global effect of infliximab cannot be determined. There are also limited data suggesting a possible role for anakinra in the treatment of Blau syndrome, with normalization of cytokines and symptomatic improvement in a patient after treatment [160].
Conclusion
The two most studied groups of PRRs, namely the TLRs and NLRs, have been shown not only to have independent effects but also to have important two-way crosstalk between these pathways. The interactions between these two major pathways are being investigated and currently hint at the complexity of the innate immune response to PRRs. PRRs can activate either TLRs or NLRs, or both, thereby initiating a more rapid and enhanced response. Monosodium urate has been shown to act in synergy with lipopolysaccharide, a ligand for TLR4, inducing an enhanced response after co-stimulation of the NLR and TLR pathway, and release of IL-1β [161]. In addition, there is evidence of alternative pathways that result in NF-κB activation and the production of cytokines, in a similar manner to TLRs and NLRs. Antineutrophil cytoplasmic antibody, an autoantibody that is directed against the enzymes located in neutrophils and monocytes, specifically against proteinase 3, primes human monocytic cells, via protease-activated receptor-2, to produce cytokines [162]. These antibodies prime the innate immune system, following an upstream event whereby the presence of bacterial components led to stimulation by TLR and NOD1/2 [163], subsequently leading to secretion of proinflammatory cytokines. Matsumoto and colleagues [164] reported that proteinase 3 is downregulated in rheumatoid arthritis patients after treatment with the anti-TNF therapy infliximab. This suggests that these mechanisms actively participate in inflammatory processes, and that these interactions may not be exclusive of one another.
Abbreviations
- ASC:
-
apoptosis-associated speck-like protein
- CAPS:
-
cryopyrin-associated periodic syndromes
- CARD:
-
caspase activation and recruitment
- CINCA:
-
chronic infantile neurologic, cutaneous and articular syndrome
- CLR:
-
C-type lectin receptor
- DAMP:
-
damage-associated molecular pattern
- IBD:
-
inflammatory bowel disease
- IKK:
-
IκB kinase
- IL:
-
interleukin
- Ipaf:
-
IL-1β converting enzyme protease activating factor
- LRR:
-
leucine-rich repeat
- MDP:
-
muramyl dipeptide
- NALP1:
-
NACHT, leucine rich repeat and pyrin domain containing 1
- NF-κB:
-
nuclear factor-κB
- NLR:
-
NOD-like receptor
- NLRC1:
-
NLR family, CARD domain containing 1
- NLRP1:
-
NLR family, pyrin domain containing 1
- NOD1:
-
nucleotide-binding oligomerization domain containing 1
- NOMID:
-
neonatal onset multisystem inflammatory disease
- PAMP:
-
pathogen-associated molecular pattern
- PRR:
-
pathogen recognition receptor
- PYD:
-
pyrin domain
- RIP:
-
receptor-interacting protein
- SNP:
-
single nucleotide polymorphism
- TLR:
-
Toll-like receptor
- TNF:
-
tumour necrosis factor.
References
Smith DE, Renshaw BR, Ketchem RR, Kubin M, Garka KE, Sims JE: Four new members expand the interleukin-1 superfamily. J Biol Chem. 2000, 275: 1169-1175. 10.1074/jbc.275.2.1169.
Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, et al: IL-33, an inter-leukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005, 23: 479-490. 10.1016/j.immuni.2005.09.015.
Dinarello CA: Biologic basis for interleukin-1 in disease. Blood. 1996, 87: 2095-2147.
Dinarello CA: Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am J Clin Nutr. 2006, 83: 447S-455S.
Janeway CA, Medzhitov R: Innate immune recognition. Annu Rev Immunol. 2002, 20: 197-216. 10.1146/annurev.immunol.20.083001.084359.
Medzhitov R, Janeway CA: Innate immunity: impact on the adaptive immune response. Curr Opin Immunol. 1997, 9: 4-9. 10.1016/S0952-7915(97)80152-5.
Poltorak A, Smirnova I, He X, Liu MY, Van Huffel C, McNally O, Birdwell D, Alejos E, Silva M, Du X, Thompson P, Chan EK, Ledesma J, Roe B, Clifton S, Vogel SN, Beutler B: Genetic and physical mapping of the Lps locus: identification of the toll-4 receptor as a candidate gene in the critical region. Blood Cells Mol Dis. 1998, 24: 340-355. 10.1006/bcmd.1998.0201.
Gazzinelli RT, Denkers EY: Protozoan encounters with Toll-like receptor signalling pathways: implications for host parasitism. Nat Rev Immunol. 2006, 6: 895-906. 10.1038/nri1978.
Trinchieri G, Sher A: Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007, 7: 179-190. 10.1038/nri2038.
Akira S, Uematsu S, Takeuchi O: Pathogen recognition and innate immunity. Cell. 2006, 124: 783-801. 10.1016/j.cell.2006.02.015.
HUGO: Nucleotide-binding domain and leucine rich repeat containing family. [http://www.genenames.org/genefamily/nlr.php]
Fritz JH, Ferrero RL, Philpott DJ, Girardin SE: Nod-like proteins in immunity, inflammation and disease. Nat Immunol. 2006, 7: 1250-1257. 10.1038/ni1412.
Sirard JC, Vignal C, Dessein R, Chamaillard M: Nod-like receptors: cytosolic watchdogs for immunity against pathogens. PLoS Pathog. 2007, 3: e152-10.1371/journal.ppat.0030152.
Sutterwala FS, Mijares LA, Li L, Ogura Y, Kazmierczak BI, Flavell RA: Immune recognition of Pseudomonas aeruginosa mediated by the IPAF/NLRC4 inflammasome. J Exp Med. 2007, 204: 3235-3245. 10.1084/jem.20071239.
Tattoli I, Travassos LH, Carneiro LA, Magalhaes JG, Girardin SE: The Nodosome: Nod1 and Nod2 control bacterial infections and inflammation. Semin Immunopathol. 2007, 29: 289-301. 10.1007/s00281-007-0083-2.
Martinon F, Burns K, Tschopp J: The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002, 10: 417-426. 10.1016/S1097-2765(02)00599-3.
Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM: Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. 2004, 430: 213-218. 10.1038/nature02664.
Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein D, Rouiller I, Reed JC: Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell. 2007, 25: 713-724. 10.1016/j.molcel.2007.01.032.
Steinman L: Therapy of autoimmune disease with monoclonal antibodies to class II gene products of the major histocompatibility complex. Prog Allergy. 1988, 45: 161-167.
McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, Mansfield E, Gadina M, Karenko L, Pettersson T, McCarthy J, Frucht DM, Aringer M, Torosyan Y, Teppo AM, Wilson M, Karaarslan HM, Wan Y, Todd I, Wood G, Schlimgen R, Kumarajeewa TR, Cooper SM, Vella JP, Amos CI, Mulley J, Quane KA, Molloy MG, Ranki A, Powell RJ, et al: Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 1999, 97: 133-144. 10.1016/S0092-8674(00)80721-7.
Ting JP, Kastner DL, Hoffman HM: CATERPILLERs, pyrin and hereditary immunological disorders. Nat Rev Immunol. 2006, 6: 183-195. 10.1038/nri1788.
Martinon F, Tschopp J: Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007, 14: 10-22. 10.1038/sj.cdd.4402038.
Chu ZL, Pio F, Xie Z, Welsh K, Krajewska M, Krajewski S, Godzik A, Reed JC: A novel enhancer of the Apaf1 apoptosome involved in cytochrome c-dependent caspase activation and apoptosis. J Biol Chem. 2001, 276: 9239-9245. 10.1074/jbc.M006309200.
Hlaing T, Guo RF, Dilley KA, Loussia JM, Morrish TA, Shi MM, Vincenz C, Ward PA: Molecular cloning and characterization of DEFCAP-L and -S, two isoforms of a novel member of the mammalian Ced-4 family of apoptosis proteins. J Biol Chem. 2001, 276: 9230-9238. 10.1074/jbc.M009853200.
Petrilli V, Papin S, Tschopp J: The inflammasome. Curr Biol. 2005, 15: R581-10.1016/j.cub.2005.07.049.
Bruey JM, Bruey-Sedano N, Luciano F, Zhai D, Balpai R, Xu C, Kress CL, Bailly-Maitre B, Li X, Osterman A, Matsuzawa S, Terskikh AV, Faustin B, Reed JC: Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell. 2007, 129: 45-56. 10.1016/j.cell.2007.01.045.
Kummer JA, Broekhuizen R, Everett H, Agostini L, Kuijk L, Martinon F, van Bruggen R, Tschopp J: Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem. 2007, 55: 443-452. 10.1369/jhc.6A7101.2006.
Jin Y, Birlea SA, Fain PR, Spritz RA: Genetic variations in NALP1 are associated with generalized vitiligo in a Romanian population. J Invest Dermatol. 2007, 127: 2558-2562. 10.1038/sj.jid.5700953.
Liu F, Lo CF, Ning X, Kajkowski EM, Jin M, Chiriac C, Gonzales C, Naureckiene S, Lock YW, Pong K, Zaleska MM, Jacobsen JS, Silverman S, Ozenberger BA: Expression of NALP1 in cerebellar granule neurons stimulates apoptosis. Cell Signal. 2004, 16: 1013-1021.
Martinon F, Agostini L, Meylan E, Tschopp J: Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004, 14: 1929-1934. 10.1016/j.cub.2004.10.027.
Solle M, Labasi J, Perregaux DG, Stam E, Petrushova N, Koller BH, Griffiths RJ, Gabel CA: Altered cytokine production in mice lacking P2X(7) receptors. J Biol Chem. 2001, 276: 125-132. 10.1074/jbc.M006781200.
Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J: Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14: 1583-1589. 10.1038/sj.cdd.4402195.
Perregaux D, Barberia J, Lanzetti AJ, Geoghegan KF, Carty TJ, Gabel CA: IL-1 beta maturation: evidence that mature cytokine formation can be induced specifically by nigericin. J Immunol. 1992, 149: 1294-1303.
Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, Parks RJ, Tschopp J: The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008, 452: 103-107. 10.1038/nature06664.
Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E: Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol. 2008, 9: 847-856. 10.1038/ni.1631.
Cassel SL, Eisenbarth SC, Iyer SS, Sadler JJ, Colegio OR, Tephly LA, Carter AB, Rothman PB, Flavell RA, Sutterwala FS: The Nalp3 inflammasome is essential for the development of silicosis. Proc Natl Acad Sci USA. 2008, 105: 9035-9040. 10.1073/pnas.0803933105.
Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J: Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008, 320: 674-677. 10.1126/science.1156995.
Hoffman HM, Wanderer AA, Broide DH: Familial cold auto-inflammatory syndrome: phenotype and genotype of an autosomal dominant periodic fever. J Allergy Clin Immunol. 2001, 108: 615-620. 10.1067/mai.2001.118790.
Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, Teillac-Hamel D, Fischer A, de Saint Basile G: Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002, 71: 198-203. 10.1086/341357.
Dowds TA, Masumoto J, Zhu L, Inohara N, Nunez G: Cryopyrin-induced interleukin 1beta secretion in monocytic cells: enhanced activity of disease-associated mutants and requirement for ASC. J Biol Chem. 2004, 279: 21924-21928. 10.1074/jbc.M401178200.
Tschopp J, Martinon F, Burns K: NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol. 2003, 4: 95-104. 10.1038/nrm1019.
Aksentijevich I, D Putnam C, Remmers EF, Mueller JL, Le J, Kolodner RD, Moak Z, Chuang M, Austin F, Goldbach-Mansky R, Hoffman HM, Kastner DL: The clinical continuum of cryopyrinopathies: novel CIAS1 mutations in North American patients and a new cryopyrin model. Arthritis Rheum. 2007, 56: 1273-1285. 10.1002/art.22491.
Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, Stein L, Russo R, Goldsmith D, Dent P, Rosenberg HF, Austin F, Remmers EF, Balow JE, Rosenzweig S, Komarow H, Shoham NG, Wood G, Jones J, Mangra N, Carrero H, Adams BS, Moore TL, Schikler K, Hoffman H, Lovell DJ, Lipnick R, Barron K, O'Shea JJ, Kastner DL, et al: De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 2002, 46: 3340-3348. 10.1002/art.10688.
Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J: NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004, 20: 319-325. 10.1016/S1074-7613(04)00046-9.
Janssen R, Verhard E, Lankester A, Ten Cate R, van Dissel JT: Enhanced interleukin-1beta and interleukin-18 release in a patient with chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 2004, 50: 3329-3333. 10.1002/art.20494.
Takada H, Ishimura M, Inada H, Ohga S, Kusuhara K, Moroi Y, Furue M, Hara T: Lipopolysaccharide-induced monocytic cell death for the diagnosis of mild neonatal-onset multisystem inflammatory disease. J Pediatr. 2008, 152: 885-887. 10.1016/j.jpeds.2008.01.038.
Dowds TA, Masumoto J, Chen FF, Ogura Y, Inohara N, Nunez G: Regulation of cryopyrin/Pypaf1 signaling by pyrin, the familial Mediterranean fever gene product. Biochem Biophys Res Commun. 2003, 302: 575-580. 10.1016/S0006-291X(03)00221-3.
Stojanov S, Kastner DL: Familial autoinflammatory diseases: genetics, pathogenesis and treatment. Curr Opin Rheumatol. 2005, 17: 586-599. 10.1097/bor.0000174210.78449.6b.
Wise CA, Gillum JD, Seidman CE, Lindor NM, Veile R, Bashiardes S, Lovett M: Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet. 2002, 11: 961-969. 10.1093/hmg/11.8.961.
McDermott MF: A common pathway in periodic fever syndromes. Trends Immunol. 2004, 25: 457-460. 10.1016/j.it.2004.07.007.
Day TG, Ramanan AV, Hinks A, Lamb R, Packham J, Wise C, Punaro M, Donn RP: Autoinflammatory genes and susceptibility to psoriatic juvenile idiopathic arthritis. Arthritis Rheum. 2008, 58: 2142-2146. 10.1002/art.23604.
Giaglis S, Mimidis K, Papadopoulos V, Thomopoulos K, Sidiropoulos P, Rafail S, Nikolopoulou V, Fragouli E, Kartalis G, Tzioufas A, Boumpas D, Ritis K: Increased frequency of mutations in the gene responsible for familial Mediterranean fever (MEFV) in a cohort of patients with ulcerative colitis: evidence for a potential disease-modifying effect?. Dig Dis Sci. 2006, 51: 687-692. 10.1007/s10620-006-3192-1.
Sari S, Egritas O, Dalgic B: The familial Mediterranean fever (MEFV) gene may be a modifier factor of inflammatory bowel disease in infancy. Eur J Pediatr. 2008, 167: 391-393. 10.1007/s00431-007-0508-x.
Omi T, Kumada M, Kamesaki T, Okuda H, Munkhtulga L, Yanagisawa Y, Utsumi N, Gotoh T, Hata A, Soma M, Umemura S, Ogihara T, Takahashi N, Tabara Y, Shimada K, Mano H, Kajii E, Miki T, Iwamoto S: An intronic variable number of tandem repeat polymorphisms of the cold-induced autoinflammatory syndrome 1 (CIAS1) gene modifies gene expression and is associated with essential hypertension. Eur J Hum Genet. 2006, 14: 1295-1305. 10.1038/sj.ejhg.5201698.
Rosengren S, Hoffman HM, Bugbee W, Boyle DL: Expression and regulation of cryopyrin and related proteins in rheumatoid arthritis synovium. Ann Rheum Dis. 2005, 64: 708-714. 10.1136/ard.2004.025577.
Mahr S, Burmester GR, Hilke D, Göbel U, Grützkau A, Häupl T, Hauschild M, Koczan D, Krenn V, Neidel J, Perka C, Radbruch A, Thiesen HJ, Müller B: Cis- and trans-acting gene regulation is associated with osteoarthritis. Am J Hum Genet. 2006, 78: 793-803. 10.1086/503849.
Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J: Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006, 440: 237-241. 10.1038/nature04516.
So A, De Smedt T, Revaz S, Tschopp J: A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 2007, 9: R28-10.1186/ar2143.
McGonagle D, Tan AL, Shankaranarayana S, Madden J, Emery P, McDermott MF: Management of treatment resistant inflammation of acute on chronic tophaceous gout with anakinra. Ann Rheum Dis. 2007, 66: 1683-1684. 10.1136/ard.2007.073759.
McGonagle D, Tan AL, Madden J, Emery P, McDermott MF: Successful treatment of resistant pseudogout with anakinra. Arthritis Rheum. 2008, 58: 631-633. 10.1002/art.23119.
Alexander T, Klotz O, Feist E, Ruther K, Burmester GR, Pleyer U: Successful treatment of acute visual loss in Muckle-Wells syndrome with interleukin 1 receptor antagonist. Ann Rheum Dis. 2005, 64: 1245-1246. 10.1136/ard.2004.032060.
Dalgic B, Egritas O, Sari S, Cuisset L: A variant Muckle-Wells syndrome with a novel mutation in CIAS1 gene responding to anakinra. Pediatr Nephrol. 2007, 22: 1391-1394. 10.1007/s00467-007-0500-8.
Gerard S, le Goff B, Maugars Y, Berthelot JM, Malard O: Lasting remission of a Muckle-Wells syndrome with CIAS-1 mutation using half-dose anakinra. Joint Bone Spine. 2007, 74: 659-10.1016/j.jbspin.2007.01.032.
Hawkins PN, Lachmann HJ, Aganna E, McDermott MF: Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum. 2004, 50: 607-612. 10.1002/art.20033.
Maksimovic L, Stirnemann J, Caux F, Ravet N, Rouaghe S, Cuisset L, Letellier E, Grateau G, Morin AS, Fain O: New CIAS1 mutation and anakinra efficacy in overlapping of Muckle-Wells and familial cold autoinflammatory syndromes. Rheumatology (Oxford). 2008, 47: 309-310. 10.1093/rheumatology/kem318.
Mirault T, Launay D, Cuisset L, Hachulla E, Lambert M, Queyrel V, Quemeneur T, Morell-Dubois S, Hatron PY: Recovery from deafness in a patient with Muckle-Wells syndrome treated with anakinra. Arthritis Rheum. 2006, 54: 1697-1700. 10.1002/art.21807.
Rynne M, Maclean C, Bybee A, McDermott MF, Emery P: Hearing improvement in a patient with variant Muckle-Wells syndrome in response to interleukin 1 receptor antagonism. Ann Rheum Dis. 2006, 65: 533-534. 10.1136/ard.2005.038091.
Yamazaki T, Masumoto J, Agematsu K, Sawai N, Kobayashi S, Shigemura T, Yasui K, Koike K: Anakinra improves sensory deafness in a Japanese patient with Muckle-Wells syndrome, possibly by inhibiting the cryopyrin inflammasome. Arthritis Rheum. 2008, 58: 864-868. 10.1002/art.23261.
Hoffman HM, Rosengren S, Boyle DL, Cho JY, Nayar J, Mueller JL, Anderson JP, Wanderer AA, Firestein GS: Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 2004, 364: 1779-1785. 10.1016/S0140-6736(04)17401-1.
Metyas SK, Hoffman HM: Anakinra prevents symptoms of familial cold autoinflammatory syndrome and Raynaud's disease. J Rheumatol. 2006, 33: 2085-2087.
O'Connell SM, O'Regan GM, Bolger T, Hoffman HM, Cant A, Irvine AD, Watson RM: Response to IL-1-receptor antagonist in a child with familial cold autoinflammatory syndrome. Pediatr Dermatol. 2007, 24: 85-89.
Ross JB, Finlayson LA, Klotz PJ, Langley RG, Gaudet R, Thompson K, Churchman SM, McDermott MF, Hawkins PN: Use of anakinra (Kineret) in the treatment of familial cold autoinflammatory syndrome with a 16-month follow-up. J Cutan Med Surg. 2008, 12: 8-16.
Thornton BD, Hoffman HM, Bhat A, Don BR: Successful treatment of renal amyloidosis due to familial cold autoinflammatory syndrome using an interleukin 1 receptor antagonist. Am J Kidney Dis. 2007, 49: 477-481. 10.1053/j.ajkd.2006.10.026.
Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, Kim HJ, Brewer C, Zalewski C, Wiggs E, Hill S, Turner ML, Karp BI, Aksentijevich I, Pucino F, Penzak SR, Haverkamp MH, Stein L, Adams BS, Moore TL, Fuhlbrigge RC, Shaham B, Jarvis JN, O'Neil K, Vehe RK, Beitz LO, Gardner G, Hannan WP, Warren RW, et al: Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006, 355: 581-592. 10.1056/NEJMoa055137.
Goldbach-Mansky R, Pucino F, Kastner DL: Treatment of patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome: comment on the article by Matsubara et al. Arthritis Rheum. 2007, 56: 2099-2101. 10.1002/art.22561. author reply 2101–2092.
Lovell DJ, Bowyer SL, Solinger AM: Interleukin-1 blockade by anakinra improves clinical symptoms in patients with neonatal-onset multisystem inflammatory disease. Arthritis Rheum. 2005, 52: 1283-1286. 10.1002/art.20953.
de Koning HD, Bodar EJ, Simon A, Hilst van der JC, Netea MG, Meer van der JW: Beneficial response to anakinra and thalidomide in Schnitzler's syndrome. Ann Rheum Dis. 2006, 65: 542-544. 10.1136/ard.2005.045245.
Alten R, Gram H, Joosten LA, Berg van den WB, Sieper J, Wassenberg S, Burmester G, van Riel P, Diaz-Lorente M, Bruin GJ, Woodworth TG, Rordorf C, Batard Y, Wright AM, Jung T: The human anti-interleukin-1b monoclonal antibody ACZ885 is effective in joint inflammation models in mice and in a proof of concept study in rheumatoid arthritis patients. Arthritis Res Ther. 2008, 10: R67-10.1186/ar2438.
Tomillero A, Moral MA: Gateways to clinical trials. Methods Find Exp Clin Pharmacol. 2008, 30: 231-251.
Mathews R, Churchman S, Church L, Coulthard L, Byryer D, Nizam S, Saleem B, Cook GP, Emery P, McDermott MF: Effects of TNF blockade by infliximab on inflammasome regulators in rheumatoid arthritis patients [Abstract]. Ann Rheum Dis. 2008, 67 (suppl 1): A25-
Karababa M, Kolly L, Tschopp J, Busso N: Crucial role of ASC-dependent inflammasome in murine arthritis [Abstract]. Arthritis Rheum. 2007, 56 (suppl 1): 1280-
Johansen C, Moeller K, Kragballe K, Iversen L: The activity of caspase-1 is increased in lesional psoriatic epidermis. J Invest Dermatol. 2007, 127: 2857-2864.
Franchi L, Stoolman J, Kanneganti TD, Verma A, Ramphal R, Nunez G: Critical role for Ipaf in Pseudomonas aeruginosa-induced caspase-1 activation. Eur J Immunol. 2007, 37: 3030-3039. 10.1002/eji.200737532.
Franchi L, Amer A, Body-Malapel M, Kanneganti TD, Ozören N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, Grant EP, Núñez G: Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat Immunol. 2006, 7: 576-582. 10.1038/ni1346.
Amer A, Franchi L, Kanneganti TD, Body-Malapel M, Ozören N, Brady G, Meshinchi S, Jagirdar R, Gewirtz A, Akira S, Núñez G: Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J Biol Chem. 2006, 281: 35217-35223. 10.1074/jbc.M604933200.
Jin Y, Mailloux CM, Gowan K, Riccardi SL, LaBerge G, Bennett DC, Fain PR, Spritz RA: NALP1 in vitiligo-associated multiple autoimmune disease. N Engl J Med. 2007, 356: 1216-1225. 10.1056/NEJMoa061592.
Murdoch S, Djuric U, Mazhar B, Seoud M, Khan R, Kuick R, Bagga R, Kircheisen R, Ao A, Ratti B, Hanash S, Rouleau GA, Slim R: Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat Genet. 2006, 38: 300-302. 10.1038/ng1740.
Steimle V, Otten LA, Zufferey M, Mach B: Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell. 1993, 75: 135-146.
Martinez A, Alvarez-Lafuente R, Mas A, Bartolome M, Garcia-Montojo M, de Las Heras V, de la Concha EG, Arroyo R, Urcelay E: Environment-gene interaction in multiple sclerosis: human herpesvirus 6 and MHC2TA. Hum Immunol. 2007, 68: 685-689. 10.1016/j.humimm.2007.05.005.
Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, Almer S, Tysk C, O'Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel JF, Sahbatou M, Thomas G: Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001, 411: 599-603. 10.1038/35079107.
Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Hafner R, Chamaillard M, Zouali H, Thomas G, Hugot JP: CARD15 mutations in Blau syndrome. Nat Genet. 2001, 29: 19-20. 10.1038/ng720.
McGonagle D, Savic S, McDermott MF: The NLR network and the immunological disease continuum of adaptive and innate immune-mediated inflammation against self. Semin Immunopathol. 2007, 29: 303-313. 10.1007/s00281-007-0084-1.
Suzuki T, Franchi L, Toma C, Ashida H, Ogawa M, Yoshikawa Y, Mimuro H, Inohara N, Sasakawa C, Nunez G: Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 2007, 3: e111-10.1371/journal.ppat.0030111.
Girardin SE, Travassos LH, Herve M, Blanot D, Boneca IG, Philpott DJ, Sansonetti PJ, Mengin-Lecreulx D: Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J Biol Chem. 2003, 278: 41702-41708. 10.1074/jbc.M307198200.
Inohara N, Koseki T, del Peso L, Hu Y, Yee C, Chen S, Carrio R, Merino J, Liu D, Ni J, Ni J, Núñez G: Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J Biol Chem. 1999, 274: 14560-14567. 10.1074/jbc.274.21.14560.
Inohara N, Koseki T, Lin J, del Peso L, Lucas PC, Chen FF, Ogura Y, Nunez G: An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem. 2000, 275: 27823-27831.
Abbott DW, Wilkins A, Asara JM, Cantley LC: The Crohn's disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol. 2004, 14: 2217-2227. 10.1016/j.cub.2004.12.032.
Pan Q, Mathison J, Fearns C, Kravchenko VV, Da Silva Correia J, Hoffman HM, Kobayashi KS, Bertin J, Grant EP, Coyle AJ, Sutterwala FS, Ogura Y, Flavell RA, Ulevitch RJ: MDP-induced interleukin-1beta processing requires Nod2 and CIAS1/NALP3. J Leukoc Biol. 2007, 82: 177-183. 10.1189/jlb.1006627.
Park JH, Kim YG, McDonald C, Kanneganti TD, Hasegawa M, Body-Malapel M, Inohara N, Nunez G: RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol. 2007, 178: 2380-2386.
Yang Y, Yin C, Pandey A, Abbott D, Sassetti C, Kelliher MA: NOD2 pathway activation by MDP or Mycobacterium tuberculosis infection involves the stable polyubiquitination of Rip2. J Biol Chem. 2007, 282: 36223-36229. 10.1074/jbc.M703079200.
Girardin SE, Tournebize R, Mavris M, Page AL, Li X, Stark GR, Bertin J, DiStefano PS, Yaniv M, Sansonetti PJ, Philpott DJ: CARD4/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep. 2001, 2: 736-742. 10.1093/embo-reports/kve155.
Philpott DJ, Yamaoka S, Israel A, Sansonetti PJ: Invasive Shigella flexneri activates NF-kappa B through a lipopolysaccharide-dependent innate intracellular response and leads to IL-8 expression in epithelial cells. J Immunol. 2000, 165: 903-914.
Kim JG, Lee SJ, Kagnoff MF: Nod1 is an essential signal transducer in intestinal epithelial cells infected with bacteria that avoid recognition by toll-like receptors. Infect Immun. 2004, 72: 1487-1495. 10.1128/IAI.72.3.1487-1495.2004.
Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Mémet S, Huerre MR, Coyle AJ, DiStefano PS, Sansonetti PJ, Labigne A, Bertin J, Philpott DJ, Ferrero RL: Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004, 5: 1166-1174. 10.1038/ni1131.
Fischer W, Puls J, Buhrdorf R, Gebert B, Odenbreit S, Haas R: Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol. 2001, 42: 1337-1348. 10.1046/j.1365-2958.2001.02714.x.
Cascales E, Christie PJ: The versatile bacterial type IV secretion systems. Nat Rev Microbiol. 2003, 1: 137-149. 10.1038/nrmicro753.
Rosenstiel P, Hellmig S, Hampe J, Ott S, Till A, Fischbach W, Sahly H, Lucius R, Fölsch UR, Philpott D, Schreiber S: Influence of polymorphisms in the NOD1/CARD4 and NOD2/CARD15 genes on the clinical outcome of Helicobacter pylori infection. Cell Microbiol. 2006, 8: 1188-1198. 10.1111/j.1462-5822.2006.00701.x.
Zilbauer M, Dorrell N, Elmi A, Lindley KJ, Schuller S, Jones HE, Klein NJ, Nunez G, Wren BW, Bajaj-Elliott M: A major role for intestinal epithelial nucleotide oligomerization domain 1 (NOD1) in eliciting host bactericidal immune responses to Campylobacter jejuni. Cell Microbiol. 2007, 9: 2404-2416. 10.1111/j.1462-5822.2007.00969.x.
Travassos LH, Carneiro LA, Girardin SE, Boneca IG, Lemos R, Bozza MT, Domingues RC, Coyle AJ, Bertin J, Philpott DJ, Plotkowski MC: Nod1 participates in the innate immune response to Pseudomonas aeruginosa. J Biol Chem. 2005, 280: 36714-36718. 10.1074/jbc.M501649200.
Welter-Stahl L, Ojcius DM, Viala J, Girardin S, Liu W, Delarbre C, Philpott D, Kelly KA, Darville T: Stimulation of the cytosolic receptor for peptidoglycan, Nod1, by infection with Chlamydia trachomatis or Chlamydia muridarum. Cell Microbiol. 2006, 8: 1047-1057. 10.1111/j.1462-5822.2006.00686.x.
Laitinen T, Daly MJ, Rioux JD, Kauppi P, Laprise C, Petäys T, Green T, Cargill M, Haahtela T, Lander ES, Laitinen LA, Hudson TJ, Kere J: A susceptibility locus for asthma-related traits on chromosome 7 revealed by genome-wide scan in a founder population. Nat Genet. 2001, 28: 87-91. 10.1038/88319.
Weidinger S, Klopp N, Rummler L, Wagenpfeil S, Novak N, Baurecht HJ, Groer W, Darsow U, Heinrich J, Gauger A, Schafer T, Jakob T, Behrendt H, Wichmann HE, Ring J, Illig T: Association of NOD1 polymorphisms with atopic eczema and related phenotypes. J Allergy Clin Immunol. 2005, 116: 177-184. 10.1016/j.jaci.2005.02.034.
Zouali H, Lesage S, Merlin F, Cézard JP, Colombel JF, Belaiche J, Almer S, Tysk C, O'Morain C, Gassull M, Christensen S, Finkel Y, Modigliani R, Gower-Rousseau C, Macry J, Chamaillard M, Thomas G, Hugot JP, EPWG group; EPIMAD group: CARD4/NOD1 is not involved in inflammatory bowel disease. Gut. 2003, 52: 71-74. 10.1136/gut.52.1.71.
Van Limbergen J, Nimmo ER, Russell RK, Drummond HE, Smith L, Anderson NH, Davies G, Arnott ID, Wilson DC, Satsangi J: Investigation of NOD1/CARD4 variation in inflammatory bowel disease using a haplotype-tagging strategy. Hum Mol Genet. 2007, 16: 2175-2186. 10.1093/hmg/ddm169.
Tremelling M, Hancock L, Bredin F, Sharpstone D, Bingham SA, Parkes M: Complex insertion/deletion polymorphism in NOD1 (CARD4) is not associated with inflammatory bowel disease susceptibility in East Anglia panel. Inflamm Bowel Dis. 2006, 12: 967-971. 10.1097/01.mib.0000234131.89971.e5.
Coto-Segura P, Mallo-Garcia S, Costa-Romero M, Arostegui JI, Yague J, Ramos-Polo E, Santos-Juanes J: A sporadic case of early-onset sarcoidosis resembling Blau syndrome due to the recurrent R334W missense mutation on the NOD2 gene. Br J Dermatol. 2007, 157: 1257-1259. 10.1111/j.1365-2133.2007.08210.x.
Kanazawa N, Matsushima S, Kambe N, Tachibana T, Nagai S, Miyachi Y: Presence of a sporadic case of systemic granulomatosis syndrome with a CARD15 mutation. J Invest Dermatol. 2004, 122: 851-852. 10.1111/j.0022-202X.2004.22341.x.
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nuñez G, Cho JH: A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001, 411: 603-606. 10.1038/35079114.
Romagnani P, Annunziato F, Baccari MC, Parronchi P: T cells and cytokines in Crohn's disease. Curr Opin Immunol. 1997, 9: 793-799. 10.1016/S0952-7915(97)80180-X.
Lesage S, Zouali H, Cézard JP, Colombel JF, Belaiche J, Almer S, Tysk C, O'Morain C, Gassull M, Binder V, Finkel Y, Modigliani R, Gower-Rousseau C, Macry J, Merlin F, Chamaillard M, Jannot AS, Thomas G, Hugot JP, EPWG-IBD Group; EPIMAD Group; GETAID Group: CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. 2002, 70: 845-857. 10.1086/339432.
Bonen DK, Ogura Y, Nicolae DL, Inohara N, Saab L, Tanabe T, Chen FF, Foster SJ, Duerr RH, Brant SR, Cho JH, Nuñez G: Crohn's disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology. 2003, 124: 140-146. 10.1053/gast.2003.50019.
Netea MG, Ferwerda G, de Jong DJ, Girardin SE, Kullberg BJ, Meer van der JW: NOD2 3020insC mutation and the pathogenesis of Crohn's disease: impaired IL-1beta production points to a loss-of-function phenotype. Neth J Med. 2005, 63: 305-308.
Ligumsky M, Simon PL, Karmeli F, Rachmilewitz D: Role of interleukin 1 in inflammatory bowel disease: enhanced production during active disease. Gut. 1990, 31: 686-689. 10.1136/gut.31.6.686.
Mitsuyama K, Sata M, Tanikawa K: Significance of interleukin-6 in patients with inflammatory bowel disease. Gastroenterol Jpn. 1991, 26: 20-28.
Monteleone G, Biancone L, Marasco R, Morrone G, Marasco O, Luzza F, Pallone F: Interleukin 12 is expressed and actively released by Crohn's disease intestinal lamina propria mononuclear cells. Gastroenterology. 1997, 112: 1169-1178. 10.1016/S0016-5085(97)70128-8.
Watanabe T, Kitani A, Murray PJ, Strober W: NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004, 5: 800-808. 10.1038/ni1092.
Watanabe T, Kitani A, Murray PJ, Wakatsuki Y, Fuss IJ, Strober W: Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen-specific colitis. Immunity. 2006, 25: 473-485. 10.1016/j.immuni.2006.06.018.
Borm ME, van Bodegraven AA, Mulder CJ, Kraal G, Bouma G: The effect of NOD2 activation on TLR2-mediated cytokine responses is dependent on activation dose and NOD2 genotype. Genes Immun. 2008, 9: 274-278. 10.1038/gene.2008.9.
Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA: Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005, 307: 731-734. 10.1126/science.1104911.
Ouellette AJ, Bevins CL: Paneth cell defensins and innate immunity of the small bowel. Inflamm Bowel Dis. 2001, 7: 43-50. 10.1097/00054725-200102000-00007.
Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ: Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol. 2000, 1: 113-118. 10.1038/77783.
Ogura Y, Lala S, Xin W, Smith E, Dowds TA, Chen FF, Zimmermann E, Tretiakova M, Cho JH, Hart J, Greenson JK, Keshav S, Nuñez G: Expression of NOD2 in Paneth cells: a possible link to Crohn's ileitis. Gut. 2003, 52: 1591-1597. 10.1136/gut.52.11.1591.
Cuthbert AP, Fisher SA, Mirza MM, King K, Hampe J, Croucher PJ, Mascheretti S, Sanderson J, Forbes A, Mansfield J, Schreiber S, Lewis CM, Mathew CG: The contribution of NOD2 gene mutations to the risk and site of disease in inflammatory bowel disease. Gastroenterology. 2002, 122: 867-874. 10.1053/gast.2002.32415.
Hampe J, Frenzel H, Mirza MM, Croucher PJ, Cuthbert A, Mascheretti S, Huse K, Platzer M, Bridger S, Meyer B, Nürnberg P, Stokkers P, Krawczak M, Mathew CG, Curran M, Schreiber S: Evidence for a NOD2-independent susceptibility locus for inflammatory bowel disease on chromosome 16p. Proc Natl Acad Sci USA. 2002, 99: 321-326. 10.1073/pnas.261567999.
Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, Feathers RW, Chu H, Lima H, Fellermann K, Ganz T, Stange EF, Bevins CL: Reduced Paneth cell alpha-defensins in ileal Crohn's disease. Proc Natl Acad Sci USA. 2005, 102: 18129-18134. 10.1073/pnas.0505256102.
Kramer M, Netea MG, de Jong DJ, Kullberg BJ, Adema GJ: Impaired dendritic cell function in Crohn's disease patients with NOD2 3020insC mutation. J Leukoc Biol. 2006, 79: 860-866. 10.1189/jlb.0805484.
Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, Günther S, Prescott NJ, Onnie CM, Häsler R, Sipos B, Fölsch UR, Lengauer T, Platzer M, Mathew CG, Krawczak M, Schreiber S: A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007, 39: 207-211. 10.1038/ng1954.
Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER, Cummings FR, Soars D, Drummond H, Lees CW, Khawaja SA, Bagnall R, Burke DA, Todhunter CE, Ahmad T, Onnie CM, McArdle W, Strachan D, Bethel G, Bryan C, Lewis CM, Deloukas P, Forbes A, Sanderson J, Jewell DP, Satsangi J, Mansfield JC, Wellcome Trust Case Control Consortium, et al: Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet. 2007, 39: 830-832. 10.1038/ng2061.
Tremelling M, Cummings F, Fisher SA, Mansfield J, Gwilliam R, Keniry A, Nimmo ER, Drummond H, Onnie CM, Prescott NJ, Sanderson J, Bredin F, Berzuini C, Forbes A, Lewis CM, Cardon L, Deloukas P, Jewell D, Mathew CG, Parkes M, Satsangi J: IL23R variation determines susceptibility but not disease phenotype in inflammatory bowel disease. Gastroenterology. 2007, 132: 1657-1664. 10.1053/j.gastro.2007.02.051.
Inoue N, Tamura K, Kinouchi Y, Fukuda Y, Takahashi S, Ogura Y, Inohara N, Núñez G, Kishi Y, Koike Y, Shimosegawa T, Shimoyama T, Hibi T: Lack of common NOD2 variants in Japanese patients with Crohn's disease. Gastroenterology. 2002, 123: 86-91. 10.1053/gast.2002.34155.
Yamazaki K, Takazoe M, Tanaka T, Kazumori T, Nakamura Y: Absence of mutation in the NOD2/CARD15 gene among 483 Japanese patients with Crohn's disease. J Hum Genet. 2002, 47: 469-472. 10.1007/s100380200067.
McGovern DP, Van Heel DA, Negoro K, Ahmad T, Jewell DP: Further evidence of IBD5/CARD15 (NOD2) epistasis in the susceptibility to ulcerative colitis. Am J Hum Genet. 2003, 73: 1465-1466. 10.1086/379745.
Manouvrier-Hanu S, Puech B, Piette F, Boute-Benejean O, Desbonnet A, Duquesnoy B, Farriaux JP: Blau syndrome of granulomatous arthritis, iritis, and skin rash: a new family and review of the literature. Am J Med Genet. 1998, 76: 217-221. 10.1002/(SICI)1096-8628(19980319)76:3<217::AID-AJMG4>3.0.CO;2-N.
van Duist MM, Albrecht M, Podswiadek M, Giachino D, Lengauer T, Punzi L, De Marchi M: A new CARD15 mutation in Blau syndrome. Eur J Hum Genet. 2005, 13: 742-747. 10.1038/sj.ejhg.5201404.
Chamaillard M, Philpott D, Girardin SE, Zouali H, Lesage S, Chareyre F, Bui TH, Giovannini M, Zaehringer U, Penard-Lacronique V, Sansonetti PJ, Hugot JP, Thomas G: Gene-environment interaction modulated by allelic heterogeneity in inflammatory diseases. Proc Natl Acad Sci USA. 2003, 100: 3455-3460. 10.1073/pnas.0530276100.
Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata-Hizume M, Nagai S, Fuji A, Yuasa T, Manki A, Sakurai Y, Nakajima M, Kobayashi H, Fujiwara I, Tsutsumi H, Utani A, Nishigori C, Heike T, Nakahata T, Miyachi Y: Early-onset sarcoidosis and CARD15 mutations with constitutive nuclear factor-kappaB activation: common genetic etiology with Blau syndrome. Blood. 2005, 105: 1195-1197. 10.1182/blood-2004-07-2972.
Schurmann M, Valentonyte R, Hampe J, Muller-Quernheim J, Schwinger E, Schreiber S: CARD15 gene mutations in sarcoidosis. Eur Respir J. 2003, 22: 748-754. 10.1183/09031936.03.00040602.
Newman B, Rubin LA, Siminovitch KA: NOD2/CARD15 gene mutation is not associated with susceptibility to Wegener's granulomatosis. J Rheumatol. 2003, 30: 305-307.
Brenmoehl J, Herfarth H, Glück T, Audebert F, Barlage S, Schmitz G, Froehlich D, Schreiber S, Hampe J, Schölmerich J, Holler E, Rogler G: Genetic variants in the NOD2/CARD15 gene are associated with early mortality in sepsis patients. Intensive Care Med. 2007, 33: 1541-1548. 10.1007/s00134-007-0722-z.
Holler E, Rogler G, Herfarth H, Brenmoehl J, Wild PJ, Hahn J, Eissner G, Scholmerich J, Andreesen R: Both donor and recipient NOD2/CARD15 mutations associate with transplant-related mortality and GvHD following allogeneic stem cell transplantation. Blood. 2004, 104: 889-894. 10.1182/blood-2003-10-3543.
Joosten LA, Heinhuis B, Abdollahi-Roodsaz S, Ferwerda G, Lebourhis L, Philpott DJ, Nahori MA, Popa C, Morre SA, Meer van der JW, Girardin SE, Netea MG, Berg van den WB: Differential function of the NACHT-LRR (NLR) members Nod1 and Nod2 in arthritis. Proc Natl Acad Sci USA. 2008, 105: 9017-9022. 10.1073/pnas.0710445105.
Pawlik A, Kurzawski M, Gawronska-Szklarz B, Drozdzik M, Herczynska M: NOD2 allele variants in patients with rheumatoid arthritis. Clin Rheumatol. 2007, 26: 868-871. 10.1007/s10067-006-0404-1.
Cummings J, Cooney R, Clarke G, Beckley J, Geremia A, Cardon L, Jewell D: The genetics of Nod-like receptors proteins in Crohn's disease. J Crohn's Colitis. 2008, 2: P191-10.1016/j.crohns.2008.03.007.
Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, NIDDK IBD Genetics Consortium, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, et al: Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008, 40: 955-962. 10.1038/ng.175.
Rutgeerts P, D'Haens G, Targan S, Vasiliauskas E, Hanauer SB, Present DH, Mayer L, Van Hogezand RA, Braakman T, DeWoody KL, Schaible TF, Van Deventer SJ: Efficacy and safety of retreatment with anti-tumor necrosis factor antibody (infliximab) to maintain remission in Crohn's disease. Gastroenterology. 1999, 117: 761-769. 10.1016/S0016-5085(99)70332-X.
Mascheretti S, Hampe J, Croucher PJ, Nikolaus S, Andus T, Schubert S, Olson A, Bao W, Folsch UR, Schreiber S: Response to infliximab treatment in Crohn's disease is not associated with mutations in the CARD15 (NOD2) gene: an analysis in 534 patients from two multicenter, prospective GCP-level trials. Pharmacogenetics. 2002, 12: 509-515. 10.1097/00008571-200210000-00002.
Vermeire S, Louis E, Rutgeerts P, De Vos M, Van Gossum A, Belaiche J, Pescatore P, Fiasse R, Pelckmans P, Vlietinck R, Merlin F, Zouali H, Thomas G, Colombel JF, Hugot JP, Belgian Group of Infliximab Expanded Access Program and Fondation Jean Dausset CEPH, Paris, rance: NOD2/CARD15 does not influence response to infliximab in Crohn's disease. Gastroenterology. 2002, 123: 106-111. 10.1053/gast.2002.34172.
Carter JD, Valeriano J, Vasey FB: Crohn disease worsened by anakinra administration. J Clin Rheumatol. 2003, 9: 276-277. 10.1097/01.RHU.0000081265.06408.e4.
Milman N, Andersen CB, Hansen A, van Overeem Hansen T, Nielsen FC, Fledelius H, Ahrens P, Nielsen OH: Favourable effect of TNF-alpha inhibitor (infliximab) on Blau syndrome in monozygotic twins with a de novo CARD15 mutation. APMIS. 2006, 114: 912-919. 10.1111/j.1600-0463.2006.apm_522.x.
Aróstegui JI, Arnal C, Merino R, Modesto C, Antonia Carballo M, Moreno P, García-Consuegra J, Naranjo A, Ramos E, de Paz P, Rius J, Plaza S, Yagüe J: NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum. 2007, 56: 3805-3813. 10.1002/art.22966.
Giamarellos-Bourboulis EJ, Mouktaroudi M, Bodar E, Ven van der J, Kullberg BJ, Netea MG, Meer van der JW: Crystals of monosodium urate monohydrate enhance LPS-induced release of interleukin-1β by mononuclear cells through a caspase-1 mediated process. Ann Rheum Dis. 2008
Uehara A, Iwashiro A, Sato T, Yokota S, Takada H: Antibodies to proteinase 3 prime human monocytic cells via protease-activated receptor-2 and NF-kappaB for Toll-like receptor- and NOD-dependent activation. Mol Immunol. 2007, 44: 3552-3562. 10.1016/j.molimm.2007.03.010.
Uehara A, Hirabayashi Y, Takada H: Antibodies to proteinase 3 (PR3-ANCA) prime human oral, lung, and kidney epithelial cells through protease-activated receptor-2 to secrete proinflammatory cytokines upon stimulation with agonists to various Toll-like receptors, NOD1 and NOD2. Clin Vaccine Immunol. 2008, 15: 1060-1066. 10.1128/CVI.00137-08.
Matsumoto T, Kaneko T, Seto M, Wada H, Kobayashi T, Nakatani K, Tonomura H, Tono Y, Ohyabu M, Nobori T, Shiku H, Sudo A, Uchida A, Kurosawa DJ, Kurosawa S: The membrane proteinase 3 expression on neutrophils was downregulated after treatment with infliximab in patients with rheumatoid arthritis. Clin Appl Thromb Hemost. 2008, 14: 186-192. 10.1177/1076029607303961.
Bhat A, Naguwa SM, Gershwin ME: Genetics and new treatment modalities for familial Mediterranean fever. Ann N Y Acad Sci. 2007, 1110: 201-208. 10.1196/annals.1423.022.
Calligaris L, Marchetti F, Tommasini A, Ventura A: The efficacy of anakinra in an adolescent with colchicine-resistant familial Mediterranean fever. Eur J Pediatr. 2008, 167: 695-696. 10.1007/s00431-007-0547-3.
Chae JJ, Wood G, Masters SL, Richard K, Park G, Smith BJ, Kastner DL: The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci USA. 2006, 103: 9982-9987. 10.1073/pnas.0602081103.
Roldan R, Ruiz AM, Miranda MD, Collantes E: Anakinra: new therapeutic approach in children with Familial Mediterranean Fever resistant to colchicine. Joint Bone Spine. 2008, 75: 504-505. 10.1016/j.jbspin.2008.04.001.
Allantaz F, Chaussabel D, Stichweh D, Bennett L, Allman W, Mejias A, Ardura M, Chung W, Wise C, Palucka K, Ramilo O, Punaro M, Banchereau J, Pascual V: Blood leukocyte microarrays to diagnose systemic onset juvenile idiopathic arthritis and follow the response to IL-1 blockade. J Exp Med. 2007, 204: 2131-2144.
Dierselhuis MP, Frenkel J, Wulffraat NM, Boelens JJ: Anakinra for flares of pyogenic arthritis in PAPA syndrome. Rheumatology (Oxford). 2005, 44: 406-408. 10.1093/rheumatology/keh479.
Bodar EJ, Hilst van der JC, Drenth JP, Meer van der JW, Simon A: Effect of etanercept and anakinra on inflammatory attacks in the hyper-IgD syndrome: introducing a vaccination provocation model. Neth J Med. 2005, 63: 260-264.
Benezra D, Maftzir G, Barak V: Blood serum interleukin-1 receptor antagonist in pars planitis and ocular Behcet disease. Am J Ophthalmol. 1997, 123: 593-598.
Duzgun N, Ayaslioglu E, Tutkak H, Aydintug OT: Cytokine inhibitors: soluble tumor necrosis factor receptor 1 and interleukin-1 receptor antagonist in Behcet's disease. Rheumatol Int. 2005, 25: 1-5. 10.1007/s00296-003-0400-6.
Imrie FR, Dick AD: Biologics in the treatment of uveitis. Curr Opin Ophthalmol. 2007, 18: 481-486. 10.1097/ICU.0b013e3282f03d42.
Carneiro LA, Travassos LH, Girardin SE: Nod-like receptors in innate immunity and inflammatory diseases. Ann Med. 2007, 39: 581-593. 10.1080/07853890701576172.
Acknowledgements
This work was supported by grants from the Sir Jules Thorn 'Seed Corn' Fund and the Charitable Foundation of the Leeds Teaching Hospitals (Dr Sprakes is currently funded by the Charitable Trustees, Leeds General Infirmary).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Mathews, R.J., Sprakes, M.B. & McDermott, M.F. NOD-like receptors and inflammation. Arthritis Res Ther 10, 228 (2008). https://doi.org/10.1186/ar2525
Published:
DOI: https://doi.org/10.1186/ar2525