
Abstract
Dendritic cells are the major antigen-presenting and antigen-priming cells of the immune system. We review the antigen-presenting and proinflammatory roles played by dendritic cells in the initiation of rheumatoid arthritis (RA) and atherosclerosis, which complicates RA. Various signals that promote the activation of NF-κB and the secretion of TNF and IL-1 drive the maturation of dendritic cells to prime self-specific responses, and drive the perpetuation of synovial inflammation. These signals may include genetic factors, infection, cigarette smoking, immunostimulatory DNA and oxidized low-density lipoprotein, with major involvement of autoantibodies. We propose that the pathogenesis of RA and atherosclerosis is intimately linked, with the vascular disease of RA driven by similar and simultaneous triggers to NF-κB.
Similar content being viewed by others
Introduction
Rheumatoid arthritis (RA) is characterized by systemic and synovial tissue chronic inflammation, and by bone and cartilage erosion and destruction [1]. Autoimmune diseases such as RA result from a process involving three distinct but related components – a break in self-tolerance, development of chronic inflammation in one or several organs, and, if ongoing, tissue destruction and its resultant detrimental effects.
Dendritic cells (DC) are essential regulators of both innate and acquired arms of the immune system [2]. Their capacity to prime naïve T lymphocytes for helper and cytotoxic function distinguishes them from other antigen-presenting cells (APC). DC are also essential accessory cells in the generation of primary antibody responses, and are powerful enhancers of natural killer T cells and of natural killer cell cytotoxicity [3]. On the other hand, DC are also involved in the maintenance of tolerance to antigens. Along with the medullary thymic epithelial cells, DC contribute to thymic central tolerance and shaping of the T-cell repertoire by presenting endogenous self-antigens to T cells and deleting those T cells that exhibit strong autoreactivity [4]. In the periphery, resting DC delete autoreactive lymphocytes and expand the population of regulatory T cells. DC therefore have potential use in protective and therapeutic strategies for tolerance restoration in autoimmune diseases (for review see [5]).
Dendritic cells play several roles in RA
DC are likely to contribute in several ways to the pathogenesis of RA. First, it is clear from autoimmune models that DC are able to prime MHC-restricted autoimmune responses in lymphoid organs [6–8]. Through this process, DC orchestrate the development of the autoantibody and chronic inflammatory pathology on which the clinical features of RA are based. Second, DC infiltrate synovial tissue and synovial fluid and here are able to take up, process and present antigen locally, contributing to disease perpetuation [9, 10]. Animal models and histological evidence show that DC drive the generation of ectopic lymphoid tissue in inflammatory environments, probably including the synovium [8, 11]. Furthermore DC, along with synoviocytes and macrophages, produce innate immune inflammatory mediators, and these mediators drive inflammatory pathology in RA [7, 12]. Finally, evidence is accumulating that DC also contribute to the complications of RA, including atherosclerosis.
In the present review we consider each of these activities of DC in RA. In any human systemic condition, evidence for these activities relies on in vitro analysis of patient cells and tissues, and on animal models of RA and other autoimmune diseases. Each of these experimental approaches contributes to our current overall understanding of RA pathogenesis. In the near future, approaches developed to image DC in situ in patients, and to use DC therapeutically, will help to validate in the clinic some of the hypotheses generated over the past 20 years of DC research in RA.
Dendritic cells respond to inflammatory signals to prime T-cell activation
DC precursors originate in bone marrow [13–15]. DC reside in peripheral uninflamed tissues, including synovial tissue in a resting or immature state [16, 17]. Immature DC efficiently capture antigens, including pathogens, particulate, and soluble foreign antigens or self-antigens [18]. After antigen uptake, DC rapidly cross the endothelium of lymphatic vessels and migrate to the draining secondary lymphoid organs, under the influence of CCR7 chemotactic ligands [19]. The uptake of immunogenic antigen or Toll-like receptor (TLR) ligands stimulates differentiation and maturation by DC. This process has been shown to drive a differentiation programme in DC, in which they downregulate their capacity to further capture antigen, but they upregulate antigen processing and presentation, and their expression of costimulatory molecules, secretion of cytokines and responsiveness to chemotactic CCR7 ligands, directing them to lymph nodes [20]. In this paradigm, after reaching secondary lymphoid organs, DC engage with and present antigen to local naïve T cells, disappearing after several days due to apoptosis and active killing by cytotoxic T cells [21]. Depending on the nature of the inflammatory signal received by immature DC, various differentiation programmes may be stimulated. The nature of the resulting T-cell response can be contributed by upstream DC signals, by the subsets of DC that participate in the immune response, and by the recruitment of other cell types that produce mediators such as prostaglandins or histamine (Table 1) [22].
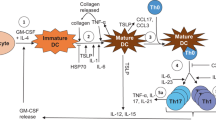
DC are important directors of immune responsiveness, through their interactions with lymphocytes and other accessory cells. Evidence broadly suggests that, under steady-state conditions, recruitment of resting DC precursors into tissues and migration into secondary lymphoid organs occurs constitutively, in the absence of inflammatory events, and may favour tolerance induction [23–25]. On the other hand, inflammation-associated stimulation of DC maturation and activation may initiate T-cell proinflammatory cytokine production, cytotoxic function and B-cell antibody production [26] (Figure 1).

Dendritic cells are important directors of immune responsiveness. (a) Under steady-state conditions, recruitment of resting dendritic cell (DC) precursors into tissues and migration into secondary lymphoid organs occurs constitutively, and may favour tolerance induction. (b) On the other hand, stimulation of DC maturation and activation may initiate T-cell proinflammatory cytokine production, cytotoxic function, and B-cell antibody production.
The DC maturation programme can be stimulated by various mechanisms, including pathogen-derived molecules (lipopolysaccharide, DNA, RNA), proinflammatory cytokines (TNF, IL-1, IL-6), tissue factors such as hyaluronan fragments, heparin sulphate and heat shock proteins, migration of DC across endothelial barriers between inflamed tissues and lymphatics, and T-cell-derived signals (CD154) [27–31]. In contrast, low-affinity T-cell signalling, anti-inflammatory signals, such as IL-10, transforming growth factor beta, prostaglandins and corticosteroids, tend to modify DC maturation and alter the T-cell outcome, deviating the immune response to a Th2-type or regulatory response [32].
NF-κB and p38 mitogen-activated protein kinase represent the two major pathways signalling the DC maturation phenotype [29]. A broad range of stimuli activate NF-κB, notably TLR ligands, including lipopolysaccharide, mycobacterial and yeast products, proinflammatory cytokines such as IL-1, TNF and IL-6, as well as other potentially harmful stimuli such as double-stranded RNA, heparan sulphate and hyaluronan derived from damaged tissues, viral proteins, free radicals, UV and γ-irradiation [33–35]. As a family, NF-κB induces a wide variety of genes, and also affects the function of other transcription factors. Many of the genes that are activated by NF-κB are important for cellular responses to stress, injury and inflammation. Triggers for these states are therefore associated with NF-κB activation [35].
In immune responses, NF-κB target genes are involved in inflammation, cellular organization and differentiation and proliferation. Tissue macrophages are the major source of NF-κB-induced proinflammatory cytokines [36–38]. NF-κB-induced cytokines such as TNF, IL-1 and IL-6 activate innate responses in RA, leading to the release of C-reactive protein (CRP) and complement, and to upregulation of adhesion molecules by endothelial cells (EC). NF-κB-induced chemokines, including IL-8, MIP-1α, MCP-1, RANTES and eotaxin, and growth factors such as granulocyte/macrophage colony-stimulating factor, mobilize and redirect myeloid cells to inflamed tissue [39–43]. A similar set of responses to those that occur in response to infection therefore also occurs in rheumatoid inflammation. NF-κB also plays an important role in lymphoid organogenesis through the induction of the chemokines CXC12, CXCL13, CCL21 and CCL19 [44–48]. Mice lacking the RelB subunit of NF-κB lack peripheral lymph nodes [49].
Two major subsets of DC, known as myeloid DC and plasmacytoid DC, are described in humans. Both subtypes have the capacity for activation, in response to particular TLR or T-cell ligands, with resulting effects on antigen presentation and cytokine production. Major subsets of myeloid DC include those in epithelial tissues, known as Langerhans cells, and those in other tissues, known as interstitial DC. All have the capacity for tolerance as well as potent antigen-presenting function. Plasmacytoid DC represent a distinct population of APC which also produce large amounts of cytokines, including TNF and IFN-α – particularly after stimulation by viruses, double-stranded RNA, CpG DNA motifs and CD154 (Table 1) [50–55].
Genetic and environmental risk factors for RA
HLA-DR gene variation in the major histocompatability locus (MHC) is the strongest gene region associated with RA. A second major association is the tyrosine phosphatase PTPN22 gene, in which a gain-of-function polymorphism reduces the T-cell activation response to antigen. This appears to be a general susceptibility polymorphism for a number of autoimmune diseases, which is hypothesized to reduce the capacity of thymocytes for negative selection towards self-antigen [56]. A weaker association of RA with the MHC class II transactivator gene (MHC2TA) – a protein clearly involved in antigen processing and presentation in the class II pathway – has been reported in several populations, but has not been consistently replicated [57]. Like some of the cytokine gene polymorphisms, one might rather predict this gene to modify RA severity. An association with a functional polymorphism with a gene encoding the peptidyl-arginine deiminase enzyme (PADI4), which catalyses citrullination of arginine, has been identified in Japanese populations [58, 59]. Citrullination is a physiological process of protein alteration occurring during apoptosis and inflammation. Citrullination has been described to occur during macrophage activation, during antigen-specific priming and as a response to smoking [60–62], and it replaces charged imino arginine side-chain groups with uncharged carbonyl groups. The RA HLA association has been mapped to the third hypervariable region of DRβ-chains, especially amino acids 70–74, encoding a conserved amino acid sequence that forms the fourth anchoring pocket (P4) in the HLA groove. This susceptibility sequence, known as the 'shared epitope', is found in multiple RA-associated DR molecules [63]. The shared epitope is positively charged and thus has the capacity to bind proteins or peptides containing a negatively charged or nonpolar amino acid.
Genetic factors contribute about two-thirds of the risk for the development of RA. Evidence for a gene–environment interaction has emerged from twin studies [64]. Significant environmental risk factors include cigarette smoking, parturition and lactation, and mineral oil exposure, and relevant protective factors include use of the oral contraceptive pill and a diet rich in fruit and vegetables [65]. Finally, Epstein–Barr virus exposure and a greater Epstein–Barr viral load is associated with RA. Epstein–Barr virus has immunomodulatory effects, including B-cell activation, and could potentially contribute cross-reactive viral peptides or antibodies [66, 67].
Anticyclic citrullinated peptide (anti-CCP) autoantibodies and rheumatoid factor are more probable in RA patients who smoke [60, 64, 68]. In view of evidence that smoking promotes citrullination of self-proteins, therefore, it has been proposed that smoking promotes anti-CCP in those with at-risk HLA genotypes [60]. Indeed, although the clinical phenotype is similar, anti-CCP-negative, shared epitope-negative RA is likely to be driven by different autoantigens, genetic and environmental factors. More than one subset of RA may constitute this group. In consideration of the multiple mechanisms driving different animal models of autoimmune arthritis, and the heterogeneity of the response to treatment among patients, mechanisms of disease may be similar to anti-CCP-positive, shared epitope-positive RA in some subsets, but very different in others [69–73]. Various roles of DC in autoimmune arthritis are described below.
Dendritic cells and the initiation of RA
'Central' tolerance defects are important contributors to spontaneous autoimmune disease. In the foetal and neonatal period, central tolerance is actively maintained in the thymus [74]. During this process, a repertoire of T cells restricted to self-MHC displayed by the thymic cortical epithelial cells is selected in each individual. In addition, those T cells reactive to self-antigen expressed and presented by medullary APC, which include medullary epithelial cells and medullary DC, are deleted by negative selection above a threshold of affinity for self-antigens presented by those APC [75]. Since an affinity threshold applies for central deletion of self-reactive T cells, circulation of low-affinity self-reactive T cells in the periphery is inevitable. Self-antigen is commonly ignored by these T cells, however, because their affinity threshold is below that required for self-antigen priming in the periphery.
In various spontaneous autoimmune animal models, defects pertaining to the interaction of APC and thymocytes interferes with the normal process of negative selection. Unlike the normal situation, this permits the release of dangerously autoreactive T cells into the periphery, where subsequent genetic or environmental proinflammatory events more readily trigger the priming of these T cells and the development of autoimmune disease [69]. An example is the skg mouse model of spontaneous arthritis, resembling RA, in which DC activated by fungal β-glucans prime autoreactive peripheral T cells, in an IL-1-dependent fashion, which can then drive the proliferation of autoantibodies and a proinflammatory arthritogenic response [76]. Alternatively, to initiate autoimmunity, peripheral DC may prime the immune system to respond to modified self-antigens, potentially generated for the first time in the periphery, either circumventing central tolerance mechanisms or compounding central defects. As described later, self-proteins modified by citrullination in the periphery are important autoantigens presented by DC in RA, and in the murine collagen-induced arthritis model.
Dendritic cell antigen presentation in induction and maintenance of RA
DC play an essential role in the priming of lymphocytes in autoimmunity [8, 77]. Presentation of viral or modified self-antigens, of which the immune system has been ignorant, represents a common theme in the initiation of autoimmunity. Several autoantigens are described in RA, including a variety of post-translationally modified citrullinated proteins. In the collagen-induced arthritis model of autoimmune arthritis, anti-CCP develop spontaneously and have been shown to play a pathogenetic role, in that they are found prior to visible clinical disease. Furthermore, monoclonal antibodies directed against citrullinated proteins were shown to bind antigens within inflamed synovium and to enhance submaximal disease. Mice tolerized with a citrulline-containing peptide demonstrated significantly reduced disease severity and incidence compared with control mice [78].
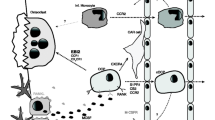
Shared epitope-encoding HLA alleles are particularly associated with anti-CCP-positive RA [60, 79, 80]. Citrullination replaces charged imino side-chain groups with an uncharged carbonyl group, increasing the affinity of citrullinated proteins with the shared epitope. Fibrin and vimentin are two citrullinated proteins identified thus far in synovial extracts from inflamed joints, and are prominent synovial candidate antigens in anti-CCP-positive RA [81, 82]. Citrullinated collagen types I and II and eukaryotic translation initiation factor 4G1 are further protein candidates [83]. Citrullinated self-proteins produced in inflamed synovial tissue are therefore probably taken up, processed and presented by activated synovial DC to prime populations of citrulline self-peptide-specific T cells in draining lymph nodes [78]. In some cases, peptides might be derived from regurgitated digestion by macrophages, as DC have limited capacity to process large, complex proteins such as type II collagen and fibrinogen [84]. Other proteins such as vimentin may be acquired by ingestion of apoptotic macrophages. Effector function, including cytokine production and B-cell and monocyte help, of autoantigen-specific memory T cells trafficking to joints would be boosted by local DC presenting citrullinated peptides. Antigen-specific T cells are critical for the promotion of autoantibody production, and for driving monocyte activation and cytokine production. These T cells would promote production of anti-CCP autoantibodies in follicular areas of RA synovial tissue and lymphoid organs (Figure 2).

A model for dendritic cell presentation of citrullinated self-antigenic peptides, and the development of chronic autoimmune inflammatory disease of joint and vascular tissues. anti-CCP, anticyclic citrullinated peptide; DC, dendritic cells; EC, endothelial cells; FDC, follicular dendritic cells; MΦ, macrophages; ox-LDL, oxidized low-density lipoprotein; RF, rheumatoid factor.
Given the disease-enhancing capacity of anti-CCP in murine models, presentation of citrullinated antigens complexed with anti-CCP antibodies may be facilitated through the opsonizing effects of antibody and complement. Cross-linking by rheumatoid factor may enhance Fc-dependent inflammatory responses [85, 86]. Autoantigenic immune complexes have been shown, in murine models of arthritis, to promote vascular permeability necessary for continued enhanced traffic of inflammatory cells into the synovial compartment [87]. Immune complexes have been demonstrated in RA for over 30 years, and have recently been described for citrullinated type II collagen [85, 86]. Citrulline-reactive T cells have been demonstrated in DRB1*0401-transgenic mice, and have also been observed after priming of naïve mice to foreign hen egg lysozyme antigen, but have not yet been convincingly determined in RA patients [61, 82].
Other autoantigenic specificities than citrulline are also described in RA, which would be presented in a similar way by DC. These include type II collagen, human cartilage gp39 in about 60% of RA, and glucose-6-phosphate isomerase in a much smaller proportion of patients [88]. It remains to be seen whether these autoimmune specificities segregate with particular HLA-DR-presenting elements.
Dendritic cells and RA synovial inflammation
Gene transcriptional activity of the NF-κB family is characteristic of the RA inflammatory lesion. There are two major pathways of NF-κB: the classical pathway (comprising homodimers and heterodimers of RelA, c-Rel and p50), and the alternate pathway (comprising RelB and p52). In DC, the classical pathway drives transcription of prosurvival and pro-inflammatory response genes, including cytokines such as IL-6, TNF and IL-12. The alternate pathway controls DC maturation for antigen-presenting function, medullary thymic epithelial cell development required for negative selection, and mature monocyte development (for review see [35]).
In B cells, signals such as TNF and TLR ligands drive classical pathway activation and B-cell activation factor of the TNF ligand family (BAFF), and CD154 drive the alternate pathway. However, TNF, TLR agonists or CD154 signal activation of both pathways uniquely in DC, through exchange of NF-κB dimers in the nucleus [89]. Moreover, DC make little or no response to BAFF.
Given its role in DC function, immunohistochemical detection of nuclear RelB is an excellent marker of functionally differentiated DC in perivascular regions of synovial tissue biopsies from patients with untreated RA, and can be used to quantitate mature DC in biopsies [16, 90, 91]. Most disease-modifying or biologic therapies block at least the classical NF-κB pathway. Since this will lead to reduction of RelB activity in DC, numbers of synovial nuclear RelB+ DC have been shown to decrease after treatment with disease-modifying antirheumatic drugs [91]. Furthermore, incidence and severity of antigen-induced arthritis was considerably reduced in RelB-deficient bone marrow chimeric mice compared with control mice [92]. In contrast to the inflammatory setting, precursors of immature myeloid DC in peripheral blood express neither RelB mRNA nor protein [90]. Nuclear RelB+ cells are also absent in normal nonlymphoid peripheral tissues, such as normal synovial tissue and epithelia [93]. RelB+ DC in rheumatoid synovial tissue closely resemble mature interdigitating lymph node DC [90, 94, 95]. Mature myeloid DC in perivascular, T-cell-enriched areas of synovial tissue are further characterized by expression of CD86, DC-LAMP and CCR7, and are associated with, and presumably attracted by, cells expressing the chemokines CCL19 (SLC) and CCL21 (ELC) [11, 16, 94]. In contrast, immature DC are also abundant in the synovial lining and sublining layers of the synovium associated with CCL20 (MIP-3α)-expressing cells, as well as in rheumatoid nodules and the synovial fluid. In synovial tissue the immature DC are characterized by CCR6 and CD1a expression, and in nodules by CMRF-44 and CD14 expression [11, 96]. Local transforming growth factor beta may play a role in the maintaining DC in an immature state or in the upregulation of CD1a expression [97].
DC and macrophages contribute very early in the development of autoimmune inflammatory lesions in mouse models, such as autoimmune diabetes and polyarthritis, to produce local cytokines, including TNF [98–100]. DC have also been shown in diabetic models to stimulate ectopic lymphoid tissue development by lymphotoxin-β receptor signalling, and blockade of this signal has been shown to be sufficient to block disease development [8, 101, 102]. While so far little studied in the joint, this research is now technically feasible with the development of CD11c-DTR mice, in which DC can be transiently depleted [103].
DC enter synovial tissue by means of inflamed synovial blood vessels and are chemoattracted there by virtue of specific chemokine receptor expression, in response to CX3CL1 (fractalkine), CCL19 (SLC), CCL21 (ELC) and CCL20 (MIP-3α). These chemokines play an important role in driving the inflammatory disease. For example, ectopic expression of CCL19 has been shown sufficient for formation of lymphoid tissue similar to that seen in rheumatoid synovial tissue [104]. Inhibition of CX3CL1 has been shown to reduce clinical scores in the murine collagen-induced arthritis model [105]. RA synovial DC have also been shown to produce high levels of CCL18 (DCCK1), a chemotactic factor for naïve T cells and a stimulator of collagen production by fibroblasts [106]. The sustained immunomodulatory effect of TNF blockade in RA relates in part to reduction of traffic of DC and other immunocytes to the inflammatory site [107].
Increased numbers of myeloid and plasmacytoid DC are observed in synovial fluid and perivascular regions of synovial tissues in patients with RA and other autoimmune rheumatic diseases, in which cells producing TNF are colocated [10, 12, 16, 108, 109]. Plasmacytoid DC are recruited to normal lymphoid organs as well as inflammatory sites including RA synovial tissue with local differentiation, but there is no recruitment to normal peripheral tissues [110] (Table 1). These DC are likely to play an important pro-inflammatory role, particularly after sensing immunostimulatory nucleic acid sequences. In contrast, myeloid DC precursors populate normal resting synovial tissues – but additional CD11c+ myeloid cellular recruitment takes place at the RA synovial inflammatory site in response to inflammatory chemokines, where RelB nuclear translocation associated with DC maturation may take place [16]. Nuclear RelB+ DC in inflamed joints are generally found closely associated with T lymphocytes [16, 90, 93], which may signal the alternate NF-κB pathway through proinflammatory cytokines, CD154 (CD40L) and lymphotoxin-β [111, 112].
Synthesis: signalling of NF-κB activation by dendritic cells and priming/induction of RA
The antigen-presenting and antigen-priming functions of DC to autoreactive T cells appear to be very proximal events and to be essential to the subsequent pathogenesis of disease, including the generation of autoantibodies in patients at risk due to genetic and environmental factors. From several different animal arthritic models, it is clear that proinflammatory stimuli driving TNF, IL-1 or NF-κB p50 are all sufficient to drive the development of autoimmune polyarthritis in susceptible strains, through the simultaneous promotion of DC or monocyte activation, priming of autoreactive lymphocytes, and sustained synovial inflammation [70, 113–115]. It is of interest when considering the environmental associations with RA that several factors, including nicotine, lactation and Epstein–Barr virus, promote NF-κB activity, associated either with B-cell activation or TNF secretion by myeloid cells including monocytes and DC [116–119].
In contrast, pregnancy and the oral contraceptive pill as well as high fruit and Mediterranean diets are RA protective. Combinations of disease-modifying antirheumatic drugs and biologics can induce RA clinical remission [120]. Many disease-modifying antirheumatic drugs and anti-inflammatory drugs and natural substances are able to suppress NF-κB, including the RelB subunit, which is critical for DC priming function. These include 1,25-dihydroxy-vitamin D, glucocorticoids and active components of turmeric, red wine, mangoes and other fruits [121]. Taken together, both human and murine evidence indicates that NF-κB activation is required to drive RA, and indicates those factors that suppress this activity are disease-suppressive or protective [38, 114, 122].
Role of dendritic cells in RA complications: atherosclerosis
Cardiovascular disease mortality and morbidity are increased in RA patients, with traditional cardiovascular risk factors insufficient to explain the increase in risk [123]. Considerable evidence demonstrates that inflammation associated with RA plays a key role in the onset and progression of atherosclerosis in these patients. RA patients also have an increased burden of subclinical vascular disease compared with matched control individuals, as demonstrated by the carotid intima-media thickness and endothelial dysfunction [124, 125]. Atherosclerotic disease is associated with erythrocyte sedimentation rate and CRP levels in RA, and the mean CRP level over time predicts peripheral endothelial function [125, 126].
The atherosclerotic lesion represents a set of highly specific inflammatory cellular and molecular responses including abundant infiltration by monocytes, macrophages and T cells, together with CRP and complement. Immune mechanisms have been postulated in atherogenesis, in view of elevated values of circulating inflammatory markers such as CRP, serum amyloid A, IL-6 and IL-1 receptor antagonist, accompanying acute coronary syndromes [127, 128].
Atherosclerosis occurs principally in large and medium-sized elastic and muscular arteries, and can lead to ischemia of various organs including the heart, the brain or extremities. This process starts as asymmetrical focal thickenings of the vascular intima, which is infiltrated with inflammatory cells as a result of stimuli such as oxidized low-density lipoprotein (LDL) or infection [129]. Circulating rheumatoid factor and other immune complexes may also cause direct injury to vascular EC with the same result [130]. Monocytes are the first cells to attach to the endothelium and migrate into the underlying subendothelial space. Initially, resident monocytes differentiate into macrophages accumulating intracellular modified forms of LDL to form fatty streak lesions [128]. This is followed by continued recruitment of monocytes, T cells and natural killer T cells, mast cells and DC to form raised fibro-fatty plaques, in which the central lipid and foamy macrophage core is surrounded by immune cells and then proliferating smooth muscle cells and a collagen-rich matrix. T cells within these plaques are characterized by a Th1-type phenotype, and produce IFN-γ and TNF. The fibrous cap prevents contact between the prothrombotic lesion and the blood. Plaques can develop a range of complications, particularly rupture and thrombosis, with clinical consequences including myocardial infarction and stroke [131].
DC have been identified in atherosclerotic plaques in humans, and in rats with diet-induced hyperlipidemia, and are thought to play an important role in atherogenesis [132]. As noted for the inflamed synovium, DC are highly migratory, and probably traffic between the blood and the arterial intima across vascular EC, across the penetrating vasa vasorum that supply the arterial wall, and to the draining lymph nodes. In support, DC can be detected between smooth muscle cells in the medial layer of vessels. They are markedly increased in media underlying atherosclerotic plaques compared with adjacent media of nonatherosclerotic areas, suggesting that some intimal vascular DC migrate through the media and adventitia to adjacent lymph node, where they could present atherosclerosis-associated antigens [133].
Of interest with respect to RA, an autoimmune hypothesis has been proposed for atherogenesis, which incorporates the concept of vascular-associated lymphoid tissue – analogous to mucosa-associated lymphoid tissue in the respiratory and gastrointestinal tracts. Vascular-associated lymphoid tissue consists of disseminated focal accumulations of immuno-competent cells, including DC, in the subendothelial layer of the arteries [134]. DC are found in healthy human arterial walls and accumulate most densely in arterial regions subjected to major haemodynamic stress under turbulent flow conditions known to predispose to development of atherosclerosis, as a consequence of chronic inflammatory stress in these regions [135]. As in the joint, more than 90% of DC in atherosclerotic lesions colocalize with T cells located in neovascularization areas associated with inflammatory infiltrates [133]. In support of the role of inflammation in this pathological process, DC function has been reported to be increased in patients with unstable angina. As in the synovium, DC are important APC and effector cells in the inflammatory process, which is associated with plaque instability and vulnerability toward rupture [136].
Endothelial dysfunction and dendritic cells
EC play a pivotal role in the inflammatory response. EC activation promotes vascular permeability, oedema and leukocyte recruitment. Endothelial dysfunction has been shown to precede both the formation of atherosclerotic plaques and specific inflammation of joints following an immune stimulus. Vascular cell adhesion molecule-1 is induced in response to EC injury, and in animal models it plays a key role in the recruitment of monocytes and other immune cells to intimal plaques [131]. Several studies have demonstrated that endothelial dysfunction is associated with high inflammatory activity in RA, is present early in the disease course, and improves after treatment with antirheumatic drugs [137, 138]. Inflammation-associated endothelial dysfunction has a significant impact on DC maturation and adherence to the endothelium. For example, DC adhesion and transmigration are markedly increased after exposing EC to hypoxia, oxidized LDL or TNF. EC express TLR2 and TLR4, which may transduce inflammatory, proatherogenic signals, including HSP-60, oxidized LDL and microorganisms [139, 140]. DC and other immune cells within plaques, as in RA synovium, show evidence of NF-κB activation, resulting from both TLRs and signals from cytokines, such as TNF [141]. It has been proposed that lymph node-migratory DC prime HSP-60, oxidized LDL or bacterial antigen-specific T cells in vascular draining lymph nodes, and that effector T cells can be restimulated by mature DC in the vascular lesion, leading to release of cytokines, which promote atherosclerotic disease [142–144].
Lipid abnormality and dendritic cells
Dyslipidemia is an important risk factor for the atherosclerotic process in general. Dyslipidemia in RA is mainly driven by a low concentration of high-density lipoprotein, associated with an unfavourable cardiovascular risk. Total cholesterol levels and high-density lipoprotein cholesterol levels in RA are inversely associated with the acute phase response, regardless of whether patients are treated with antirheumatic drugs [145]. Of importance, the acute phase response promotes oxidative modification of LDL. Oxidized LDL in turn promotes mature DC generation from monocytes, and probably provides a source of atherogenic autoantigen [146, 147]. Low levels of high-density lipoprotein have also been shown to impair DC migration to draining lymph nodes in a mouse model, with implications for the local proinflammatory activity of oxidized LDL-activated DC in the atherosclerotic lesion [148].
Smoking and dendritic cells
Cigarette smoking increases the risk of RA, as discussed above, and of cardiovascular disease in RA [149, 150]. Nicotine promotes progression of advanced atherosclerotic plaques, but also activates NF-κB, with augmented APC function and proinflammatory cytokine secretion [151–153]. Nicotine significantly enhances the recruitment of DC to atherosclerotic lesions in a mouse model. Smoking is also an important contributor to the association of RA and cardiovascular disease, either reflecting its role as a risk factor in its own right or because it is associated with more severe rheumatoid disease. While CRP and rheumatoid factor are associated with more severe atherosclerotic disease in RA, no association has been shown to date for anti-CCP, despite the association of smoking with anti-CCP in RA. This may relate to very specific roles played by CRP and rheumatoid factor at the vascular endothelium or within plaques.
Conclusion: NF-κB activation links RA and complicating atherosclerosis
DC play critical antigen-presenting and antigen-priming roles in the initiation of RA and atherosclerosis, as well as proinflammatory roles in RA and atherosclerosis. Various signals that promote the activation of NF-κB and the secretion of TNF and IL-1 drive the maturation of DC to prime self-specific responses, and drive the perpetuation of synovial and vascular inflammation. These signals may include infection, cigarette smoking, immunostimulatory DNA, oxidized LDL and primary genetic lesions. The pathogenesis of RA and atherosclerosis is intimately linked, with the vascular disease of RA driven by similar and simultaneous triggers. Understanding this link has implications for discovery of NF-κB response genes that might modify the risk or expression of RA in an individual exposed to environmental factors, as well as the ability of a given treatment regimen to halt disease progression in joints or vasculature. Finally, discovery of the key role of NF-κB to DC function has opened the door for new antigen-specific strategies using NF-κB-inhibitory drugs to target DC with antigen, avoiding systemic toxicity associated with such compounds.
Note
This review is part of a series on Cells of the synovium in rheumatoid arthritis edited by Gary Firestein.
Other articles in this series can be found at http://arthritis-research.com/articles/review-series.asp?series=ar_Cells
Abbreviations
- anti-CCP:
-
= anticyclic citrullinated peptide
- APC:
-
= antigen-presenting cells
- CRP:
-
= C-reactive protein
- DC:
-
= dendritic cells
- EC:
-
= endothelial cells
- Fc:
-
= crystallizable fragment
- IFN:
-
= interferon
- IL:
-
= interleukin
- LDL:
-
= low-density lipoprotein
- MHC:
-
= major histocompatability complex
- NF:
-
= nuclear factor
- RA:
-
= rheumatoid arthritis
- TLR:
-
= Toll-like receptor
- TNF:
-
= tumour necrosis factor.
References
Weyand CM: New insights into the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford). 2000, 39: 3-8.
Banchereau J, Steinman RM: Dendritic cells and the control of immunity. Nature. 1998, 392: 245-252. 10.1038/32588.
Kitamura H, Iwakabe K, Yahata T, Nishimura S, Ohta A, Ohmi Y, Sato M, Takeda K, Okumura K, Van Kaer L, et al: The natural killer T (NKT) cell ligand alpha-galactosylceramide demonstrates its immunopotentiating effect by inducing interleukin (IL)-12 production by dendritic cells and IL-12 receptor expression on NKT cells. J Exp Med. 1999, 189: 1121-1128. 10.1084/jem.189.7.1121.
Brocker T: Survival of mature CD4 T lymphocytes is dependent on major histocompatibility complex class II-expressing dendritic cells. J Exp Med. 1997, 186: 1223-1232. 10.1084/jem.186.8.1223.
Lutzky V, Thomas R: Dendritic cells. Contemporary Targeted Therapies in Rheumatology. Edited by: Smolen JS, Lipsky PE. 2007, London: Informa Healthcare, 63-78.
Dittel BN, Visintin I, Merchant RM, Janeway CA: Presentation of the self antigen myelin basic protein by dendritic cells leads to experimental autoimmune encephalomyelitis. J Immunol. 1999, 163: 32-39.
Leung BP, Conacher M, Hunter D, McInnes IB, Liew FY, Brewer JM: A novel dendritic cell-induced model of erosive inflammatory arthritis: distinct roles for dendritic cells in T cell activation and induction of local inflammation. J Immunol. 2002, 169: 7071-7077.
Ludewig B, Odermatt B, Landmann S, Hengartner H, Zinkernagel RM: Dendritic cells induce autoimmune diabetes and maintain disease via de novo formation of local lymphoid tissue. J Exp Med. 1998, 188: 1493-1501. 10.1084/jem.188.8.1493.
Thomas R, Davis LS, Lipsky PE: Rheumatoid synovium is enriched in mature antigen-presenting dendritic cells. J Immunol. 1994, 152: 2613-2623.
Thomas R, Lipsky PE: Could endogenous self-peptides presented by dendritic cells initiate rheumatoid arthritis?. Immunol Today. 1996, 17: 559-564. 10.1016/S0167-5699(96)20030-1.
Page G, Lebecque S, Miossec P: Anatomic localization of immature and mature dendritic cells in an ectopic lymphoid organ: correlation with selective chemokine expression in rheumatoid synovium. J Immunol. 2002, 168: 5333-5341.
Cavanagh LL, Boyce A, Smith L, Padmanabha J, Filgueira L, Pietschmann P, Thomas R: Rheumatoid arthritis synovium contains plasmacytoid dendritic cells. Arthritis Res Ther. 2005, 7: R230-R240. 10.1186/ar1467.
Caux C, Dezutter-Dambuyant C, Schmitt D, Banchereau J: GM-CSF and TNF-α cooperate in the generation of dendritic Langerhans cells. Nature. 1992, 360: 258-261. 10.1038/360258a0.
Caux C, Massacrier C, Dezutter-Dambuyant C, Vanbervliet B, Jacquet C, Schmitt D, Banchereau J: Human dendritic Langer-hans cells generated in vitro from CD34+ progenitors can prime naive CD4+ T cells and process soluble antigen. J Immunol. 1995, 155: 5427-5435.
Inaba K, Inaba M, Romani N, Aya A, Deguchi M, Ikehara S, Muramatsu S, Steinman RM: Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992, 176: 1693-1702. 10.1084/jem.176.6.1693.
Pettit AR, MacDonald KPA, O'Sullivan B, Thomas R: Differentiated dendritic cells expressing nuclear RelB are predominantly located in rheumatoid synovial tissue perivascular mononuclear cell aggregates. Arthritis Rheum. 2000, 43: 791-800. 10.1002/1529-0131(200004)43:4<791::AID-ANR9>3.0.CO;2-E.
Larsen CP, Steinman RM, Witmer-Pack M, Hankins DF, Morris PJ, Austyn JM: Migration and maturation of Langerhans cells in skin transplants and explants. J Exp Med. 1990, 172: 1483-1493. 10.1084/jem.172.5.1483.
Steinman RM, Swanson J: The endocytic activity of dendritic cells. J Exp Med. 1995, 182: 283-288. 10.1084/jem.182.2.283.
Sallusto F, Palermo B, Lenig D, Miettinen M, Matikainen S, Julkunen I, Forster R, Burgstahler R, Lipp M, Lanzavecchia A: Distinct patterns and kinetics of chemokine production regulate dendritic cell function. Eur J Immunol. 1999, 29: 1617-1625. 10.1002/(SICI)1521-4141(199905)29:05<1617::AID-IMMU1617>3.0.CO;2-3.
Sallusto F, Lanzavecchia A: Understanding dendritic cell and T-lymphocyte traffic through the analysis of chemokine receptor expression. Immunol Rev. 2000, 177: 134-140. 10.1034/j.1600-065X.2000.17717.x.
Hermans IF, Ritchie DS, Yang J, Roberts JM, Ronchese F: CD8+ T cell-dependent elimination of dendritic cells in vivo limits the induction of antitumor immunity. J Immunol. 2000, 164: 3095-3101.
Kalinski P, Vieira P, Schuitemaker JH, Cai Q, Kapsenberg M: Generation of human type 1- and type 2-polarized dendritic cells from peripheral blood. Methods Mol Biol. 2003, 215: 427-436.
Kurts C, Kosaka H, Carbone FR, Allison J, Miller JFAP, Heath WR: Class I-restricted cross presentation of exogenous self antigens leads to deletion of autoreactive CD8+ T cells. J Exp Med. 1997, 186: 239-245. 10.1084/jem.186.2.239.
Steptoe RJ, Ritchie JM, Wilson NS, Villadangos JA, Lew AM, Harrison LC: Cognate CD4+ help elicited by resting dendritic cells does not impair the induction of peripheral tolerance in CD8+T cells. J Immunol. 2007, 178: 2094-2103.
Steptoe RJ, Ritchie JM, Jones LK, Harrison LC: Autoimmune diabetes is suppressed by transfer of proinsulin-encoding Gr-1+ myeloid progenitor cells that differentiate in vivo into resting dendritic cells. Diabetes. 2005, 54: 434-442. 10.2337/diabetes.54.2.434.
Sallusto F, Lanzavecchia A: Mobilizing dendritic cells for tolerance, priming and chronic inflammation. J Exp Med. 1999, 189: 611-614. 10.1084/jem.189.4.611.
Caux C, Massacrier C, Vanbervliet B, Dubois B, van Kooten C, Durand I, Banchereau J: Activation of human dendritic cells through CD40 cross-linking. J Exp Med. 1994, 180: 1263-1272. 10.1084/jem.180.4.1263.
Cella M, Salio M, Sakakibara Y, Langen H, Julkunen I, Lanzavecchia A: Maturation, activation, and protection of dendritic cells induced by double-stranded RNA. J Exp Med. 1999, 189: 821-829. 10.1084/jem.189.5.821.
O'Sullivan BJ, Thomas R: CD40 Ligation conditions dendritic cell antigen-presenting function through sustained activation of NF-kappaB. J Immunol. 2002, 168: 5491-5498.
Kodaira Y, Nair SK, Wrenshall LE, Gilboa E, Platt JL: Phenotypic and functional maturation of dendritic cells mediated by heparan sulfate. J Immunol. 2000, 165: 1599-1604.
Yan M, Peng J, Jabbar IA, Liu X, Filgueira L, Frazer IH, Thomas R: Activation of dendritic cells by human papillomavirus-like particles through TLR4 and NF-kappaB-mediated signalling, moderated by TGF-beta. Immunol Cell Biol. 2005, 83: 83-91. 10.1111/j.1440-1711.2004.01291.x.
Reis e Sousa C: Dendritic cells in a mature age. Nat Rev Immunol. 2006, 6: 476-483. 10.1038/nri1845.
Siebenlist U, Franzoso G, Brown K: Structure, regulation and function of NF-kappa B. Annu Rev Cell Biol. 1994, 10: 405-455. 10.1146/annurev.cb.10.110194.002201.
Ghosh S, May MJ, Kopp EB: NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998, 16: 225-260. 10.1146/annurev.immunol.16.1.225.
O'Sullivan B, Thompson A, Thomas R: NF-kappa B as a therapeutic target in autoimmune disease. Expert Opin Ther Targets. 2007, 11: 111-122. 10.1517/14728222.11.2.111.
Bondeson J, Foxwell B, Brennan F, Feldmann M: Defining therapeutic targets by using adenovirus: blocking NF-kappaB inhibits both inflammatory and destructive mechanisms in rheumatoid synovium but spares anti-inflammatory mediators. Proc Natl Acad Sci USA. 1999, 96: 5668-5673. 10.1073/pnas.96.10.5668.
Bondeson J, Browne KA, Brennan FM, Foxwell BM, Feldmann M: Selective regulation of cytokine induction by adenoviral gene transfer of IkappaBalpha into human macrophages: lipopolysaccharide-induced, but not zymosan-induced, proinflammatory cytokines are inhibited, but IL-10 is nuclear factor-kappaB independent. J Immunol. 1999, 162: 2939-2945.
Foxwell B, Browne K, Bondeson J, Clarke C, de Martin R, Brennan F, Feldmann M: Efficient adenoviral infection with IkappaB alpha reveals that macrophage tumor necrosis factor alpha production in rheumatoid arthritis is NF-kappaB dependent. Proc Natl Acad Sci USA. 1998, 95: 8211-8215. 10.1073/pnas.95.14.8211.
Koch AE, Burrows JC, Haines GK, Carlos TM, Harlan LM, Leibovich SJ: Immunolocalization of endothelial and leukocyte adhesion molecules in human rheumatoid and osteoarthritic synovial tissues. Lab Invest. 1991, 64: 313-320.
Koch AE, Kunkel SL, Harlow LA, Johnson B, Evanoff HL, Haines GK, Burdick MD, Pope RM, Strieter RM: Enhanced production of monocyte chemoattractant protein-1 in rheumatoid arthritis. J Clin Invest. 1992, 90: 772-779.
Koch AE, Kunkel SL, Harlow LA, Mazarakis DD, Haines GK, Burdick MD, Pope RM, Strieter RM: Macrophage inflammatory protein-1 alpha. A novel chemotactic cytokine for macrophages in rheumatoid arthritis. J Clin Invest. 1994, 93: 921-928.
Bischof RJ, Zafiropoulos D, Hamilton JA, Campbell IK: Exacerbation of acute inflammatory arthritis by the colony-stimulating factors CSF-1 and granulocyte macrophage (GM)-CSF: evidence of macrophage infiltration and local proliferation. Clin Exp Immunol. 2000, 119: 361-367. 10.1046/j.1365-2249.2000.01125.x.
Chu CQ, Field M, Allard S, Abney E, Feldmann M, Maini RN: Detection of cytokines at the cartilage/pannus junction in patients with rheumatoid arthritis: implications for the role of cytokines in cartilage destruction and repair. Br J Rheumatol. 1992, 31: 653-661. 10.1093/rheumatology/31.10.653.
Xia Y, Pauza ME, Feng L, Lo D: RelB regulation of chemokine expression modulates local inflammation. Am J Pathol. 1997, 151: 375-387.
Hilliard B, Samoilova EB, Liu TS, Rostami A, Chen Y: Experimental autoimmune encephalomyelitis in NF-kappa B-deficient mice: roles of NF-kappa B in the activation and differentiation of autoreactive T cells. J Immunol. 1999, 163: 2937-2943.
Pahl HL: Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999, 18: 6853-6866. 10.1038/sj.onc.1203239.
Poljak L, Carlson L, Cunningham K, Kosco-Vilbois MH, Siebenlist U: Distinct activities of p52/NF-kappa B required for proper secondary lymphoid organ microarchitecture: functions enhanced by Bcl-3. J Immunol. 1999, 163: 6581-6588.
Lo JC, Basak S, James ES, Quiambo RS, Kinsella MC, Alegre ML, Weih F, Franzoso G, Hoffmann A, Fu YX: Coordination between NF-kappaB family members p50 and p52 is essential for mediating LTbetaR signals in the development and organization of secondary lymphoid tissues. Blood. 2006, 107: 1048-1055. 10.1182/blood-2005-06-2452.
Burkly L, Hession C, Ogata L, Reilly C, Marconi LA, Olson D, Tizard R, Cate R, Lo D: Expression of relB is required for the development of thymic medulla and dendritic cells. Nature. 1995, 373: 531-536. 10.1038/373531a0.
Banchereau J, Paczesny S, Blanco P, Bennett L, Pascual V, Fay J, Palucka AK: Dendritic cells: controllers of the immune system and a new promise for immunotherapy. Ann N Y Acad Sci. 2003, 987: 180-187.
Krug A, Towarowski A, Britsch S, Rothenfusser S, Hornung V, Bals R, Giese T, Engelmann H, Endres S, Krieg AM, et al: Toll-like receptor expression reveals CpG DNA as a unique microbial stimulus for plasmacytoid dendritic cells which synergizes with CD40 ligand to induce high amounts of IL-12. Eur J Immunol. 2001, 31: 3026-3037. 10.1002/1521-4141(2001010)31:10<3026::AID-IMMU3026>3.0.CO;2-H.
Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, Liu YJ: Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001, 194: 863-869. 10.1084/jem.194.6.863.
Hochrein H, Shortman K, Vremec D, Scott B, Hertzog P, O'Keeffe M: Differential production of IL-12, IFN-alpha, and IFN-gamma by mouse dendritic cell subsets. J Immunol. 2001, 166: 5448-5455.
Cella M, Facchetti F, Lanzavecchia A, Colonna M: Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat Immunol. 2000, 1: 305-310. 10.1038/79747.
Penna G, Sozzani S, Adorini L: Cutting edge: selective usage of chemokine receptors by plasmacytoid dendritic cells. J Immunol. 2001, 167: 1862-1866.
Bottini N, Vang T, Cucca F, Mustelin T: Role of PTPN22 in type 1 diabetes and other autoimmune diseases. Semin Immunol. 2006, 18: 207-213. 10.1016/j.smim.2006.03.008.
Iikuni N, Ikari K, Momohara S, Tomatsu T, Hara M, Yamanaka H, Okamoto H, Kamatani N: MHC2TA is associated with rheumatoid arthritis in Japanese patients. Ann Rheum Dis. 2007, 66: 274-275. 10.1136/ard.2006.063347.
Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M, Nagasaki M, Nakayama-Hamada M, Kawaida R, Ono M, et al: Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. 2003, 34: 395-402. 10.1038/ng1206.
Mori M, Yamada R, Kobayashi K, Kawaida R, Yamamoto K: Ethnic differences in allele frequency of autoimmune-disease-associated SNPs. J Hum Genet. 2005, 50: 264-266. 10.1007/s10038-005-0246-8.
Klareskog L, Stolt P, Lundberg K, Kallberg H, Bengtsson C, Grunewald J, Ronnelid J, Harris HE, Ulfgren AK, Rantapaa-Dahlqvist S, et al: A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006, 54: 38-46. 10.1002/art.21575.
Ireland J, Herzog J, Unanue ER: Cutting edge: unique T cells that recognize citrullinated peptides are a feature of protein immunization. J Immunol. 2006, 177: 1421-1425.
Gyorgy B, Toth E, Tarcsa E, Falus A, Buzas EI: Citrullination: a posttranslational modification in health and disease. Int J Biochem Cell Biol. 2006, 38: 1662-1677. 10.1016/j.biocel.2006.03.008.
du Montcel ST, Michou L, Petit-Teixeira E, Osorio J, Lemaire I, Lasbleiz S, Pierlot C, Quillet P, Bardin T, Prum B, et al: New classification of HLA-DRB1 alleles supports the shared epitope hypothesis of rheumatoid arthritis susceptibility. Arthritis Rheum. 2005, 52: 1063-1068. 10.1002/art.20989.
Silman AJ, Newman J, MacGregor AJ: Cigarette smoking increases the risk of rheumatoid arthritis. Results from a nationwide study of disease-discordant twins. Arthritis Rheum. 1996, 39: 732-735. 10.1002/art.1780390504.
Oliver JE, Silman AJ: Risk factors for the development of rheumatoid arthritis. Scand J Rheumatol. 2006, 35: 169-174. 10.1080/03009740600718080.
Balandraud N, Meynard JB, Auger I, Sovran H, Mugnier B, Reviron D, Roudier J, Roudier C: Epstein–Barr virus load in the peripheral blood of patients with rheumatoid arthritis: accurate quantification using real-time polymerase chain reaction. Arthritis Rheum. 2003, 48: 1223-1228. 10.1002/art.10933.
Balandraud N, Roudier J, Roudier C: Epstein–Barr virus and rheumatoid arthritis. Autoimmun Rev. 2004, 3: 362-367. 10.1016/j.autrev.2004.02.002.
Padyukov L, Silva C, Stolt P, Alfredsson L, Klareskog L: A gene–environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum. 2004, 50: 3085-3092. 10.1002/art.20553.
Yoshitomi H, Sakaguchi N, Kobayashi K, Brown GD, Tagami T, Sakihama T, Hirota K, Tanaka S, Nomura T, Miki I, et al: A role for fungal β-glucans and their receptor Dectin-1 in the induction of autoimmune arthritis in genetically susceptible mice. J Exp Med. 2005, 201: 949-960. 10.1084/jem.20041758.
Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, Ikuse T, Asano M, Iwakura Y: Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. 2000, 191: 313-320. 10.1084/jem.191.2.313.
Matsumoto I, Maccioni M, Lee DM, Maurice M, Simmons B, Brenner M, Mathis D, Benoist C: How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat Immunol. 2002, 3: 360-365. 10.1038/ni772.
Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D: Organ-specific disease provoked by systemic autoimmunity. Cell. 1996, 87: 811-822. 10.1016/S0092-8674(00)81989-3.
Courtenay JS, Dallman MJ, Dayan AD, Martin A, Mosedale B: Immunisation against heterologous type II collagen induces arthritis in mice. Nature. 1980, 283: 666-668. 10.1038/283666a0.
Ardavin C: Thymic dendritic cells. Immunol Today. 1997, 18: 350-361. 10.1016/S0167-5699(97)01090-6.
Kappler JW, Roehm N, Marrack P: T cell tolerance by clonal elimination in the thymus. Cell. 1987, 49: 273-280. 10.1016/0092-8674(87)90568-X.
Sakaguchi N, Takahashi T, Hata H, Nomura T, Tagami T, Yamazaki S, Sakihama T, Matsutani T, Negishi I, Nakatsuru S, et al: Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature. 2003, 426: 454-460. 10.1038/nature02119.
Drakesmith H, O'Neil D, Schneider SC, Binks M, Medd P, Sercarz E, Beverley P, Chain B: In vivo priming of T cells against cryptic determinants by dendritic cells exposed to interleukin 6 and native antigen. Proc Natl Acad Sci USA. 1998, 95: 14903-14908. 10.1073/pnas.95.25.14903.
Kuhn KA, Kulik L, Tomooka B, Braschler KJ, Arend WP, Robinson WH, Holers VM: Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest. 2006, 116: 961-973. 10.1172/JCI25422.
van Gaalen F, Ioan-Facsinay A, Huizinga TW, Toes RE: The devil in the details: the emerging role of anticitrulline autoimmunity in rheumatoid arthritis. J Immunol. 2005, 175: 5575-5580.
van Gaalen FA, van Aken J, Huizinga TW, Schreuder GM, Breedveld FC, Zanelli E, van Venrooij WJ, Verweij CL, Toes RE, de Vries RR: Association between HLA class II genes and autoantibodies to cyclic citrullinated peptides (CCPs) influences the severity of rheumatoid arthritis. Arthritis Rheum. 2004, 50: 2113-2121. 10.1002/art.20316.
Hida S, Miura NN, Adachi Y, Ohno N: Influence of arginine deimination on antigenicity of fibrinogen. J Autoimmun. 2004, 23: 141-150. 10.1016/j.jaut.2004.06.002.
Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E: Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J Immunol. 2003, 171: 538-541.
Dessen A, Lawrence CM, Cupo S, Zaller DM, Wiley DC: X-ray crystal structure of HLA-DR4 (DRA* 0101, DRB1*0401) complexed with a peptide from human collagen II. Immunity. 7: 473-481. 10.1016/S1074-7613(00)80369-6.
Michaelsson E, Holmdahl M, Engstrom A, Burkhardt H, Scheynius A, Holmdahl R: Macrophages, but not dendritic cells, present collagen to T cells. Eur J Immunol. 1995, 25: 2234-2241. 10.1002/eji.1830250818.
Andreis M, Hurd ER, Lospalluto J, Ziff M: Comparison of the presence of immune complexes in Felty's syndrome and rheumatoid arthritis. Arthritis Rheum. 1978, 21: 310-315. 10.1002/art.1780210304.
Yoshida M, Tsuji M, Kurosaka D, Yasuda J, Ito Y, Nishizawa T, Yamada A: Autoimmunity to citrullinated type II collagen in rheumatoid arthritis. Mod Rheumatol. 2006, 16: 276-281. 10.1007/s10165-006-0498-y.
Binstadt BA, Patel PR, Alencar H, Nigrovic PA, Lee DM, Mahmood U, Weissleder R, Mathis D, Benoist C: Particularities of the vasculature can promote the organ specificity of autoimmune attack. Nat Immunol. 2006, 7: 284-292. 10.1038/ni1306.
Baeten D, Steenbakkers PG, Rijnders AM, Boots AM, Veys EM, De Keyser F: Detection of major histocompatibility complex/human cartilage gp-39 complexes in rheumatoid arthritis synovitis as a specific and independent histologic marker. Arthritis Rheum. 2004, 50: 444-451. 10.1002/art.20012.
Saccani S, Pantano S, Natoli G: Modulation of NF-kappaB activity by exchange of dimers. Mol Cell. 2003, 11: 1563-1574. 10.1016/S1097-2765(03)00227-2.
Pettit AR, Quinn C, MacDonald KP, Cavanagh LL, Thomas G, Townsend W, Handel M, Thomas R: Nuclear localization of RelB is associated with effective antigen-presenting cell function. J Immunol. 1997, 159: 3681-3691.
Pettit AR, Weedon H, Ahern S, Zehntner S, Frazer IH, Slavotinek J, Au V, Smith MD, Thomas R: Association of clinical, radiological and synovial immunopathological response to antirheumatic treatment in rheumatoid arthritis. Rheumatology. 2001, 40: 1243-1255. 10.1093/rheumatology/40.11.1243.
Lawlor KE, Campbell IK, O'Donnell K, Wu L, Wicks IP: Molecular and cellular mediators of interleukin-1-dependent acute inflammatory arthritis. Arthritis Rheum. 2001, 44: 442-450. 10.1002/1529-0131(200102)44:2<442::AID-ANR63>3.0.CO;2-M.
Thompson AG, Pettit AR, Padmanabha J, Mansfield H, Frazer IH, Strutton GM, Thomas R: Nuclear RelB+ cells are found in normal lymphoid organs and in peripheral tissue in the context of inflammation, but not under normal resting conditions. Immunol Cell Biol. 2002, 80: 164-169. 10.1046/j.1440-1711.2002.01070.x.
Thomas R, Quinn C: Functional differentiation of dendritic cells in rheumatoid arthritis: role of CD86 in the synovium. J Immunol. 1996, 156: 3074-3086.
Thomas R, Lipsky PE: Dendritic cells: origin and differentiation. Stem Cells. 1996, 14: 196-206.
Highton J, Kean A, Hessian PA, Thomson J, Rietveld J, Hart DN: Cells expressing dendritic cell markers are present in the rheumatoid nodule. J Rheumatol. 2000, 27: 339-346.
Summers KL, O'Donnell JL, Heiser A, Highton J, Hart DN: Synovial fluid transforming growth factor beta inhibits dendritic cell–T lymphocyte interactions in patients with chronic arthritis. Arthritis Rheum. 1999, 42: 507-518. 10.1002/1529-0131(199904)42:3<507::AID-ANR16>3.0.CO;2-Y.
Jansen A, Delarch-Homo F, Hooijkaas H, Leenen PJ, Dardenne M, Drexhage HA: Immunohistochemical characterization of monocytes–macrophages and dendritic cells involved in the initiation of the insulitis and B-cell destruction in NOD mice. Diabetes. 1994, 43: 667-675. 10.2337/diabetes.43.5.667.
Dahlen E, Dawe K, Ohlsson L, Hedlund G: Dendritic cells and macrophages are the first and major producers of TNF-alpha in pancreatic islets in the nonobese diabetic mouse. J Immunol. 1998, 160: 3585-3593.
Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, Yoshikawa H, Nagata S: Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. 2006, 443: 998-1002. 10.1038/nature05245.
Takemura S, Braun A, Crowson C, Kurtin PJ, Cofield RH, O'Fallon WM, Goronzy JJ, Weyand CM: Lymphoid neogenesis in rheumatoid synovitis. J Immunol. 2001, 167: 1072-1080.
Lee Y, Chin RK, Christiansen P, Sun Y, Tumanov AV, Wang J, Chervonsky AV, Fu YX: Recruitment and activation of naive T cells in the islets by lymphotoxin beta receptor-dependent tertiary lymphoid structure. Immunity. 2006, 25: 499-509. 10.1016/j.immuni.2006.06.016.
Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, et al: In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 2002, 17: 211-220. 10.1016/S1074-7613(02)00365-5.
Fan L, Reilly CR, Luo Y, Dorf ME, Lo D: Cutting edge: ectopic expression of the chemokine TCA4/SLC is sufficient to trigger lymphoid neogenesis. J Immunol. 2000, 164: 3955-3959.
Nanki T, Urasaki Y, Imai T, Nishimura M, Muramoto K, Kubota T, Miyasaka N: Inhibition of fractalkine ameliorates murine collagen-induced arthritis. J Immunol. 2004, 173: 7010-7016.
van Lieshout AW, van der Voort R, le Blanc LM, Roelofs MF, Schreurs BW, van Riel PL, Adema GJ, Radstake TR: Novel insights in the regulation of CCL18 secretion by monocytes and dendritic cells via cytokines, toll-like receptors and rheumatoid synovial fluid. BMC Immunol. 2006, 7: 23-10.1186/1471-2172-7-23.
Paleolog EM, Hunt M, Elliott MJ, Feldmann M, Maini RN, Woody JN: Deactivation of vascular endothelium by monoclonal anti-tumor necrosis factor alpha antibody in rheumatoid arthritis. Arthritis Rheum. 1996, 39: 1082-1091. 10.1002/art.1780390703.
Harding B, Knight SC: The distribution of dendritic cells in the synovial fluids of patients with arthritis. Clin Exp Immunol. 1986, 63: 594-600.
Van Krinks CH, Matyszak MK, Gaston JS: Characterization of plasmacytoid dendritic cells in inflammatory arthritis synovial fluid. Rheumatology (Oxford). 2004, 43: 453-460. 10.1093/rheumatology/keh115.
Jahnsen FL, Lund-Johansen F, Dunne JF, Farkas L, Haye R, Brandtzaeg P: Experimentally induced recruitment of plasmacytoid (CD123high) dendritic cells in human nasal allergy. J Immunol. 2000, 165: 4062-4068.
Thomas R, MacDonald KPA, Pettit AR, Cavanagh LL, Padmanabha J, Zehntner S: Dendritic cells and the pathogenesis of rheumatoid arthritis. J Leukoc Biol. 1999, 66: 286-292.
Muller JR, Siebenlist U: Lymphotoxin beta receptor induces sequential activation of distinct NF-kappa B factors via separate signaling pathways. J Biol Chem. 2003, 278: 12006-12012. 10.1074/jbc.M210768200.
Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G: Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991, 10: 4025-4031.
Campbell IK, Gerondakis S, O'Donnell K, Wicks IP: Distinct roles for the NF-kappaB1 (p50) and c-Rel transcription factors in inflammatory arthritis. J Clin Invest. 2000, 105: 1799-1806.
Tak PP, Gerlag DM, Aupperle KR, van de Geest DA, Overbeek M, Bennett BL, Boyle DL, Manning AM, Firestein GS: Inhibitor of nuclear factor kappaB kinase beta is a key regulator of synovial inflammation. Arthritis Rheum. 2001, 44: 1897-1907. 10.1002/1529-0131(200108)44:8<1897::AID-ART328>3.0.CO;2-4.
Izumi KM, Kieff ED: The Epstein–Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc Natl Acad Sci USA. 1997, 94: 12592-12597. 10.1073/pnas.94.23.12592.
Yang SR, Chida AS, Bauter MR, Shafiq N, Seweryniak K, Maggirwar SB, Kilty I, Rahman I: Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol. 2006, 291: L46-L57. 10.1152/ajplung.00241.2005.
Brand JM, Frohn C, Cziupka K, Brockmann C, Kirchner H, Luhm J: Prolactin triggers proinflammatory immune responses in peripheral immune cells. Eur Cytokine Netw. 2004, 15: 99-104.
Pai S, O'Sullivan BJ, Jabbar IA, Peng J, Connoly G, Khanna R, Thomas R: Nasopharyngeal carcinoma-associated Epstein–Barr virus encoded oncogene latent membrane protein 1 potentiates regulatory T cell function. Immunol Cell Biol. 2007, 85: 370-377. 10.1038/sj.icb.7100046.
Quinn MA, Conaghan PG, O'Connor PJ, Karim Z, Greenstein A, Brown A, Brown C, Fraser A, Jarret S, Emery P: Very early treatment with infliximab in addition to methotrexate in early, poor-prognosis rheumatoid arthritis reduces magnetic resonance imaging evidence of synovitis and damage, with sustained benefit after infliximab withdrawal: results from a twelve-month randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2005, 52: 27-35. 10.1002/art.20712.
Palanki MS: Inhibitors of AP-1 and NF-kappa B mediated transcriptional activation: therapeutic potential in autoimmune diseases and structural diversity. Curr Med Chem. 2002, 9: 219-227.
Tomita T, Takeuchi E, Tomita N, Morishita R, Kaneko M, Yamamoto K, Nakase T, Seki H, Kato K, Kaneda Y, et al: Suppressed severity of collagen-induced arthritis by in vivo transfection of nuclear factor kappaB decoy oligodeoxynucleotides as a gene therapy. Arthritis Rheum. 1999, 42: 2532-2542. 10.1002/1529-0131(199912)42:12<2532::AID-ANR5>3.0.CO;2-2.
Dessein P, Joffe B, Veller M, Stevens B, Tobias M, Reddi K, Stanwix A: Traditional and nontraditional cardiovascular risk factors are associated with atherosclerosis in rheumatoid arthritis. J Rheumatol. 2005, 32: 435-442.
Park YB, Ahn CW, Choi HK, Lee SH, In BH, Lee HC, Nam CM, Lee SK: Atherosclerosis in rheumatoid arthritis: morphologic evidence obtained by carotid ultrasound. Arthritis Rheum. 2002, 46: 1714-1719. 10.1002/art.10359.
Vaudo G, Marchesi S, Gerli R, Allegrucci R, Giordano A, Siepi D, Pirro M, Shoenfeld Y, Schillaci G, Mannarino E: Endothelial dysfunction in young patients with rheumatoid arthritis and low disease activity. Ann Rheum Dis. 2004, 63: 31-35. 10.1136/ard.2003.007740.
Del Rincon I, Williams K, Stern MP, Freeman GL, O'Leary DH, Escalante A: Association between carotid atherosclerosis and markers of inflammation in rheumatoid arthritis patients and healthy subjects. Arthritis Rheum. 2003, 48: 1833-1840. 10.1002/art.11078.
Ridker PM, Hennekens CH, Buring JE, Rifai N: C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000, 342: 836-843. 10.1056/NEJM200003233421202.
Hansson GK, Libby P: The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006, 6: 508-519. 10.1038/nri1882.
Willerson J, Ridker P: Inflammation as a cardiovascular risk factor. Circulation. 2004, 109 (21 Suppl 1): II-2-I-10. 10.1161/01.CIR.0000129535.04194.38.
Van Doornum S, McColl G, Wicks I: Accelerated atherosclerosis. An extraarticulatr feature of rheumatoid arthritis?. Arthritis Rheum. 2002, 46: 862-873. 10.1002/art.10089.
Murray CJ, Lopez AD: Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet. 1997, 349: 1436-1442. 10.1016/S0140-6736(96)07495-8.
Ozmen J, Bobryshev Y, Lord R, Ashwell K: Identification of dendritic cells in aortic atherosclerotic lesions in rats with diet-induced hypercholesteraemia. Histol Histopathol. 2002, 17: 223-237.
Bobryshev Y, Lord R: Mapping of vascular dendritic cells in atherosclerotic arteries suggests their involvement in local immune-inflammatory reactions. Cardiovasc Res. 1998, 37: 799-810. 10.1016/S0008-6363(97)00229-0.
Wick G, Romen M, Amberger A, Metzler B, Mayr M, Falkensammer G, Xu Q: Atherosclerosis, autoimmunity, and vascular-associated lymphoid tissue. FASEB J. 1997, 11: 1199-1207.
Jongstra-Bilen J, Haidari M, Zhu S-N, Chen M, Guha D, Cybulsky M: Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J Exp Med. 2006, 203: 2073-2083. 10.1084/jem.20060245.
Ranjit S, Dazhu L, Qiutang Z, Yibo F, Yushu L, Xiang W, Shen C, Yuan T: Differentiation of dendritic cells in monocyte cultures isolated from patients with unstable angina. Int J Cardiol. 2004, 97: 551-555. 10.1016/j.ijcard.2004.05.022.
Bergholm R, Leirisalo-Repo M, Vehkavaara S, Makimattila S, Taskinen M, Yki-Jarvinen H: Impaired responsiveness to NO in newly diagnosed patients with rheumatoid arthritis. Arterioscler Thromb Vasc Biol. 2002, 22: 1637-1641. 10.1161/01.ATV.0000033516.73864.4E.
Hürlimann D, Forster A, Noll G, Enseleit F, Chenevard R, Distler O, Béchir M, Spieker L, Neidhart M, Michel B, et al: Anti-tumor necrosis factor-alpha treatment improves endothelial function in patients with rheumatoid arthritis. Circulation. 2002, 106: 2184-2187. 10.1161/01.CIR.0000037521.71373.44.
Xu XH, Shah PK, Faure E, Equils O, Thomas L, Fishbein MC, Luthringer D, Xu XP, Rajavashisth TB, Yano J, et al: Toll-like receptor-4 is expressed by macrophages in murine and human lipid-rich atherosclerotic plaques and upregulated by oxidized LDL. Circulation. 2001, 104: 3103-3108.
Miller YI, Viriyakosol S, Binder CJ, Feramisco JR, Kirkland TN, Witztum JL: Minimally modified LDL binds to CD14, induces macrophage spreading via TLR4/MD-2, and inhibits phagocytosis of apoptotic cells. J Biol Chem. 2003, 278: 1561-1568. 10.1074/jbc.M209634200.
Edfeldt K, Swedenborg J, Hansson GK, Yan ZQ: Expression of toll-like receptors in human atherosclerotic lesions: a possible pathway for plaque activation. Circulation. 2002, 105: 1158-1161.
Pober J, Collins T, Gimbrone M, Libby P, Reiss C: Inducible expression of class II major histocompatibility complex antigens and the immunogenicity of vascular endothelium. Transplantation. 1986, 41: 141-146. 10.1097/00007890-198602000-00001.
de Boer OJ, van der Wal AC, Houtkamp MA, Ossewaarde JM, Teeling P, Becker AE: Unstable atherosclerotic plaques contain T-cells that respond to Chlamydia pneumoniae. Cardiovasc Res. 2000, 48: 402-408. 10.1016/S0008-6363(00)00195-4.
Stemme S, Faber B, Holm J, Wiklund O, Witztum JL, Hansson GK: T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc Natl Acad Sci USA. 1995, 92: 3893-3897. 10.1073/pnas.92.9.3893.
Situnayake R, Kitas G: Dyslipidemia and rheumatoid arthritis. Ann Rheum Dis. 1997, 56: 341-342.
Perrin-Cocon L, Coutant F, Agaugue S, Deforges S, Andre P, Lotteau V: Oxidized low-density lipoprotein promotes mature dendritic cell transition from differentiating monocyte. J Immunol. 2001, 167: 3785-3791.
Mandal K, Jahangiri M, Xu Q: Autoimmune mechanisms of atherosclerosis. Handb Exp Pharmacol. 2005, 170: 723-743.
Angeli V, Llodra J, Rong JX, Satoh K, Ishii S, Shimizu T, Fisher EA, Randolph GJ: Dyslipidemia associated with atherosclerotic disease systemically alters dendritic cell mobilization. Immunity. 2004, 21: 561-574. 10.1016/j.immuni.2004.09.003.
Heliövaara M, Aho K, Aromaa A, Knekt P, Reunanen A: Smoking and risk of rheumatoid arthritis. J Rheumatol. 1993, 20: 1830-1835.
Maradit-Kremers H, Nicola PJ, Crowson CS, Ballman KV, Gabriel SE: Cardiovascular death in rheumatoid arthritis: a population-based study. Arthritis Rheum. 2005, 52: 722-732. 10.1002/art.20878.
Ueno H, Pradhan S, Schlessel D, Hirasawa H, Sumpio BE: Nicotine enhances human vascular endothelial cell expression of ICAM-1 and VCAM-1 via protein kinase C, p38 mitogen-activated protein kinase, NF-kappaB, and AP-1. Cardiovasc Toxicol. 2006, 6: 39-50. 10.1385/CT:6:1:39.
Lau PP, Li L, Merched AJ, Zhang AL, Ko KW, Chan L: Nicotine induces proinflammatory responses in macrophages and the aorta leading to acceleration of atherosclerosis in low-density lipoprotein receptor(-/-) mice. Arterioscler Thromb Vasc Biol. 2006, 26: 143-149. 10.1161/01.ATV.0000193510.19000.10.
Heeschen C, Jang J, Weis M, Pathak A, Kaji S, Hu R, Tsao P, Johnson F, Cooke JP: Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat Med. 2001, 7: 833-839. 10.1038/89961.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Lutzky, V., Hannawi, S. & Thomas, R. Cells of the synovium in rheumatoid arthritis. Dendritic cells. Arthritis Res Ther 9, 219 (2007). https://doi.org/10.1186/ar2200
Published:
DOI: https://doi.org/10.1186/ar2200