
Abstract
In the past few years considerable advances have been made in the genetics of susceptibility to rheumatoid arthritis (RA). For decades the HLA-DRB1 alleles were the only extensively replicated genetic factor, but more genetic risk factors have now been identified that predispose to RA. Interestingly, several of the observed genetic variants conferred risk to anticitrulline-peptide antibody (ACPA)-positive RA and two variants may be restricted to ACPA-negative RA, pointing to the need for subclassification of RA. The current manuscript reviews recently identified genetic factors predisposing to ACPA-positive RA and ACPA-negative RA. Additionally, although being scarcely explored, genetic variants affecting the severity of disease course are discussed.
Similar content being viewed by others

Introduction
Rheumatoid arthritis (RA) is a chronic potentially destructive arthritis classified by the presence of four out of the seven criteria developed by the American College of Rheumatology (ACR) in 1987. The criteria were formulated by experts who compared characteristics of patients with longstanding classical RA (mean disease duration 8 years). These ACR criteria have been criticized during past years as they have a low discriminative ability in patients presenting with recent-onset arthritis [1–4]. This is not surprising considering the components of the ACR criteria. One of the criteria is the presence of erosions on the radiographs of hands and wrists. In the early phases of RA only 13% of the patients have erosive disease [5]. Additionally, erosions often initially present in the small joints of the feet, and appear in the small joints of the hands at a later point in the disease course [6]. Also, rheumatoid nodules are very rare in the early phases of RA, and rheumatoid factor is present in only 50% of the patients with early RA [7].
Conversely, the serological factor that has the strongest association with RA, anticitrulline-peptide antibody (ACPA), is not part of the classification criteria. A taskforce has therefore been formed by the European League Against Rheumatism to develop classification criteria for early inflammatory arthritis. This taskforce is guided by the European League Against Rheumatism Standing Committee on Epidemiology as well as the ACR Quality Measurement Committee. It was agreed that this was an appropriate goal and that a proposal outlining the work to be done should be submitted to the Executive Board of the European League Against Rheumatism and the ACR for consideration of support. This exercise should not be seen as an attempt to redefine criteria for established RA (as classifiable by the original 1987 ACR criteria), but as an attempt to develop criteria for early (actual or potential) RA.
Considering the classification of RA, several questions can be raised. Do we consider RA one disease or is it a disorder composed of several (sub)entities? Second, should classification be based on clinical features or also on pathophysiological characteristics? These questions are pressing, as current evidence indicates that the identified genetic risk factors do not predispose to all RA patients but only to a specific subset.
The present manuscript highlights the latest advances in the genetics of RA susceptibility in relation to subdivision of the disease based on autoantibodies, in particular ACPA. The genetic risk factors that predispose to ACPA-positive RA and ACPA-negative RA are reviewed. Additionally, the current knowledge on genetic variants involved in the severity of RA is evaluated.
Genetic factors predisposing to anticitrulline-peptide antibody-positive RA
Human leucocyte antigens class II
The most important genetic risk factor for RA was identified three decades ago and consists of the human leucocyte antigen (HLA) class II molecules. There is extensive evidence showing that certain frequently occurring HLA-DRB1 alleles (HLA-DRB1*0101, HLA-DRB1*0102, HLA-DRB1*0401, HLA-DRB1*0404, HLA-DRB1*0405, HLA-DRB1*0408, HLA-DRB1*0410 HLA-DRB1*1001, HLA-DRB1*1402) are associated with susceptibility to RA. The indicated alleles share a conserved amino acid sequence (QKRAA, QRRAA or RRRAA) – also called the shared epitope (SE) – at position 70 to 74 in the third hypervariable region of the DRβ1 chain. These residues are part of an α-helical domain forming one side of the antigen-presenting binding site.
The SE hypothesis postulates that the SE motif itself is directly involved in the pathogenesis of RA by allowing the presentation of a peptide to arthritogenic T cells [8]. No specific arthritogenic peptides that bind to the HLA-DRB1 proteins and subsequently activate T cells have so far been identified, hence the SE hypothesis is not functionally proven. Nevertheless, this hypothesis is robust because of its consistent association (although quantitatively varying between alleles and populations) among various ethnic populations. It is estimated that the heritability of susceptibility to RA is 50% to 60% and that the SE alleles account for at least 30% of the total genetic effect [9, 10].
The role of the HLA-DRB1 SE alleles as well as of ACPA in RA susceptibility was recently studied in greater depth, and it was observed that the SE alleles associate only with RA patients who carry ACPA and not with ACPA-negative RA patients [11, 12]. This finding led to the hypothesis that the SE alleles confer risk to ACPA rather than to (ACPA-positive) RA. To investigate this hypothesis, the progression from recent-onset undifferentiated arthritis to RA was studied in relation to the SE alleles and autoantibodies [11, 13]. In patients who presented with undifferentiated arthritis, the presence of ACPA was associated with progression to RA, both in SE-positive undifferentiated arthritis patients and in SE-negative undifferentiated arthritis patients. In contrast, both in ACPA-positive undifferentiated arthritis patients and ACPA-negative undifferentiated arthritis patients, the association between the SE alleles and the development of RA was lost, indicating that the predictive value of the SE alleles is lost once the ACPA response has developed [12]. These data indicate that the SE alleles particularly confer risk to ACPA and that these antibodies explain the association between the SE alleles and RA. Moreover, these observations provided the first evidence that the etiopathology of ACPA-positive RA is different from that of ACPA-negative RA.
Protein tyrosine phosphatase nonreceptor 22
The second risk factor for RA, identified in 2004, is non-HLA linked and concerns the C1858T single nucleotide polymorphism (SNP) in the gene encoding for the protein tyrosine phosphatase nonreceptor 22 (PTPN22) [14]. An association between PTPN22 and RA has presently been demonstrated in several populations [15–17], and several studies have revealed that the PTPN22 T allele not only confers risk to RA but also to other autoimmune diseases such as lupus, type 1 diabetes and Graves disease. Intriguingly, PTPN22 is associated with ACPA-positive RA but not with ACPA-negative RA [17]. Moreover, in three independent cohorts of RA patients a gene-gene interaction for the HLA SE alleles and PTPN22 was shown for ACPA-positive RA, but not for ACPA-negative RA [18]. This observation strengthens the assumption that ACPA-positive RA and ACPA-negative RA have a different pathogenesis.
PTPN22 encodes for a lymphoid tyrosine phosphatase that affects the threshold for T-cell receptor signaling through binding to a Csk kinase. In vitro experiments have shown that the PTPN22 T-allele-encoded protein binds less efficiently to Csk, suggesting that the T cells expressing the T allele are hyperresponsive [14]. Knocking out the murine homologue of PTPN22 resulted in lower thresholds of T-cell activation and induced an increased expansion and function of the effector/memory T-cell pool, which was associated with elevated levels of serum antibodies [19]. Both of these studies indicate that the PTPN22 risk allele is associated with a reduced downregulation of T-cell activation. In contrast to this loss of function, a gain of function has also been described in carriers of the PTPN22 T allele [20]. Hence the biological mechanisms underlying the association between PTPN22 and RA are incompletely understood.
Complement component 5-TNF receptor-associated factor 1
Only very recently was the third genetic risk factor for RA identified. Interestingly, the observation was made in three countries at the same time using different approaches. In a study performed in the Netherlands, a candidate-gene approach showed an association between RA susceptibility and complement component 5-TNF receptor-associated factor 1 (C5-TRAF1) [21]. In this study the complement C5 region on chromosome 9q33-34 was investigated as a candidate because mice studies revealed that mice deficient in complement factors are resistant to arthritis and that targeting C5 by antibodies prevents the onset of arthritis and reduces the clinical severity of arthritis in mice [22, 23]. Other studies showed that C5a receptor-deficient mice are resistant to arthritis induction [24]. These data combined with the observation that high levels of C5a are found in synovial fluid of RA patients suggested these mediators play a central role in arthritis [25]. Investigating the SNPs spanning this region revealed significant associations; the most significant SNP was not located within C5 but between C5 and TRAF1 [21]. The protein encoded by TRAF1 is a member of the TNF receptor-associated factor 1 protein family, which mediates the signal transduction from various receptors of the TNF receptor superfamily, including the receptor for TNFα.
Interestingly, as this candidate gene study was completed an association between the C5-TRAF1 region was also demonstrated in two independent genome-wide association studies in the USA and Sweden [26]. The demonstration of similar results in independent studies making use of different approaches provides strong evidence for the C5-TRAF1 region as a true RA-associated genetic variant. Moreover, additional evaluation in the Dutch study revealed that this genetic risk factor specifically predisposes to ACPA-positive RA and not to ACPA-negative RA [21]. The two genome-wide association studies showing an association between C5-TRAF1 and RA only investigated ACPA-positive patients [26].
At present it is not clear whether the most significant SNP in C5-TRAF1 is causative since other proxies in high-linkage disequilibrium with this SNP can also be responsible for the observed association. Additionally, the functional consequence of carrying the C5-TRAF1 polymorphism is as yet unknown.
Cytotoxic T lymphocyte antigen 4
A genetic risk factor that has been investigated frequently in relation to RA, providing inconsistent results, is a noncoding variant in the 3' end of the gene encoding for cytotoxic T lymphocyte antigen 4 (CTLA4) [27, 28]. Combined data from the USA and Sweden have more recently suggested that CTLA4 confers risk only to the subset of ACPA-positive RA patients and not to ACPA-negative disease [29]. This association, however, was only found in the Swedish cohort and not in the American cohort. Although this may be seen as support for another genetic risk factor that mainly contributes to ACPA-positive disease, it may also be a false-positive finding or a population-specific effect.
The CTLA4 protein plays an important role in downregulation of T-cell activation. To be fully activated the T cell requires the recognition of an antigen bound to HLA and a costimulatory signal between CD80 or CD86 on the antigen-presenting cell and CD28 on the T cell. This costimulatory signal can be inhibited by CTLA4, which is expressed on T cells as CTLA4 binds to CD80/CD86 with higher affinity.
STAT4
Very recently, a linkage peak on the long arm of chromosome 2 was investigated thoroughly. The study confirmed the association between CTLA4 and RA but also observed an association with an unlinked SNP in the region encoding for STAT4 [30]. This finding was observed in a Swedish as well as an American case-control study. Although the majority of the RA patients of the American cohort were ACPA-positive, such data on the Swedish cohort were not provided. Based on this study, therefore, no information is available on whether STAT4 predisposes to ACPA-positive RA and/or ACPA-negative RA. Additionally, it cannot be excluded that this study provided false-positive results. Multiple replication studies are thus required before this polymorphism can be added to the list of identified risk factors for RA.
TNFα-induced protein 3-oligodendrocyte lineage transcription factor 3 (TNFAIP3-OLIG3)
The Wellcome Trust Case Control Consortium performed a genome-wide association study in almost 2,000 RA patients. They reported moderately associated regions on chromosome 6 (6q23) located between the genes encoding for TNFAIP3-OLIG3, which were unequivocally replicated in a follow-up study [31]. In total, combining the Wellcome Trust Case Control Consortium data with the validation cohort, 2,515 ACPA-positive RA patients and 1,039 ACPA-negative RA patients were compared with 6,629 healthy control individuals, revealing only significant results in the ACPA-positive patients [31].
Another SNP, located 3.8 kb from the abovementioned SNP, was found by an independent group that evaluated only ACPA-positive patients. The effects of this SNP on susceptibility to RA were reported to be statistically independent from the SNP initially described by the Wellcome Trust Case Control Consortium [32]. Although the function of these intergenic SNPs as well as the definite haplotypes that predispose or protect to RA are not fully elucidated, these data do suggest that the gene(s) in the region encoding for TNFAIP3-OLIG3 are of importance for susceptibility to ACPA-positive RA.
Fc-receptor-like gene 3
A functional promoter polymorphism (-169T → T) in the Fc-receptor-like gene 3 at chromosome 1q21-23 was first demonstrated to be associated with RA in two independent Japanese case-control studies [33, 34]. Subsequently a number of studies evaluated this polymorphism, showing contrasting results. Two meta-analyses revealed recently that this SNP has a role in the risk for RA amongst East Asians, but there is no compelling evidence for an association of this SNP with RA in Caucasians [35, 36]. Similarly, although Kochi and colleagues suggested this SNP influences the level of autoantibodies, a recent study from Norway found no association between the Fc-receptor-like gene 3 genotype and the level of ACPA [37]. These data demonstrate that associations between genetic variants and disease may differ in diverse ethnic populations.
Peptidylarginine deiminase 4
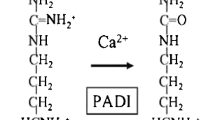
Another genetic variant, the potency of which differs between populations, is peptidylarginine deiminase 4 (PADI4). Large-scale linkage disequilibrium studies and subsequent replication studies implicated a variant of PADI4 as a risk factor for RA in Japanese and Korean populations [38, 39]. This finding that was not observed in the majority of studies in populations of European descent. Intriguingly, the Japanese/Korean association is with a haplotype rather than with a SNP with a proposed function. Another SNP linked to this haplotype may therefore be the causative risk factor.
Interestingly, a German study revealed an association between PADI4 and RA but the haplotypes providing this association were different to the haplotypes that conferred risk in the Asian populations [40]. This may indicate that PADI4 or a factor linked to PADI4 may predispose to RA but that the actual causing genetic variant is not elucidated. Possible other explanations for this discrepancy are the presence of other genetic or environmental factors that interact with the genetic factor in a specific population, thereby affecting disease susceptibility or enrichment of genetic variants in one population but not in the other population. Although the peptidylarginine deiminases are implicated in the generation of ACPA, there is no compelling evidence supporting PADI4 genotypes correlating with ACPA levels – or ACPA-positive disease in particular.
Genetic factors predisposing to anticitrulline-peptide antibody-negative RA
Human leucocyte antigens class II
The assumption that the etiopathology of ACPA-positive RA is different from that of ACPA-negative RA is strengthened when genetic risk factors are found that exclusively associate with RA characterized by the absence of ACPA. Two independent studies showed that the HLA-DR3 alleles predispose only to ACPA-negative RA but not to ACPA-positive RA [12, 41]. Whether this association is attributable to the HLA-DR3 gene itself or to genes linked to this locus is not known, however, as HLA-DR3 is part of a conserved ancestral haplotype (A1;B8;DRB1*03). The major histo-compatibility complex class III region – encoding for, amongst others, TNF and lymphotoxin α – is also part of this ancestral haplotype and has been described to influence the susceptibility to RA.
Nonhuman leucocyte antigens
Recently described in a Swedish cohort and a Dutch cohort of patients was a second risk factor for ACPA-negative RA – interferon regulatory factor 5 (IRF5) [42]. Whereas IRF5 is consistently associated with susceptibility to lupus, the correlation of IRF5 with RA is less clear as French and Spanish studies observed no association between IRF5 and RA [43, 44]. Nevertheless these two studies did not investigate the effect of this risk factor on ACPA-negative RA separately, and thus these findings may be false negative. Future studies may further increase comprehension of the role of IRF5 in RA.
The fact that more genetic risk factors for ACPA-positive disease have thus far been identified than for ACPA-negative RA does not necessarily indicate that the contribution of genetic factors to the etiopathology of ACPA-positive RA is larger than that to the etiopathology of ACPA-negative RA. In contrast, many important studies that revealed new genetic risk factors used cohorts constituting of mainly ACPA-positive RA patients [14, 30–32]. Part of the research conducted is thus biased towards locating genetic variants predisposing to ACPA-positive RA.
Pathophysiological differences between anticitrulline-peptide antibody-positive RA and anticitrulline-peptide antibody-negative RA
The reports demonstrating dissimilar genetic risk factors for ACPA-positive RA and ACPA-negative RA strongly suggest that ACPA-positive RA and ACPA-negative RA are separate subsets of RA with pathophysiological differences. Nonetheless, the effects of the described polymorphisms on gene function are still uncertain. Although the first structural difference between ACPA-positive RA and ACPA-negative RA – a difference in synovial tissue infiltrates between ACPA-positive RA patients and ACPA-negative RA-patients with active arthritis – was recently observed [45], the complete spectrum of pathophysiologic differences between ACPA-positive RA and ACPA-negative RA is as yet incompletely identified.
Recent advances in the genetics of RA severity
Altogether the genetics of RA susceptibility has made enormous progress during the past years, several more genetic risk factors will be recognized – considering the enormous efforts that are currently being made for RA susceptibility. In contrast, the field of the genetics of RA severity is relatively unexplored. Radiological joint damage is conceived as the most objective measure to determine the severity of RA. The number of studies investigating the relation between joint destruction and genetic variants is limited. Some SNPs are observed to associate with the disease outcome, but none of these associations are convincingly replicated in independent cohorts. Moreover, although a recent twin study indicated that genetic factors play a role in determining the severity of RA [46], the heritability of the level of joint destruction (the variance of joint destruction that can be ascribed to genetic factors) is still unknown.
Joint damage in RA is highly variable between patients and the cumulative level is associated with the level of (persistent) inflammation. Nonetheless, several studies have provided evidence for incomplete coupling between inflammation and destruction, which suggests that other individual factors also play a role. This suggestion leads to the hypothesis that candidate genes for the severity of RA are, on the one hand, genes that regulate the level of (local) inflammation and, on the other, genes that mediate the process of bone/cartilage destruction or bone regeneration. The contemporary data on genetics of RA severity are summarized below.
Human leucocyte antigens class II
The HLA-SE alleles, and the DR4 alleles in particular, are known to predispose to a destructive disease course. It is now also known that the SE alleles primarily associate with the presence of ACPA and that these autoantibodies are a strong predictor for a severe disease outcome. This knowledge results in the question of whether the SE alleles contribute to a destructive disease course independent of the disease's effect on ACPA; a question that remains unsolved.
The HLA-DRB1 alleles not only encode for risk factors but are also protective factors. The HLA-DRB1 alleles encoding for the amino acids DERAA protect against a destructive disease course, an effect that is independent of the presence/absence of SE alleles but is most evident in the ACPA-positive group [47].
Genetic variants in interleukins
Within a number of cytokines such as TNFα, IL1, IL4, IL-1α, IL-1β or IL-1RN, variations have been reported and associations have been found only in small studies that are not independently replicated [48–50]. Future studies will reveal whether the observed associations will be observed in replication studies.
Genetic variants in complement components
The SNPs located in the region encoding for C5 and TRAF1 observed to associate with susceptibility to RA in three different cohorts [21, 26] were evaluated for their correlation with disease severity in one study [21]. This provided the first indication that carrying the C5-TRAF1 risk allele relates to a destructive disease course. This association is not yet replicated, however, and it has not been investigated whether the correlation with a severe disease course was independent of ACPA.
Genetic variants in mediators of bone/cartilage destruction
The destruction of bone and cartilage in RA is mediated by proteolytic enzymes that belong to the family of matrix metalloproteinases (MMPs). The most intensively studied proteinases are MMP3 (stromelysin) and MMP1 (collagenase). The levels of these MMPs are increased in both serum and synovial fluid of patients with RA, and the serum levels of MMP3 have been correlated with joint destruction in RA. Studies assessing polymorphism within the MMP3 gene promoter region revealed correlations between MMP3 haplotypes and joint damage; however, the results on which haplotypes conferred an increased risk were inconsistent [51–53]. Further evaluation of the effect of genetic variation in MMP3 on the level of joint damage in RA is therefore required.
The increasing comprehension of the role of genetics in disease outcome in RA may promote the development of personalized medicine. Although the prospects of RA patients on a group level have improved dramatically due to the growing arsenal of new aggressive antirheumatic drugs and recent data demonstrating that combination therapy is more effective than monotherapy, these developments do not automatically lead to benefit on the individual level. At present, > 95% of newly diagnosed RA patients start with methotrexate monotherapy, which is ineffective in 66% of patients [54]. Subsequently, methotrexate is switched to another disease-modifying antoretroviral drug or a disease-modifying antoretroviral drug is added to the methotrexate therapy; if this strategy also fails, targeted therapies such as TNF blockers may be prescribed. The disease course in RA is highly variable and this commonly used step-up therapeutic strategy implies that a proportion of newly diagnosed RA patients are insufficiently treated for a specific time, resulting in the development of erosions that might have been prevented if a more vigorous treatment strategy had been applied. Clinical and serological characteristics solely are insufficient to predict the disease outcome in individual patients [55]. Additional knowledge on genetic variants will increase the predictive ability.
Conclusion
Several genetic factors are associated with susceptibility to RA. Interestingly, the majority of the identified genetic factors conferred risk to ACPA-positive RA, whereas two genetic variants may be restricted to ACPA-negative RA (Table 1). This observation illustrates that knowledge of genetic variants may contribute to the understanding of RA and that sub-classification of RA into ACPA-positive and ACPA-negative entities is advocated. Nonetheless, the complete spectrum of pathophysiologic differences between ACPA-positive RA and ACPA-negative RA is incompletely identified and the effects of the described polymorphisms on gene function are still uncertain. Further studies elucidating these effects will enhance the understanding of the mechanisms of disease and will thereby promote the development of targeted therapies and the translation from genetics to clinical practice.
In contrast to the recent advances in the field of susceptibility to RA, genetic variants affecting the severity of the disease course in RA are scarcely explored. It is hoped that future studies will identify genetic risk factors that predict a severe disease course and that these result will allow personalized medicine in RA, thereby reducing avoidable joint destruction due to undertreatment and also lessening complications due to overtreatment.
Abbreviations
- ACPA:
-
anticitrulline-peptide antibody
- ACR:
-
American College of Rheumatology
- C5-TRAF1 :
-
= complement component 5-TNF receptor-associated factor 1
- CTLA4 :
-
= cytotoxic T lymphocyte antigen 4
- HLA:
-
human leucocyte antigen
- IL:
-
interleukin
- IRF5 :
-
= interferon regulatory factor 5
- MMP:
-
matrix metalloproteinase
- PADI4 :
-
= peptidylarginine deiminase 4
- PTPN22 :
-
= protein tyrosine phosphatase nonreceptor 22
- RA:
-
rheumatoid arthritis
- SE:
-
shared epitope
- SNP:
-
single nucleotide polymorphism
- TNF:
-
tumor necrosis factor.
References
Harrison BJ, Symmons DP, Barrett EM, Silman AJ: The performance of the 1987 ARA classification criteria for rheumatoid arthritis in a population based cohort of patients with early inflammatory polyarthritis. American Rheumatism Association. J Rheumatol. 1998, 25: 2324-2330.
Harrison B, Symmons D: Early inflammatory polyarthritis: results from the Norfolk Arthritis Register with a review of the literature. II. Outcome at three years. Rheumatology (Oxford). 2000, 39: 939-949. 10.1093/rheumatology/39.9.939.
Symmons DP, Hazes JM, Silman AJ: Cases of early inflammatory polyarthritis should not be classified as having rheumatoid arthritis. J Rheumatol. 2003, 30: 902-904.
Green M, Marzo-Ortega H, McGonagle D, Wakefield R, Proud-man S, Conaghan P, Gooi J, Emery P: Persistence of mild, early inflammatory arthritis: the importance of disease duration, rheumatoid factor, and the shared epitope. Arthritis Rheum. 1999, 42: 2184-2188. 10.1002/1529-0131(199910)42:10<2184::AID-ANR20>3.0.CO;2-2.
Machold KP, Stamm TA, Eberl GJ, Nell VK, Dunky A, Uffmann M, Smolen JS: Very recent onset arthritis – clinical, laboratory, and radiological findings during the first year of disease. J Rheumatol. 2002, 29: 2278-2287.
van der Heijde DM, van Leeuwen MA, van Riel PL, van de Putte LB: Radiographic progression on radiographs of hands and feet during the first 3 years of rheumatoid arthritis measured according to Sharp's method (van der Heijde modification). J Rheumatol. 1995, 22: 1792-1796.
Nell VP, Machold KP, Stamm TA, Eberl G, Heinzl H, Uffmann M, Smolen JS, Steiner G: Autoantibody profiling as early diagnostic and prognostic tool for rheumatoid arthritis. Ann Rheum Dis. 2005, 64: 1731-1736. 10.1136/ard.2005.035691.
Gregersen PK, Silver J, Winchester RJ: The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30: 1205-1213. 10.1002/art.1780301102.
MacGregor AJ, Antoniades L, Matson M, Andrew T, Spector TD: Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000, 43: 30-37. 10.1002/1529-0131(200001)43:1<30::AID-ANR5>3.0.CO;2-B.
Deighton CM, Walker DJ, Griffiths ID, Roberts DF: The contribution of HLA to rheumatoid arthritis. Clin Genet. 1989, 36: 178-182.
Huizinga TW, Amos CI, van der Helm-van Mil AH, Chen W, van Gaalen FA, Jawaheer D, Schreuder GM, Wener M, Breedveld FC, Ahmad N, Lum RF, de Vries RR, Gregersen PK, Toes RE, Criswell LA: Refining the complex rheumatoid arthritis phenotype based on specificity of the HLA-DRB1 shared epitope for antibodies to citrullinated proteins. Arthritis Rheum. 2005, 52: 3433-3438. 10.1002/art.21385.
Irigoyen P, Lee AT, Wener MH, Li W, Kern M, Batliwalla F, Lum RF, Massarotti E, Weisman M, Bombardier C, Remmers EF, Kastner DL, Seldin MF, Criswell LA, Gregersen PK: Regulation of anti-cyclic citrullinated peptide antibodies in rheumatoid arthritis: contrasting effects of HLA-DR3 and the shared epitope alleles. Arthritis Rheum. 2005, 52: 3813-3818. 10.1002/art.21419.
van der Helm-van Mil AH, Verpoort KN, Breedveld FC, Huizinga TW, Toes ER, de Vries RR: The HLA-DRB1 shared epitope alleles are primarily a risk factor for anti-CCP antibodies and are not an independent risk factor to develop RA. Arthritis Rheum. 2006, 54: 1117-1121. 10.1002/art.21739.
Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM, Conn MT, Chang M, Chang SY, Saiki RK, Catanese JJ, Leong DU, Garcia VE, McAllister LB, Jeffery DA, Lee AT, Batli-walla F, Remmers E, Criswell LA, Seldin MF, Kastner DL, Amos CI, Sninsky JJ, Gregersen PK: A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phos-phatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. 2004, 75: 330-337. 10.1086/422827.
van Oene M, Wintle RF, Liu X, Yazdanpanah M, Gu X, Newman B, Kwan A, Johnson B, Owen J, Greer W, Mosher D, Maksymowych W, Keystone E, Rubin LA, Amos CI, Siminovitch KA: Association of the lymphoid tyrosine phosphatase R620W variant with rheumatoid arthritis, but not Crohn's disease, in Canadian populations. Arthritis Rheum. 2005, 52: 1993-1998. 10.1002/art.21123.
Gomez LM, Anaya JM, Gonzalez CI, Pineda-Tamayo R, Otero W, Arango A, Martin J: PTPN22 C1858T polymorphism in Colombian patients with autoimmune diseases. Genes Immun. 2005, 6: 628-631. 10.1038/sj.gene.6364261.
Wesoly J, van der Helm-van Mil AH, Toes RE, Chokkalingam AP, Carlton VE, Begovich AB, Huizinga TW: Association of the PTPN22 C1858T single-nucleotide polymorphism with rheumatoid arthritis phenotypes in an inception cohort. Arthritis Rheum. 2005, 52: 2948-2950. 10.1002/art.21294.
Kallberg H, Padyukov L, Plenge RM, Ronnelid J, Gregersen PK, van der Helm-van Mil AH, Toes RE, Huizinga TW, Klareskog L, Alfredsson L, Epidemiological Investigation of Rheumatoid Arthritis study group: Gene-gene and gene-environment interactions involving HLA-DR, PTPN22 and smoking in two subsets of rheumatoid arthritis. Am J Hum Genet. 2007, 80: 867-875. 10.1086/516736.
Hasegawa K, Martin F, Huang G, Tumas D, Diehl L, Chan AC: PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells. Science. 2004, 303: 685-689. 10.1126/science.1092138.
Vang T, Congia M, Macis MD, Musumeci L, Orrú V, Zavattari P, Nika K, Tautz L, Taskén K, Cucca F, Mustelin T, Bottini N: Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nat Genet. 2005, 37: 1317-1319. 10.1038/ng1673.
Kurreeman FA, Padyukov L, Marques RB, Schrodi SJ, Sed-dighzadeh M, Stoeken-Rijsbergen G, van der Helm-van Mil AH, Allaart CF, Verduyn W, Houwing-Duistermaat J, Alfredsson L, Begovich AB, Klareskog L, Huizinga TW, Toes RE: A candidate gene approach identifies the TRAF1/C5 region as a risk factor for rheumatoid arthritis. PLoS Med. 2007, 4: e278-10.1371/journal.pmed.0040278.
Wang Y, Kristan J, Hao L, Lenkoski CS, Shen Y, Matis LA: A role for complement in antibody-mediated inflammation: C5-deficient DBA/1 mice are resistant to collagen-induced arthritis. J Immunol. 2000, 164: 4340-4347.
Wang Y, Rollins SA, Madri JA, Matis LA: Anti-C5 monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proc Natl Acad Sci USA. 1995, 92: 8955-8959. 10.1073/pnas.92.19.8955.
Grant EP, Picarella D, Burwell T, Delaney T, Croci A, Avitahl N, Humbles AA, Gutierrez-Ramos JC, Briskin M, Gerard C, Coyle AJ: Essential role for the C5a receptor in regulating the effector phase of synovial infiltration and joint destruction in experimental arthritis. J Exp Med. 2002, 196: 1461-1471. 10.1084/jem.20020205.
Høgåsen K, Mollnes TE, Harboe M, Götze O, Hammer HB, Oppermann M: Terminal complement pathway activation and low lysis inhibitors in rheumatoid arthritis synovial fluid. J Rheumatol. 1995, 22: 24-28.
Plenge RM, Seielstad M, Padyukov L, Lee AT, Remmers EF, Ding B, Liew A, Khalili H, Chandrasekaran A, Davies LR, Li W, Tan AK, Bonnard C, Ong RT, Thalamuthu A, Pettersson S, Liu C, Tian C, Chen WV, Carulli JP, Beckman EM, Altshuler D, Alfredsson L, Criswell LA, Amos CI, Seldin MF, Kastner DL, Klareskog L, Gregersen PK: TRAF1-C5 as a risk locus for rheumatoid arthritis – a genomewide study. N Engl J Med. 2007, 357: 1199-1209. 10.1056/NEJMoa073491.
Rodriguez MR, Nunez-Roldan A, Aguilar F, Valenzuela A, Garcia A, Gonzalez-Escribano MF: Association of the CTLA4 3'untranslated region polymorphism with the susceptibility to rheumatoid arthritis. Hum Immunol. 2002, 63: 76-81. 10.1016/S0198-8859(01)00358-5.
Vaidya B, Pearce SH, Charlton S, Marshall N, Rowan AD, Griffiths ID, Kendall-Taylor P, Cawston TE, Young-Min S: An association between the CTLA4 exon 1 polymorphism and early rheumatoid arthritis with autoimmune endocrinopathies. Rheumatology (Oxford). 2002, 41: 180-183. 10.1093/rheumatology/41.2.180.
Plenge RM, Padyukov L, Remmers EF, Purcell S, Lee AT, Karlson EW, Wolfe F, Kastner DL, Alfredsson L, Altshuler D, Gregersen PK, Klareskog L, Rioux JD: Replication of putative candidate-gene associations with rheumatoid arthritis in >4,000 samples from North America and Sweden: association of susceptibility with PTPN22, CTLA4, and PADI4. Am J Hum Genet. 2005, 77: 1044-1060. 10.1086/498651.
Remmers EF, Plenge RM, Lee AT, Graham RR, Hom G, Behrens TW, de Bakker PI, Le JM, Lee HS, Batliwalla F, Li W, Masters SL, Booty MG, Carulli JP, Padyukov L, Alfredsson L, Klareskog L, Chen WV, Amos CI, Criswell LA, Seldin MF, Kastner DL, Gregersen PK: STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med. 2007, 357: 977-986. 10.1056/NEJMoa073003.
Thomson W, Barton A, Ke X, Eyre S, Hinks A, Bowes J, Donn R, Symmons D, Hider S, Bruce IN, Wellcome Trust Case Control Consortium, Wilson AG, Marinou I, Morgan A, Emery P, YEAR Consortium, Carter A, Steer S, Hocking L, Reid DM, Wordsworth P, Harrison P, Strachan D, Worthington J: Rheumatoid arthritis association at 6q23. Nat Genet. 2007, 39: 1431-1433. 10.1038/ng.2007.32.
Plenge RM, Cotsapas C, Davies L, Price AL, de Bakker PI, Maller J, Pe'er I, Burtt NP, Blumenstiel B, DeFelice M, Parkin M, Barry R, Winslow W, Healy C, Graham RR, Neale BM, Izmailova E, Roubenoff R, Parker AN, Glass R, Karlson EW, Maher N, Hafler DA, Lee DM, Seldin MF, Remmers EF, Lee AT, Padyukov L, Alfredsson L, Coblyn J, Weinblatt ME, Gabriel SB, Purcell S, Klareskog L, Gregersen PK, Shadick NA, Daly MJ, Altshuler D: Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007, 39: 1477-1482. 10.1038/ng.2007.27.
Kochi Y, Yamada R, Suzuki A, Harley JB, Shirasawa S, Sawada T, Bae SC, Tokuhiro S, Chang X, Sekine A, Takahashi A, Tsunoda T, Ohnishi Y, Kaufman KM, Kang CP, Kang C, Otsubo S, Yumura W, Mimori A, Koike T, Nakamura Y, Sasazuki T, Yamamoto K: A functional variant in FCRL3, encoding Fc receptor-like 3, is associated with rheumatoid arthritis and several autoimmunities. Nat Genet. 2005, 37: 478-485. 10.1038/ng1540.
Ikari K, Momohara S, Nakamura T, Hara M, Yamanaka H, Tomatsu T, Kamatani N: Supportive evidence for a genetic association of the FCRL3 promoter polymorphism with rheumatoid arthritis. Ann Rheum Dis. 2006, 65: 671-673. 10.1136/ard.2005.043489.
Begovich AB, Chang M, Schrodi SJ: Meta-analysis evidence of a differential risk of the FCRL3 -169T → C polymorphism in white and East Asian rheumatoid arthritis patients. Arthritis Rheum. 2007, 56: 3168-3171. 10.1002/art.22857.
Takata Y, Inoue H, Sato A, Tsugawa K, Miyatake K, Hamada D, Shinomiya F, Nakano S, Yasui N, Tanahashi T, Itakura M: Replication of reported genetic associations of PADI4, FCRL3, SLC22A4 and RUNX1 genes with rheumatoid arthritis: results of an independent Japanese population and evidence from meta-analysis of East Asian studies. J Hum Genet. 2007
Eike MC, Nordang GB, Karlsen TH, Boberg KM, And The Ib Vatn, Dahl-Jørgensen K, Rønningen KS, Joner G, Flatø B, Bergquist A, Thorsby E, Førre O, Kvien TK, Undlien DE, Lie BA: The FCRL3 -169T>Cpolymorphism is associated with rheumatoid arthritis and shows suggestive evidence of involvement with juvenile idiopathic arthritisin a Scandinavian panel of autoimmune diseases. Ann Rheum Dis. 2007
Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, Nagasaki M, Nakayama-Hamada M, Kawaida R, Ono M, Ohtsuki M, Furukawa H, Yoshino S, Yukioka M, Tohma S, Matsubara T, Wakitani S, Teshima R, Nishioka Y, Sekine A, Iida A, Takahashi A, Tsunoda T, Nakamura Y, Yamamoto K: Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. 2003, 34: 395-402. 10.1038/ng1206.
Kang CP, Lee HS, Ju H, Cho H, Kang C, Bae SC: A functional haplotype of the PADI4 gene associated with increased rheumatoid arthritis susceptibility in Koreans. Arthritis Rheum. 2006, 54: 90-96. 10.1002/art.21536.
Hoppe B, Haupl T, Gruber R, Kiesewetter H, Burmester GR, Salama A, Dorner T: Detailed analysis of the variability of pep-tidylarginine deiminase type 4 in German patients with rheumatoid arthritis: a case-control study. Arthritis Res Ther. 2006, 8: R34-10.1186/ar1889.
Verpoort KN, van Gaalen FA, van der Helm-van Mil AH, Schreuder GM, Breedveld FC, Huizinga TW, de Vries RR, Toes RE: Association of HLA-DR3 with anti-cyclic citrullinated peptide antibody-negative rheumatoid arthritis. Arthritis Rheum. 2005, 52: 3058-3062. 10.1002/art.21302.
Sigurdsson S, Padyukov L, Kurreeman FA, Liljedahl U, Wiman AC, Alfredsson L, Toes R, Ronnelid J, Klareskog L, Huizinga TW, Alm G, Syvanen AC, Ronnblom L: Association of a haplotype in the promoter region of the interferon regulatory factor 5 gene with rheumatoid arthritis. Arthritis Rheum. 2007, 56: 2202-2210. 10.1002/art.22704.
Garnier S, Dieude P, Michou L, Barbet S, Tan A, Lasbleiz S, Bardin T, Prum B, Cornelis F: IRF5 rs2004640-T allele, the new genetic factor for systemic lupus erythematosus, is not associated with rheumatoid arthritis. Ann Rheum Dis. 2007, 66: 828-831. 10.1136/ard.2006.061390.
Rueda B, Reddy MV, González-Gay MA, Balsa A, Pascual-Salcedo D, Petersson IF, Eimon A, Paira S, Scherbarth HR, Pons-Estel BA, González-Escribano MF, Alarcón-Riquelme ME, Martín J: Analysis of IRF5 gene functional polymorphisms in rheumatoid arthritis. Arthritis Rheum. 2006, 54: 3815-3819. 10.1002/art.22271.
van Oosterhout M, Bajema I, Levarht EW, Toes RE, Huizinga TW, van Laar JM: Differences in synovial tissue infiltrates between anti-cyclic citrullinated peptide-positive rheumatoid arthritis and anti-cyclic citrullinated peptide-negative rheumatoid arthritis. Arthritis Rheum. 2007, 58: 53-60. 10.1002/art.23148.
van der Helm-van Mil AH, Kern M, Gregersen PK, Huizinga TW: Variation in radiologic joint destruction in rheumatoid arthritis differs between monozygotic and dizygotic twins and pairs of unrelated patients. Arthritis Rheum. 2006, 54: 2028-2030. 10.1002/art.21872.
van der Helm-van Mil AH, Huizinga TW, Schreuder GM, Breed-veld FC, de Vries RR, Toes RE: An independent role of protective HLA class II alleles in rheumatoid arthritis severity and susceptibility. Arthritis Rheum. 2005, 52: 2637-2644. 10.1002/art.21272.
Marinou I, Healy J, Mewar D, Moore DJ, Dickson MC, Binks MH, Montgomery DS, Walters K, Wilson AG: Association of interleukin-6 and interleukin-10 genotypes with radiographic damage in rheumatoid arthritis is dependent on autoantibody status. Arthritis Rheum. 2007, 56: 2549-2556. 10.1002/art.22814.
Prots I, Skapenko A, Wendler J, Mattyasovszky S, Yoné CL, Spriewald B, Burkhardt H, Rau R, Kalden JR, Lipsky PE, Schulze-Koops H: Association of the IL4R single-nucleotide polymorphism I50V with rapidly erosive rheumatoid arthritis. Arthritis Rheum. 2006, 54: 1491-1500. 10.1002/art.21832.
Lard LR, van Gaalen FA, Schonkeren JJ, Pieterman EJ, Stoeken G, Vos K, Nelissen RG, Westendorp RG, Hoeben RC, Breedveld FC, Toes RE, Huizinga TW: Association of the -2849 interleukin-10 promoter polymorphism with autoantibody production and joint destruction in rheumatoid arthritis. Arthritis Rheum. 2003, 48: 1841-1848. 10.1002/art.11160.
Mattey DL, Nixon NB, Dawes PT, Ollier WE, Hajeer AH: Association of matrix metalloproteinase 3 promoter genotype with disease outcome in rheumatoid arthritis. Genes Immun. 2004, 5: 147-149. 10.1038/sj.gene.6364050.
Dörr S, Lechtenböhmer N, Rau R, Herborn G, Wagner U, Müller-Myhsok B, Hansmann I, Keyszer G: Association of a specific haplotype across the genes MMP1 and MMP3 with radiographic joint destruction in rheumatoid arthritis. Arthritis Res Ther. 2004, 6: R199-R207. 10.1186/ar1164.
Constantin A, Lauwers-Cances V, Navaux F, Abbal M, van Meer-wijk J, Mazieres B, Cambon-Thomsen A, Cantagrel A: Stromelysin 1 (matrix metalloproteinase 3) and HLA-DRB1 gene polymorphisms: association with severity and progression of rheumatoid arthritis in a prospective study. Arthritis Rheum. 2002, 46: 1754-1762. 10.1002/art.10336.
van der Kooij SM, de Vries-Bouwstra JK, Goekoop-Ruiterman YP, vanZeben D, Kerstens PJ, Gerards AH, van Groenendael JH, Hazes JM, Breedveld FC, Allaart CF, Dijkmans BA: Limited efficacy of conventional DMARDs after initial methotrexate failure in patients with recent-onset rheumatoid arthritis treated according to the Disease Activity Score. Ann Rheum Dis. 2007
de Vries-Bouwstra J, Le Cessie S, Allaart C, Breedveld F, Huizinga T: Using predicted disease outcome to provide differentiated treatment of early rheumatoid arthritis. J Rheumatol. 2006, 33: 1747-1753.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
About this article
Cite this article
van der Helm-van Mil, A.H., Huizinga, T.W. Advances in the genetics of rheumatoid arthritis point to subclassification into distinct disease subsets. Arthritis Res Ther 10, 205 (2008). https://doi.org/10.1186/ar2384
Published:
DOI: https://doi.org/10.1186/ar2384