
Abstract
Background
Dietary flavonols may play an important role in the adjunct therapy of chronic inflammation. The availability of therapeutic formulations of pentahydroxyflavone glycoside, rutoside (RU), led us to investigate the ability of this molecule to modulate the release of various proinflammatory mediators from human activated macrophages in vitro and to ameliorate arthritic markers in a rat model.
Methods
RU was added simultaneously to human macrophages during their activation. Cells were then analyzed for inflammation-related gene expression using a specific array, and cell supernatants were collected to measure inflammatory mediators. RU was also injected into adjuvant-induced arthritic rats, and disease progression and body weight were evaluated until 50 days after injection. Sera and peritoneal macrophages were also collected to quantify the RU effect on various inflammatory markers.
Results
RU inhibited inflammation-related gene expression in activated human macrophages and the release of nitric oxide, tumor necrosis factor-alpha, interleukin (IL)-1, and IL-6 from these cells. In a rat model, RU inhibited clinical signs of chronic arthritis, correlating with decreased levels of inflammatory cytokines detected in rat sera and macrophage supernatants.
Conclusion
Thus, RU may have clinical value in reducing inflammatory manifestations in human arthritis and other inflammatory diseases.
Similar content being viewed by others


Introduction
The immune system has evolved to protect the host from microbial infection. Nevertheless, a breakdown in the immune system often results in infection, cancer, and autoimmune diseases. Multiple sclerosis, rheumatoid arthritis (RA), type 1 diabetes, inflammatory bowel disease, myocarditis, thyroiditis, uveitis, systemic lupus erythromatosis, and myasthenia gravis are organ-specific autoimmune diseases that afflict more than 5% of the population worldwide. Although their etiology is not known and a cure is still wanting, promising data raised in human RA implied macrophage mediators in disease progression [1, 2]. Macrophages are the major source of inflammatory mediators during immune response once activated by auto-antibodies or by antigen-specific Th1 cell-derived lymphokines [2, 3]. Though essential for the elimination of invasive antigens, chronic expression of the above mediators can induce a variety of inflammatory disorders, including RA and many other autoimmune diseases [2]. During RA, patients have an increased number of monocytes, particularly inflammatory monocytes, circulating in peripheral blood [4–6] and have an elevated number of macrophages in the joints [5]. These cells are highly activated and are one of the main producers of interleukin (IL)-1β and tumor necrosis factor-alpha (TNF-α), two essential proinflammatory cytokines required for the progression of RA because they are capable of inducing other proinflammatory cytokines and activating matrix metalloproteinases in autocrine and paracrine fashions [2]. Inhibitors of IL-1 and TNF-α cause a reduction in synovial inflammation, bone destruction, and macrophage infiltration in patients with RA [7, 8]. A critical role of TNF-α and IL-1 during RA pathogenesis was confirmed by the recent development of appropriate therapeutic counterstructures [9].
In patients with autoimmune diseases, the use of dietary supplements is on the rise, mainly because they are effective, inexpensive, and relatively safe [10]. Recent studies indicate that two main flavonols, quercetin and its glycosylated form, rutin (or rutoside, RU), attenuate various inflammatory functions of macrophages in human or animal models [11–15]. Flavonols are compounds isolated from various plants that traditionally have been used for pain and vascular protection [11]. Quercetin inhibits inflammatory reactions by regulating the generation of inflammatory cytokines such as IL-6, TNF-α, and interferon-gamma and associated activation protein-1 (AP-1) and nuclear factor-kappa-B (NF-κB) signaling pathways in immune cells in vitro and in vivo [10, 15]. RU has similar in vitro effects on immune cells but differs from quercetin by its higher therapeutic index and the absence of a modulatory effect on the cell cycle and apoptosis [16, 17].
Various RU formulations for systemic use have been commercially available for more than 40 years and are used primarily as treatment for edema related to venous insufficiency [11]. Oral administration of RU attenuated bowel inflammatory syndrome [18] and a variety of other acute and chronic inflammations in murine models [19, 20]. The scavenging property of rutin led to a decrease of oxygen radical overproduction of leukocytes of patients with RA in vitro [21]. Meanwhile, the exact anti-inflammatory mechanism(s) of RU and its cellular target(s) were not elucidated even though a decrease of nitric oxide (NO) and IL-1β production has recently been suggested in mice [19].
This led us to investigate the anti-inflammatory potential of RU on purified human activated macrophages, key effector cells in inflammatory diseases. Macrophage-related inflammatory responses were then analyzed at transcriptomic and proteic levels in vitro in order to clarify the anti-inflammatory effect of RU in human cells. RU was subsequently tested in vivo at preventive or postarthritic levels in a rat model of chronic arthritis. Data point out the inhibitory effect of RU on inflammatory cytokines, corroborating its ability to significantly reduce clinical signs in arthritic rats.
Materials and methods
Reagents
For in vitro experiments, 3,3',4',5,7-pentahydroxyflavone-3-rutinoside trihydrate (RU) (>97% purity powder; Acros Organics, Noisy-le-grand, France) was used after suspension in distilled water. For in vivo subcutaneous (s.c.) injections in rats, RU was suspended in saline.
Human cells
Peripheral blood samples were obtained from healthy volunteers with their informed consent after the approval of this study by the institutional ethics committee. These samples were pretested for the absence of HIV or hepatitis virus infections. Peripheral blood-derived mononuclear leukocytes (PBLs) were obtained by Ficoll gradient separation, and monocytes were subsequently separated from other leukocytes by adherence to CD14 beads (Miltenyi Biotec, Paris, France). CD14- PBLs were used for the hematotoxicity test of various RU preparations. Briefly, cells were incubated in medium alone or with 10-6 M phytohemagglutinin-P (PHA) (5 μg/mL; Murex Biotech Ltd, Dartford, UK) in order to induce lymphocyte proliferation. RU was added at different concentrations, cells were harvested 4 days later, and apoptotic/necrotic versus total cells were counted as indicated below. CD14+ cells were then suspended in RPMI 1640 medium supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM glutamine, and 10% fetal calf serum (FCS) (all from Gibco-Europe, Paisley, Scotland). The above culture medium, chemicals, and FCS were endotoxin-free and tested for the absence of direct activation effects on human monocytes (CD23 expression and TNF-α production as activation markers). After these procedures, more than 95% of cells expressed CD14 antigen and displayed cytochemical characteristics of monocytes/macrophages [22].
Experimental arthritis and rats
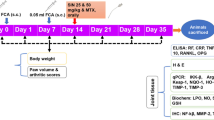
Female Lewis rats (Janvier, Le Genest St Isle, France) were housed under standard laboratory conditions with free access to food and water. The temperature was kept at 22°C ± 2°C, and a 12-hour light/dark schedule was maintained. The Animal Research Committee of the Agriculture Ministry approved this investigation. All animal procedures were performed in strict accordance with the guidelines issued by the European Economic Community (directive 86/609). Adjuvant-induced arthritis (AIA) was obtained in 6-week-old animals by s.c. injection at the base of the tail of 300 μL (1.8 mg) of inactivated Mycobacterium butyricum (Difco Laboratories Inc., now part of Becton Dickinson and Company, Franklin Lakes, NJ, USA) diluted in an emulsion of 8 mL of Vaseline oil, 1 mL of polysorbate 80, and 1 mL of phosphate-buffered saline (PBS) (Laboratoires Eurobio, Courtaboeuf, France). Rats were boosted 1 week later with the same dose of antigen and observed for up to 50 days after immunization for clinical signs of chronic arthritis. Evaluation of AIA severity was performed by two independent observers with no knowledge of the treatment protocol. The severity of AIA in each paw was quantified daily by an arbitrary clinical score measurement from 0 to 2 as follows: no signs of inflammation (0), swelling alone (0.5) for each paw, immobility (0.5) for each paw, 2 being the highest score with both paws swelling and immobile. Weight evolution of the animals was measured daily. Rats were treated with five injections (every 2 days) of RU, saline, or hydrocortisone (HC) (Sigma-Aldrich, Saint Quentin Fallavier, France) as indicated in the Results section. Injections were initiated 1 day after the appearance of the first arthritic symptoms (therapeutic) or on the first day of immunization (preventive). For macrophage collection, animals were anaesthetized with ether and the peritoneal cavity was washed with 10 mL of cold PBS (pH 7.4). After centrifugation at 300 g for 10 minutes at 4°C, cells were collected, counted, and adjusted to 106 cells per milliliter with culture medium. After 96-hour incubation in medium alone, cell supernatants from disease-free and AIA rats were collected for mediator quantification. To activate the production of various inflammatory mediators from rat peritoneal macrophages, cells were incubated with lipopolysaccharide (10 μg/mL; Sigma-Aldrich). RU was added simultaneously to cell activation. Cells or their supernatants were then tested for the presence of various inflammatory mediators.
Human cell activation
Monocytes/macrophages were activated through the physiological CD23 pathway [22]. PBL-derived adherent cells must be preactivated to express surface CD23 as described [22] following their incubation with various cytokines, including recombinant human IL-4 (50 ng/mL), during 24 hours at 37°C. After washing, cells were tested for their surface CD23 expression (>80% CD23+) and then incubated in the presence of crosslinking CD23-MAb (clones 135, IgG1k, 20 μg/mL) during 48 to 96 hours or of IgE and anti-IgE as described [22, 23]. Cells may then be analyzed for their NOx (nitrite/nitrate) content (see below) and cell supernatants for the presence of various inflammatory mediators. To detect cell apoptosis, externalization of inner membrane phosphatidylserine and DNA content was investigated by flow cytometry using a fluorescein-conjugated annexin V and propidium iodide kit (Immunotech, Marseille, France).
RNA preparations and transcriptomic arrays
After cultures, total RNA was extracted using Trizol (Invitrogen, Cergy Pontoise, France) and was subsequently purified on RNeasy columns (Qiagen, Hilden, Germany). Synthesis of biotin-labelled cRNA, purification, and hybridization (6 μg) to custom array membranes were performed according to the manufacturer's recommendations (OHS-011; SuperArray Bioscience Corporation, Frederick, MD, USA). For detailed gene content and housekeeping controls, see reference [24]. After local background subtraction, average signal intensity from duplicate spots was compared with values obtained for housekeeping genes using Alpha Imager HP automatic image capture software (Alpha-Innotec, San Leandro, CA, USA). For each gene, modulation was defined as the relative expression value for stimulated versus control sample.
Quantification of inflammatory mediators
For intracellular NO measurements, cells (>105) were incubated with 10 μM DAF-FM-DA (4-amino-5-methylamino-2',7'-difluoro-fluorescein diacetate; Molecular Probes, now part of Invitrogen) for 1 hour at 37°C in 1 mL of culture medium. Cells were then washed in PBS and incubated in 0.5 mL of PBS for 30 minutes at 37°C. Intracellular NO content was then investigated by flow cytometry. Cell supernatants (48 to 72 hours) were assayed for the stable end product of NO, NO2- using the Griess reaction modified as detailed elsewhere [25]. The inhibitory analog of L-arginine, N(6)-(1-iminoethyl)-L-lysine/2HCl (L-NIL) (Coger SA, Paris, France) [26], was used to inhibit inducible nitric oxide synthase (iNOS)-mediated NO generation at a concentration of 1 mM. To detect human cytokine levels, we have used a human multi-assay Th1/Th2 II plex kit (Bender MedSystems, Vienna, Austria) and flow cytometry. Inflammatory mediators in rat sera or cell supernatants were quantified using appropriate enzyme-linked immunosorbent assay kits in accordance with the manufacturer's recommendations for prostaglandin E2 (PGE2) (R&D Systems Europe, Lille, France), monocyte chemoattractant protein-1 (MCP-1) (Tebu, Le Perray-en Yveline, France), TNF-α, and IL-1β (Biosource, Montrouge, France).
Statistical analysis
Comparisons were assessed using the Fisher exact test for proportions and the Mann-Whitney U test for quantitative values. A p value of less than 0.05 was considered significant. For some rat in vitro experiments, results were analyzed and compared using the Student t test for paired data.
Results
Inhibition of gene transcription in human macrophage by rutoside
In vitro, RU has generally been shown to display its activities at concentrations of 30 to 200 μM [19]. Prior to RU use, we first tested its effect on human normal or PHA-mediated cycling PBLs in vitro. We had no significant increase of the number of apoptotic (annexin+) or necrotic (propidium iodide+) cells after 96 hours of cell incubation in the presence of 1 to 200 μM RU (range from -5% to +7% of cells). RU was then used at concentrations of less than or equal to 100 μM in the following experiments. After their separation, human monocyte-derived macrophages were activated in the presence of 100 μM RU. The transcription of inflammatory genes was then analyzed using a macrophage-specific macroarray. Compared with resting cells, activated macrophages acquire a significant expression of 20 new mRNAs encoding various inflammatory mediators, chemotactic factors, and their receptors (Figure 1). In addition, we observed a significant increase in mRNA expression for genes encoding IL-1β, IL-8, TNF-α, TNF-R1, and macrophage migration inhibitory factor (MIF) (>110%; p < 0.05), known for their critical role during inflammatory response [2, 27]. Treatment of macrophages with RU inhibited the expression of most of the above genes (19/20 were totally suppressed), including IL-1β, TNF-α, TNF-R1, and many chemoattracting factors, or decreased the expression of genes such as MIF and IL-8 (p < 0.05) (Figure 1). Surprisingly, the transcription of the gene encoding IL-10, known as Th2 type cytokine, was significantly increased (>230%; p < 0.05). These findings indicate that RU is a potent inhibitor of the transcription of proinflammatory genes in human macrophages with the exception of that encoding IL-10.

Rutoside (RU) mediates the inhibition of inflammatory gene transcription in activated human macrophages. Human monocyte-derived macrophages were activated (10 μg/mL CD23-McAb) alone or in the presence of 100 μM RU. After 24 hours of incubation, cellular mRNAs were extracted and analyzed by inflammation-specific macroarray. (a) RU inhibits gene expression by activated human macrophages, with the exception of interleukin (IL)-10. A representative array from two distinct experiments is shown. (b) Inflammatory genes detected on each membrane, in addition to controls. MIF, macrophage migration inhibitory factor; TNF, tumor necrosis factor.
Rutoside modulates the generation of inflammatory mediators from activated macrophages
Transcriptomic data led us to investigate the effect of RU on cytokine production at the protein level. Quantification of various cytokines in macrophage cell supernatants indicates that the addition of RU to human activated macrophages significantly (p < 0.02) decreased the concentrations of TNF-α, IL-1β, and IL-6 (Figure 2a), three critical proinflammatory cytokines. We failed to detect significant modulation of IL-10 levels in contrast to mRNA data (Figures 1b and 2a).

Inhibition of inflammatory mediator generation from activated human macrophages. (a) Rutoside (RU) decreased the generation of tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1β, and IL-6 but not IL-10 from activated human macrophages. (b) Inhibition of nitric oxide (NO) generation from human activated macrophages after their incubation with various RU concentrations. RU decreases both intracellular NO (upper panel) and extracellular nitrites (lower panel). Specific inducible nitric oxide synthase inhibitor (L-NIL, 1 mM) was used as control. Results from three different cell preparations ± standard deviation are shown. *P value obtained as compared to activated cells. L-NIL, N(6)-(1-iminoethyl)-L-lysine/2HCl.
In addition to the above cytokines, inflammatory macrophages produce various non-proteic mediators, including iNOS-dependent NO [22]. Upon their activation, human macrophages produce NO, detected at the intracellular level by specific probe (DF-FM) and at the extracellular level through the quantification of nitrites, the final metabolites of NO in cell supernatants. Data in Figure 2b show that RU partly inhibited NO generation in a dose-dependent manner at both the intracellular and extracellular levels. Generation of NO from human macrophages was also reversed by L-NIL (Figure 2b), a specific inhibitor of iNOS [26], suggesting the ability of RU to reverse iNOS-mediated NO production.
Rutoside decreases and prevents arthritic signs in rats
In vitro observations in human cells led us to investigate the therapeutic anti-inflammatory effect of RU in vivo in rat AIA, an experimental model with many clinical and histopathological features of chronic human RA [28]. RU formulations and doses used in this work were based upon various in vivo studies [29] and our preliminary analysis showing the absence of an apparent toxic effect of up to 3,000 mg/kg total doses (data not shown). AIA Lewis rats thus were treated with RU suspension at 133 mg/kg s.c. doses (one dose every 2 days × 5). As therapeutic positive control, rats were treated with HC at 150 mg/kg total dose [30]. We found that administering five doses of RU, beginning the day after the appearance of the first arthritic symptoms, significantly improved the clinical course, including arthritic scores and weight progression of AIA rats as compared with the control groups (Figure 3a; p < 0.0001). The effect of RU was superior to that of HC in inhibiting arthritic scores (p < 0.001) and remained stable after treatment (Figure 3a). Figure 3b shows the external aspect of inflamed pads in RU-treated rats compared with untreated rats. No obvious toxicity was observed resulting from the treatment in the rats (for example, the weight of the treated normal rats was close to untreated controls). Hence, these experiments indicate that the RU effectively ameliorates AIA.

The evolution of arthritis severity scores of adjuvant-induced arthritis (AIA) in rats following their treatment with rutoside (RU). (a) Arthritis clinical scores (upper panel) and body weight (lower panel) of young AIA rats following their treatment with 133 mg/kg subcutaneous doses of RU suspension, every 2 days × 5, as indicated by arrows starting after the appearance of clinical symptoms. As positive control, AIA rats received five injections of 30 mg/kg hydrocortisone (HC). Results are means from five rats from each group (standard deviation [SD] less than 25% for all groups). (b) External aspects of rat paws showing swelling (left), swelling + immobility (central), and healed paws following RU treatment (right). (c) Amelioration of arthritis severity scores (upper panel) and body weight (lower panel) of AIA rats following preventive treatment with 5 × 133 mg/kg doses of RU suspension, starting the first day of immunization, before AIA development. Results are means from five rats (SD less than 25% for all groups). (d) Cumulative AIA clinical scores from untreated rats, hydrocortisone (HC)-treated rats, or those treated by RU prior to AIA development (Pre-AIA) or following arthritis development (Post-AIA). Bars represents cumulative AIA severity scores over the course of 40 days of observation (days 0 to 40) of five rats from each group. *P value compared to untreated AIA rats.
In a second set of experiments, we tested the ability of RU to prevent AIA establishment in rats. We injected RU every 2 days for a total of five injections, beginning on the day of adjuvant injection to the rats and before the appearance of any arthritic signs. Notably, we found that pretreatment with RU could significantly reduce the severity of arthritis and growth delay observed in control groups (Figure 3c; p < 0.0002). Hence, as shown in Figure 3d, RU treatment was effective either at disease onset or prevention of severe disease establishment, whereas no such activity was obtained with controls.
Rutoside and rat macrophage inflammatory mediators ex vivo
To confirm the anti-inflammatory effects of RU and the role of macrophages, we tested the levels of selected mediators in the sera from AIA animals following their treatment with or without RU. Data (Figure 4a) show that RU significantly reduced the levels of TNF-α, IL-1β, and MCP-1 in animal sera (p < 0.02), whereas the level of PGE2, an indirect marker of COX-2 (cyclooxygenase-2) activity, was not affected by this treatment. These data reinforced human findings as they revealed that RU decreased the level of inflammatory cytokines, critical for RA pathogenesis in vivo. Although known as a major source of the above mediators [2], the role of macrophages during RU treatment required more direct ex vivo analysis. Peritoneal macrophages were then isolated from RU-treated or untreated arthritic rats and promptly incubated in medium alone. After 48 hours of incubation, cell supernatants were analyzed for the levels of TNF-α and nitrites as inflammatory markers. Our results (Figure 4b) clearly show that increased inflammatory macrophage-derived TNF-α and NO in AIA rats were attenuated after treatment with RU.

Effect of rutoside (RU) treatment on rat inflammatory mediators. (a) Sera were collected at day 50 after immunization and tested for their concentrations in tumor necrosis factor-alpha (TNF-α), prostaglandin E2 (PGE2), interleukin-1-beta (IL-1β), and monocyte chemoattractant protein-1 (MCP-1). Results are shown as mean percentage of modulation of cytokine levels compared to healthy rats. Mean percentage + standard deviation from three rats in each group is shown. P value compared to untreated adjuvant-induced arthritis (AIA) rats. (b) Freshly isolated peritoneal macrophages from untreated or RU-treated rats were collected on day 50 after immunization. They were incubated in medium alone during 48 hours, and their supernatants were collected and tested for their TNF-α or nitrite levels. P value compared to untreated AIA rat cells. (c) Macrophages from normal rats were incubated in medium alone or activated by lipopolysaccharide. RU was added (50 μM) simultaneously to cell activation. Cell supernatants were harvested 48 hours after incubation and tested for their concentrations of TNF-α, PGE2, IL-1β, and MCP-1. P value compared to activated cells. (d) RU inhibits the release of nitric oxide from activated rat macrophages in a dose-dependent manner. Bars show mean + standard deviation from three different rat cell preparations. P value compared to activated cells.
Finally, we tested the ability of RU to reduce rat macrophage inflammatory responses in vitro. Peritoneal macrophages were isolated from healthy rats and activated without various doses of RU. Our data show (Figure 4d) that the levels of NO, as indicated by the concentration of nitrites in cell supernatants, decreased after the addition of RU in a dose-dependent manner (p < 0.006). As in human cells, L-NIL had a similar inhibitory effect compared with RU, suggesting its ability to downregulate iNOS-mediated NO generation in macrophages. Inhibition reached a plateau at 50 μM RU, a concentration that was subsequently used to investigate the RU effect on other mediators. As in in vivo findings, RU decreased the level of inflammatory cytokines (TNF-α, IL-1β, and MCP-1) generated from activated rat macrophages (Figure 4c). As in ex vivo data, the production of PGE2 was not modified after the addition of similar RU dilution.
Discussion
Macrophages arise as an interesting target for modulation of inflammatory disease. Inflammatory mediators derived from these cells have a critical role during synovial inflammation and bone destruction in some patients with RA [7, 8]. Obtained using various approaches, our results clearly indicate that RU, a molecule already used in vascular diseases, inhibited the activation of human macrophages and the subsequent secretion of proinflammatory mediators from these cells. RU was shown to inhibit the transcription of more than 20 genes encoding critical proinflammatory factors, including TNF-α, IL-1, IL-8, TNF-α, MIF, and chemoattracting factors. This effect was confirmed by decreased concentrations of IL-1β, TNF-α, and IL-6 observed in cell supernatants.
NO has been identified as another proinflammatory mediator in human arthritis and experimental animal studies [3, 31]. Increased concentrations of nitrites, stable metabolites of NO, have been observed in the serum and the synovial fluid of patients with RA and osteoarthritis [3, 32]. Increased iNOS activity and NO production have also been detected in the blood mononuclear cells of patients with RA and correlated with the tender and swollen joint counts [33]. Here, we show that RU decreased the production of iNOS-mediated NO by human macrophages in a dose-dependent manner. Of particular interest, and by contrast to the above mediators, RU induced a slight but significant increase of IL-10 mRNA. However, we failed to detect an increase of IL-10 protein level. This may be due to the absorption of IL-10 by macrophages, as yet observed in CD23-activated human epithelial cells [34]. This Th2 type cytokine is well known as a downregulator of the above-mentioned Th1 mediators in human macrophages [35]. These preliminary data suggest that RU preferentially lowers Th1-like cytokine generation from human macrophages.
To support the therapeutic interest of RU, we investigated its effects in a rat model of adjuvant-induced chronic arthritis, well known to mimic Th1 type pathogenic signs of RA [28]. RU significantly reversed growth delay and severe AIA development in rats with persistent partial or complete long-term recovery, not observed in rats treated with HC. These data confirm early observations in experimental acute inflammation models [18–20, 36] and further revealed the preventive property of RU in arthritic rats. Ex vivo analysis of rat sera and macrophages confirmed RU-mediated inhibition of critical proarthritic factors such as TNF-α, IL-1, MCP-1, and NO in vivo. This property correlated with RU-mediated inhibition of murine macrophage activation and inflammatory mediator release in vitro (Figures 3 and 4). In contrast to quercetin [37], RU did not mediate the inhibition of the PGE2 pathway.
Together, the above data provide new insight into the possible mechanism of anti-inflammatory effects of RU in macrophages. Human and murine analyses of cytokine expression clearly show that RU suppresses the major inflammatory and proarthritic mediators of macrophages. The ability of RU to decrease MCP-1 levels in vivo and in vitro may add to its beneficial effects because this cytokine is a potent stimulator of monocyte recruitment into the site of inflammation [38]. We have previously shown that the nonglycosylated derivative of RU, quercetin, inhibits the production of TNF-α and NO from activated human macrophages [39]. These flavonols inhibit the phosphorylation and activation of Jun N-terminal kinase/stress-activated protein kinase, leading to the suppression of AP-1 activation. They also decrease the activation of NF-κB in both human and experimental models [12, 40]. These observations may explain the anti-inflammatory property of RU because both nuclear factors are necessary for the generation of most inflammatory mediators analyzed in the present study [41–43].
However, the exact mechanism underlying the improvement in the arthritis model requires more experimental clarifications. Despite the direct relationship between arthritis signs and macrophage inflammatory markers in the AIA rat model, we must not exclude the simultaneous effect of RU on other inflammatory partners (such as lymphocytes) that may directly or indirectly reduce arthritis manifestations. In addition, AIA is a good model for Th1-mediated and macrophage inflammatory response but is a poor model for Th2-mediated immune reactions. Further analysis of RU activities in the presence of various human lymphocyte populations is required to clarify these points.
Finally, in vivo use of quercetin as medicine suffers from the lack of approved formulation despite a first preliminary assay as adjunct treatment of prostatitis/chronic pelvic pain in humans [44]. In contrast, formulations containing either RU or its derivatives are currently used in the treatment and prevention of venous circulation disorders [11, 45].
Conclusion
RU appears as an interesting cost-effective therapeutic tool in inflammatory diseases and represents an alternative to immunosuppressor agents well known for their multiple side effects.
Abbreviations
- AIA:
-
= adjuvant-induced arthritis
- AP-1:
-
= activation protein-1
- FCS:
-
= fetal calf serum
- HC:
-
= hydrocortisone
- IL:
-
= interleukin
- iNOS:
-
= inducible nitric oxide synthase
- L-NIL:
-
= N(6)-(1-iminoethyl)-L-lysine/2HCl
- MCP-1:
-
= monocyte chemoattractant protein-1
- MIF:
-
= macrophage migration inhibitory factor
- NF-κB:
-
= nuclear factor-kappa-B
- NO:
-
= nitric oxide
- PBL:
-
= peripheral blood-derived mononuclear leukocyte
- PBS:
-
= phosphate-buffered saline
- PGE2:
-
= prostaglandin E2
- PHA:
-
= phytohemagglutinin-P
- RA:
-
= rheumatoid arthritis
- RU:
-
= rutoside
- s.c.:
-
= subcutaneous
- TNF-α:
-
= tumor necrosis factor-alpha.
References
Feldmann M, Maini RN: Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned?. Annu Rev Immunol. 2001, 19: 163-196. 10.1146/annurev.immunol.19.1.163.
Ma Y, Pope RM: The role of macrophages in rheumatoid arthritis. Curr Pharm Des. 2005, 11: 569-580. 10.2174/1381612053381927.
Farrell AJ, Blake DR, Palmer RM, Moncada S: Increased concentrations of nitrite in synovial fluid and serum samples suggest increased nitric oxide synthesis in rheumatic diseases. Ann Rheum Dis. 1992, 51: 1219-1222.
Iwahashi M, Yamamura M, Aita T, Okamoto A, Ueno A, Ogawa N, Akashi S, Miyake K, Godowski PJ, Makino H: Expression of Toll-like receptor 2 on CD16+ blood monocytes and synovial tissue macrophages in rheumatoid arthritis. Arthritis Rheum. 2004, 50: 1457-1467. 10.1002/art.20219.
Kawanaka N, Yamamura M, Aita T, Morita Y, Okamoto A, Kawashima M, Iwahashi M, Ueno A, Ohmoto Y, Makino H: CD14+, CD16+ blood monocytes and joint inflammation in rheumatoid arthritis. Arthritis Rheum. 2002, 46: 2578-2586. 10.1002/art.10545.
Mulherin D, Fitzgerald O, Bresnihan B: Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheum. 1996, 39: 115-124. 10.1002/art.1780390116.
Nuki G, Bresnihan B, Bear MB, McCabe D: Long-term safety and maintenance of clinical improvement following treatment with anakinra (recombinant human interleukin-1 receptor antagonist) in patients with rheumatoid arthritis: extension phase of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002, 46: 2838-2846. 10.1002/art.10578.
De Rycke L, Baeten D, Foell D, Kruithof E, Veys EM, Roth J, De Keyser F: Differential expression and response to anti-TNFalpha treatment of infiltrating versus resident tissue macrophage subsets in autoimmune arthritis. J Pathol. 2005, 206: 17-27. 10.1002/path.1758.
Olsen NJ, Stein CM: New drugs for rheumatoid arthritis. N Engl J Med. 2004, 350: 2167-2179. 10.1056/NEJMra032906.
Kumazawa Y, Kawaguchi K, Takimoto H: Immunomodulating effects of flavonoids on acute and chronic inflammatory responses caused by tumor necrosis factor alpha. Curr Pharm Des. 2006, 12: 4271-4279. 10.2174/138161206778743565.
Wadworth AN, Faulds D: Hydroxyethylrutosides. A review of its pharmacology, and therapeutic efficacy in venous insufficiency and related disorders. Drugs. 1992, 44: 1013-1032. 10.2165/00003495-199244060-00009.
Comalada M, Camuesco D, Sierra S, Ballester I, Xaus J, Galvez J, Zarzuelo A: In vivo quercitrin anti-inflammatory effect involves release of quercetin, which inhibits inflammation through down-regulation of the NF-kappaB pathway. Eur J Immunol. 2005, 35: 584-592. 10.1002/eji.200425778.
Mamani-Matsuda M, Rambert J, Malvy D, Lejoly-Boisseau H, Daulouede S, Thiolat D, Coves S, Courtois P, Vincendeau P, Mossalayi MD: Quercetin induces apoptosis of Trypanosoma brucei gambiense and decreases the proinflammatory response of human macrophages. Antimicrob Agents Chemother. 2004, 48: 924-929. 10.1128/AAC.48.3.924-929.2004.
Khanna D, Sethi G, Ahn KS, Pandey MK, Kunnumakkara AB, Sung B, Aggarwal A, Aggarwal BB: Natural products as a gold mine for arthritis treatment. Curr Opin Pharmacol. 2007, 7: 344-351. 10.1016/j.coph.2007.03.002.
Chen CC, Chow MP, Huang WC, Lin YC, Chang YJ: Flavonoids inhibit tumor necrosis factor-alpha-induced up-regulation of intercellular adhesion molecule-1 (ICAM-1) in respiratory epithelial cells through activator protein-1 and nuclear factor-kappaB: structure-activity relationships. Mol Pharmacol. 2004, 66: 683-693. 10.1124/mol.104.001206.
Volate SR, Davenport DM, Muga SJ, Wargovich MJ: Modulation of aberrant crypt foci and apoptosis by dietary herbal supplements (quercetin, curcumin, silymarin, ginseng and rutin). Carcinogenesis. 2005, 26: 1450-1456. 10.1093/carcin/bgi089.
Zhou SF, Xue CC, Yu XQ, Wang G: Metabolic activation of herbal and dietary constituents and its clinical and toxicological implications: an update. Curr Drug Metab. 2007, 8: 526-553. 10.2174/138920007781368863.
Cruz T, Galvez J, Ocete MA, Crespo ME, Sanchez de Medina LHF, Zarzuelo A: Oral administration of rutoside can ameliorate inflammatory bowel disease in rats. Life Sci. 1998, 62: 687-695. 10.1016/S0024-3205(97)01164-8.
Kwon KH, Murakami A, Tanaka T, Ohigashi H: Dietary rutin, but not its aglycone quercetin, ameliorates dextran sulfate sodium-induced experimental colitis in mice: attenuation of pro-inflammatory gene expression. Biochem Pharmacol. 2005, 69: 395-406. 10.1016/j.bcp.2004.10.015.
Rotelli AE, Guardia T, Juarez AO, de la Rocha NE, Pelzer LE: Comparative study of flavonoids in experimental models of inflammation. Pharmacol Res. 2003, 48: 601-606. 10.1016/S1043-6618(03)00225-1.
Ostrakhovitch EA, Afanas'ev IB: Oxidative stress in rheumatoid arthritis leukocytes: suppression by rutin and other antioxidants and chelators. Biochem Pharmacol. 2001, 62: 743-746. 10.1016/S0006-2952(01)00707-9.
Vouldoukis I, Riveros-Moreno V, Dugas B, Ouaaz F, Becherel P, Debre P, Moncada S, Mossalayi MD: The killing of Leishmania major by human macrophages is mediated by nitric oxide induced after ligation of the Fc epsilon RII/CD23 surface antigen. Proc Natl Acad Sci USA. 1995, 92: 7804-7808. 10.1073/pnas.92.17.7804.
Bécherel PA, Mossalayi MD, Ouaaz F, Le Goff L, Dugas B, Paul-Eugène N, Frances C, Chosidow O, Kilchherr E, Guillosson JJ, Debré P, Aock M: Involvement of cyclic AMP and nitric oxide in immunoglobulin E-dependent activation of Fc epsilon RII/CD23+ normal human keratinocytes. J Clin Invest. 1994, 93: 2275-2279. 10.1172/JCI117227.
SuperArray Bioscience Corporation homepage. [http://www.superarray.com]
Kolb JP, Paul-Eugene N, Damais C, Yamaoka K, Drapier JC, Dugas B: Interleukin-4 stimulates cGMP production by IFN-gamma-activated human monocytes. Involvement of the nitric oxide synthase pathway. J Biol Chem. 1994, 269: 9811-9816.
Stenger S, Donhauser N, Thuring H, Rollinghoff M, Bogdan C: Reactivation of latent leishmaniasis by inhibition of inducible nitric oxide synthase. J Exp Med. 1996, 183: 1501-1514. 10.1084/jem.183.4.1501.
Feldmann M, Steinman L: Design of effective immunotherapy for human autoimmunity. Nature. 2005, 435: 612-619. 10.1038/nature03727.
Koga T, Pearson CM: Immunogenicity and arthritogenicity in the rat of an antigen from Mycobacterium tuberculosis wax D. J Immunol. 1973, 111: 599-608.
Willems AM, Bruynzeel AM, Kedde MA, van Groeningen CJ, Bast A, van der Vijgh WJ: A phase I study of monohydroxyethylrutoside in healthy volunteers. Cancer Chemother Pharmacol. 2006, 57: 678-684. 10.1007/s00280-005-0083-7.
Mazzon E, Serraino I, Li JH, Dugo L, Caputi AP, Zhang J, Cuzzocrea S: GPI a poly (ADP-ribose) polymerase inhibitor, exhibits an anti-inflammatory effect in rat models of inflammation. Eur J Pharmacol. 6150, 415: 85-94. 10.1016/S0014-2999(01)00809-3.
Miyasaka N, Hirata Y: Nitric oxide and inflammatory arthritides. Life Sci. 1997, 61: 2073-2081. 10.1016/S0024-3205(97)00585-7.
Ueki Y, Miyake S, Tominaga Y, Eguchi K: Increased nitric oxide levels in patients with rheumatoid arthritis. J Rheumatol. 1996, 23: 230-236.
St Clair EW, Wilkinson WE, Lang T, Sanders L, Misukonis MA, Gilkeson GS, Pisetsky DS, Granger DI, Weinberg JB: Increased expression of blood mononuclear cell nitric oxide synthase type 2 in rheumatoid arthritis patients. J Exp Med. 1996, 184: 1173-1178. 10.1084/jem.184.3.1173.
Becherel PA, LeGoff L, Frances C, Chosidow O, Guillosson JJ, Debre P, Mossalayi MD, Arock M: Induction of IL-10 synthesis by human keratinocytes through CD23 ligation: a cyclic adenosine 3',5'-monophosphate-dependent mechanism. J Immunol. 1997, 159: 5761-5765.
Miagkov AV, Varley AW, Munford RS, Makarov SS: Endogenous regulation of a therapeutic transgene restores homeostasis in arthritic joints. J Clin Invest. 2002, 109: 1223-1229. 10.1172/JCI200214536.
Shen SC, Lee WR, Lin HY, Huang HC, Ko CH, Yang LL, Chen YC: In vitro and in vivo inhibitory activities of rutin, wogonin, and quercetin on lipopolysaccharide-induced nitric oxide and prostaglandin E(2) production. Eur J Pharmacol. 2002, 446: 187-194. 10.1016/S0014-2999(02)01792-2.
O'Leary KA, de Pascual-Tereasa S, Needs PW, Bao YP, O'Brien NM, Williamson G: Effect of flavonoids and vitamin E on cyclooxygenase-2 (COX-2) transcription. Mutat Res. 2004, 551: 245-254.
Rot A, von Andrian UH: Chemokines in innate and adaptive host defense: basic chemokinese grammar for immune cells. Annu Rev Immunol. 2004, 22: 891-928. 10.1146/annurev.immunol.22.012703.104543.
Mamani-Matsuda M, Kauss T, Al-Kharrat A, Rambert J, Fawaz F, Thiolat D, Moynet D, Coves S, Malvy D, Mossalayi MD: Therapeutic and preventive properties of quercetin in experimental arthritis correlate with decreased macrophage inflammatory mediators. Biochem Pharmacol. 2006, 72: 1304-1310. 10.1016/j.bcp.2006.08.001.
Wadsworth TL, McDonald TL, Koop DR: Effects of Ginkgo biloba extract (EGb 761) and quercetin on lipopolysaccharide-induced signaling pathways involved in the release of tumor necrosis factor-alpha. Biochem Pharmacol. 2001, 62: 963-974. 10.1016/S0006-2952(01)00734-1.
Stancovski I, Baltimore D: NF-kappaB activation: the I kappaB kinase revealed?. Cell. 1997, 91: 299-302. 10.1016/S0092-8674(00)80413-4.
Garg AK, Aggarwal BB: Reactive oxygen intermediates in TNF signaling. Mol Immunol. 2002, 39: 509-517. 10.1016/S0161-5890(02)00207-9.
Kleinert H, Pautz A, Linker K, Schwarz PM: Regulation of the expression of inducible nitric oxide synthase. Eur J Pharmacol. 2004, 500: 255-266. 10.1016/j.ejphar.2004.07.030.
Shoskes DA, Zeitlin SI, Shahed A, Rajfer J: Quercetin in men with category III chronic prostatitis: a preliminary prospective, double-blind, placebo-controlled trial. Urology. 1999, 54: 960-963. 10.1016/S0090-4295(99)00358-1.
MacLennan WJ, Wilson J, Rattenhuber V, Dikland WJ, Vanderdonckt J, Moriau M: Hydroxyethylrutosides in elderly patients with chronic venous insufficiency: its efficacy and tolerability. Gerontology. 1994, 40: 45-52.
Acknowledgements
This work was supported by the Conseil Régional d'Aquitaine and Oseo-ANVAR. JR was a fellow from the Association de recherche sur la polyarthrite.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
TK contributed to the acquisition of data in human and murine cells and in vivo. DM contributed to the conception and design of the study and to manuscript drafting. JR and AA-K contributed to the acquisition of data in human cells. SB and DT contributed to the acquisition of data in vivo. RE contributed to the analysis and interpretation of data. FF contributed to toxicological analysis and interpretation of data. MDM contributed to the conception and design of the study, analysis and interpretation of data, and manuscript drafting. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Kauss, T., Moynet, D., Rambert, J. et al. Rutoside decreases human macrophage-derived inflammatory mediators and improves clinical signs in adjuvant-induced arthritis. Arthritis Res Ther 10, R19 (2008). https://doi.org/10.1186/ar2372
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar2372