
Abstract
Cell therapy, pioneered for the treatment of malignancies in the form of bone marrow transplantation, has subsequently been tested and successfully employed in autoimmune diseases. Autologous haemopoietic stem cell transplantation (HSCT) has become a curative option for conditions with very poor prognosis such as severe forms of scleroderma, multiple sclerosis, and lupus, in which targeted therapies have little or no effect. The refinement of the conditioning regimens has virtually eliminated transplant-related mortality, thus making HSCT a relatively safe choice. Although HSCT remains a nonspecific approach, the knowledge gained in this field has led to the identification of new avenues. In fact, it has become evident that the therapeutic efficacy of HSCT cannot merely be the consequence of a high-dose immuno-suppression, but rather the result of a resetting of the abnormal immune regulation underlying autoimmune conditions. The identification of professional and nonprofessional immunosuppressive cells and their biological properties is generating a huge interest for their clinical exploitation. Regulatory T cells, found abnormal in several autoimmune diseases, have been proposed as central to achieve long-term remissions. Mesenchymal stem cells of bone marrow origin have more recently been shown not only to be able to differentiate into multiple tissues, but also to exert a potent antiproliferative effect that results in the inhibition of immune responses and prolonged survival of haemopoietic stem cells. All of these potential resources clearly need to be investigated at the preclinical level but support a great deal of enthusiasm for cell therapy of autoimmune diseases.
Similar content being viewed by others
Rationale for cell therapy for autoimmune diseases
Chronic inflammatory autoimmune diseases (AD) impart a massive burden on health services world wide. Efforts to define new targeted therapies have met with considerable success [1], yet these approaches are expensive and none of the new-generation biological agents consistently lead to prolonged periods of drug-free remission [2, 3].
Therapeutic strategies have historically centred on unconditional systemic immune suppression by virtue of small molecule inhibitors or immunosuppressive agents. At one time there was optimism that biological agents that targeted T cells, such as anti-CD4 or anti-CD3, might be both safer and have more durable effects for treating patients with diseases such as rheumatoid arthritis (RA). These agents target indiscriminately, however, eliminating, or at least modulating, T cells within the pool of regulatory cells and pathogen-reactive T cells, as well as the autoreactive lymphocyte populations, and their efficacy in placebo-controlled clinical trials turned out to be disappointing [4, 5]. Therapeutic depletion of a subset of CD20-expressing B cells, which does not include long-lived autoantibody-producing plasma cells, has been more promising in a growing number of AD [6, 7], where it has become evident that durability relates to the efficiency of the depletion phase and the timing of the re-emergence of pathogenic clonotypes. Nonetheless, even with prolonged cellular depletion, extended periods of remission are the exception rather than the rule [8].
Curative therapy therefore remains a major unmet need in the management of chronic inflammatory disease because it requires resetting of immune tolerance. This would necessitate depleting the expanded pool of autoreactive T lymphocytes and B lymphocytes, retarding the process of immune senescence in the residual lymphocyte populations, restoring the integrity of regulatory networks, and, at the same time, preserving a pool of memory cells capable of responding to environmental pathogens. Since many programmes of cellular activation and differentiation are imprinted through epigenetic mechanisms [9], this process of resetting is not trivial, and is relatively refractory to external manipulation. Switching established type 1 T-helper effector responses to a type 2 response is a good example. Moreover, terminally differentiated lymphocytes and plasma cells have shortened telomeres with drastically reduced replicative capacity [10], and therefore therapeutic approaches aimed at targeting cell division are also likely to fail.
What are the realistic options for achieving a cure in substantial numbers of patients with established disease? The emerging paradigm for the treatment of chronic inflammatory diseases such as RA is early aggressive therapy with tight control of disease activity aimed at robust suppression of inflammation [11, 12]. More sophisticated manipulation of effector cell populations including antigen-presenting cells, T cells, and B cells remains a possibility, but will be limited to some extent by the re-emergence of pathogenic clones. Now that technologies for cell purification and protocols for expanding specific subsets are more advanced, there are opportunities for achieving immune homeostasis by infusion of regulatory cell populations, some of which may harbour the capacity to repair tissues at sites of inflammation. Reconstitution of the immune system is now a realistic alternative. In the present article we review and discuss the current and future prospects for such cell-based therapies.
Hematopoietic stem cell transplantation for autoimmune diseases
Hematopoietic stem cell transplantation (HSCT) is a treatment aimed at resetting the deregulated immune system of patients with severe AD [13]. Recent studies have confirmed that HSCT induces alterations of the immune system that are beyond the effects of a dose-escalating immunosuppressive approach. HSCT differs from the so-called targeted therapies in that HSCT nonspecifically targets a wide array of immuno-competent cells, and creates space for a new immunological repertoire, generated from the reinfused and/or residual hematopoietic stem cells [14].
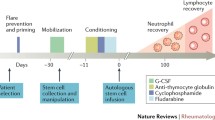
The acceptance of HSCT in the clinical arena followed successful studies in experimental animal models of AD [15] and observations of long-term remissions of AD in patients treated with HSCT for haematological malignancies [16]. Various protocols have been employed depending on the underlying disease and on individual experience of transplant centres, but most involved three consecutive steps. The first step is the mobilization of peripheral blood progenitor cells using bolus infusions of cyclophosphamide plus subcutaneous injections of granulocyte colony-stimulating factor. The second step is 'conditioning' using high-dose chemotherapy with or without lympho-depleting antibodies or total body irradiation. The final step is reinfusion of the (autologous) graft with or without prior manipulation ex vivo. The second step is the key therapeutic component, yet the initial and final steps may affect the safety and effectiveness of the procedure. For example, the addition of cyclophos-phamide to granulocyte colony-stimulating factor (first step) has been shown to diminish the risk of flares of AD [17].
With data from nearly 800 transplant cases registered, the feasibility of HSCT in human AD has now been firmly established [18]. The risks of HSCT have decreased significantly, as illustrated by the gradual drop in transplant-related mortality in patients with severe systemic sclerosis (SSc): from 17% in the first cohort of 41 patients from the European Blood and Marrow Transplant/European League Against Rheumatism (EBMT/EULAR) registry [19] to 8.7% in a more recent analysis of 65 patients (which included the 41 first cohort patients) [20], and 2.5% in the transplant arm of the ongoing Autologous Stem cell Transplantation International Scleroderma (ASTIS) trial [21], which is discussed below.
A similar trend has been observed in multiple sclerosis, the disease that accounts for most cases in the EBMT/EULAR database. Few unexpected toxicities such as lymphoma and opportunistic infections have occurred. Nevertheless, major adverse events have been observed, most notably in SSc, systemic lupus erythematosus (SLE), and juvenile idiopathic arthritis. These included respiratory insufficiency during conditioning (SSc) [22], graft failure (SLE) [17], and macrophage activation syndrome (juvenile idiopathic arthritis) [23], which accounted for the majority of transplant-related mortality in these diseases. This has led to adjustment of protocols; for example, less intense T-cell depletion in juvenile idiopathic arthritis, lung shielding with total body irradiation in SSc, and exclusion of patients with advanced disease and irreversible organ dysfunction.
There has been a striking difference between the disease targeted, the response to intervention, and toxicity, although differences in regimens and protocols may have acted as a potential confounder [24]. In general, more intense regimens were associated with higher transplant-related mortality but only a slightly lower probability of relapse. Marked improvements of disease activity, functional ability, and quality of life were seen in the majority of juvenile idiopathic arthritis patients, resulting in restoration of growth after corticosteroid therapy was discontinued [25]. Nevertheless, late relapses have occurred. In SSc, durable skin softening in patients with established generalized skin thickening has been observed in two-thirds of patients transplanted, defying conventional wisdom that fibrotic skin abnormalities are irreversible [19, 20]. In SLE patients, disease activity as measured by the Systemic Lupus Erythematosus Disease Activity Index improved markedly [26, 27]; and in those patients with pulmonary abnormalities, lung function tests showed significant improvements in the years following HSCT [28]. In contrast, most RA patients showed only transient responses, as measured by scores of disease activity, functional ability, quality of life, and rate of joint destruction, although the disease appeared more amenable to antirheumatic medication post HSCT [29, 30].
Two cases of syngeneic HSCT have been reported, one with long-lasting remission [31] and the other with rapid relapse [32], while allogeneic HSCT in another patient also resulted in a remission of RA [33]. Allogeneic HSCT offers the theoretical benefit of replacing the autoaggressive immune system and utilizing the hypothesized 'graft versus autoimmunity' effect [34] in analogy with the established curative graft versus leukaemia phenomenon, and phase I/II studies are being planned [35]. Allogeneic HSCT has become less acutely toxic due to the introduction of nonmyeloablative conditioning regimens, but the limited availability of matched donors (siblings), the risk of treatment-related toxicity (graft-versus-host disease (GvHD)), and mortality (10–30%) put constraints on the application of this modality.
Building on the experiences from pilot studies, prospective, multicentre trials have been launched in Europe and the United States to further investigate the therapeutic value of autologous HSCT in AD. The first of these, the ASTIS trial [21], started in 2001 under the auspices of the EBMT/EULAR to compare the safety and efficacy of HSCT versus conventional pulse therapy cyclophosphamide in patients with severe SSc at risk of early mortality. At the time of writing the present article (December 2006), 81 patients from 20 European centres had been randomized to either HSCT (n = 38) or to the control arm (n = 43). No unexpected toxicities or graft failures have been observed to date in either arm. One patient with heart involvement in the transplant arm died from progressive heart failure after conditioning, categorized as a probable transplant-related mortality by the independent data-monitoring committee.
The North American counterpart of the ASTIS trial, sponsored by the National Institutes of Health – the 'Scleroderma: Cyclophosphamide or Transplantation' trial – compares safety and efficacy of a different transplant regimen versus intravenous pulse therapy cyclophosphamide. The protocols of the ASTIS and 'Scleroderma: Cyclophosphamide or Transplantation' trials are matched with respect to entry criteria, study parameters, endpoints, and the control arm to facilitate future analyses [36]. Long-term follow-up of patients from these trials is crucial in order to monitor potentially late sequelae or discover delayed diverging trends in (event-free) survival. Prospective trials in SLE, multiple sclerosis, and Crohn's disease are in progress or are being planned. These trials will determine whether HSCT yields superior clinical benefit over conventional treatment, and will address open issues such as the role of post-transplant immunosuppression, the timing of HSCT, and constituents of the conditioning regimen (for example, myeloablative versus nonmyeloablative agents).
The profound immunosuppression resulting from HSCT has provided an opportunity to study the dynamics of the reconstituting immune system in relationship with the disease course. Nevertheless, it has been difficult to relate findings from immunological monitoring to the disease status, mainly because of the autologous setting of most transplants, which makes it impossible to determine the origin of mature lymphocytes after HSCT (for example, from reinfused versus residual stem cells, or expanded lymphocytes). Some patterns have emerged, however: specific autoantibodies did not always disappear after HSCT despite long-term remissions. This has been consistently observed for Scl-70 autoantibodies in SSc patients, indicating that these auto-antibodies were produced by nondividing long-lived plasma cells. Titres of IgM rheumatoid factor dropped in RA patients after HSCT, but failed to normalize and returned to pre-treatment levels before relapse. In SLE patients, antinuclear antibody and antidouble-stranded DNA antibodies disappeared in many patients after HSCT and returned to detectable levels during relapse.
HSCT has been shown not only to affect B-cell populations, but also to profoundly perturb the T-cell compartment, as illustrated by the normalization of the deregulated T-cell-receptor repertoires in multiple sclerosis [37].
In the past decade HSCT has evolved from an experimental concept to a clinically feasible and powerful therapy for selected patients with severe AD. Multicentre efforts have shifted from pilot studies and registry analyses to prospective, controlled trials. These pivotal trials will establish the position of HSCT in the treatment of AD, will possibly lead to changes of treatment paradigms, and will help us better understand pathogenetic mechanisms involved in AD.
Emerging cell therapies
The immune system has developed several strategies to control unwanted immune responses. During ontogeny, clonal deletion of autoreactive T cells is the major mechanism by which the T-cell repertoire is selected [38]. The affinity of the T-cell receptor for self-peptide–MHC ligands is the crucial parameter that drives developmental outcome in the thymus. While progenitor T cells with no affinity or high affinity for self-peptide–MHC ligands die, those with a low affinity survive. Potentially autoreactive T cells therefore persist after thymic selection and further control systems in the periphery are required to keep them in check. Although peripheral clonal deletion [39] and anergy [40] contribute to limit unwanted immune responses, active regulation is the central mechanism of immunological tolerance in adult life.
Several T-cell subsets have been identified with the ability to suppress immune responses to a variety of self-antigens and nonself-antigens. Furthermore, other nonprofessional suppressor cells have recently been shown to play important roles in chronic inflammation as well as in tumour immunosurveillance. Both professional and nonprofessional suppressor cells have potential for therapeutic exploitation and are being explored in HSCT to prevent or to treat related complications, but the suppressor cells have also been investigated in several animal models of AD. We briefly discuss the main biological features of each cell type.
Regulatory T cells
Natural regulatory T (Treg) cells are a subpopulation of thymus-derived CD4+ T cells that constitutively express the IL-2 receptor α chain (CD25) [41]. The expression of the forkhead box P3 gene product is currently the best distinctive marker for Treg cells [42]. The Treg cells play a crucial role in the maintenance of peripheral tolerance and they modulate susceptibility to autoimmune disease [41] and tumour immunity [43], as well as playing a role in the induction of transplantation tolerance [44] and in the regulation of responses to microbes. There is accumulating evidence that two subsets of CD4+CD25+ Treg cells exist: a cytokine-independent and antigen-independent naturally occurring population, and another cell type that is recruited by the cognate antigen and immunoregulatory cytokines and thus named adaptive Treg cell [45]. While the former population derives directly from the thymus, the second derives from CD4+CD25- T-cell precursors in the periphery.
Several studies have indicated that quantitative or qualitative abnormalities of Treg cells contribute to the pathogenesis of AD. Regulatory CD4+CD25+ T cells isolated from patients with active RA, although displaying an anergic phenotype, are unable to inhibit proinflammatory cytokine secretion from activated T cells and monocytes [46]. In experimental models, the depletion of Treg cells has been shown to exacerbate chronic inflammatory diseases whereas their adoptive transfer has been shown to prevent a wide range of experimental AD. Treg cells have been successfully tested in HSCT for their ability to control GvHD in animal models, whereby Treg cells have been shown to prevent GvHD or to increase host survival when GvHD has been established [47–49]. The administration of antigen-specific Treg cells generated ex vivo has similarly been shown to be very effective as sole immunosuppressive treatment at inducing specific tolerance to bone marrow allografts [50, 51].
The effect of HSCT on Treg cells is largely unknown but there is evidence that Treg cells are selectively resistant to lymphodepletion and in fact expand, while the expansion of potentially pathogenic T cells is prevented as a result of clonal competition for self-ligands [52]. The numbers of functionally active CD4+CD25+ Treg cells in juvenile idiopathic arthritis increase after HSCT, demonstrating that transplantation restores immunoregulatory mechanisms [53]. This observation is in keeping with preclinical data in a mouse model of multiple sclerosis, showing that bone marrow transplantation resulted in increased numbers of CD4+CD25high Treg cells, increased forkhead box P3 expression, a shift in T-cell epitope recognition, and a strong reduction of autoantibodies [54].
Natural killer T cells
Another T-cell subset has been identified in mice and humans with regulatory properties that exhibits natural killer cell markers. These natural killer T cells use an invariant T-cell receptor that interacts with synthetic glycolipids such as α-galactosylceramide in the context of the monomorphic CD1d antigen-presenting molecule [55]. Invariant natural killer T cells have the unique capacity to rapidly produce large amounts of both T-helper 1 and T-helper 2 cytokines, through which they play important roles in the regulation of autoimmune, allergic, antimicrobial, and antitumour immune responses [56]. The in vivo activation of invariant natural killer T cells with α-galactosylceramide has been tested with some success in animal models of various AD such as type 1 diabetes, experimental autoimmune encephalomyelitis, arthritis, and SLE [57].
Myelo-monocytes
Although cells of the monocyte lineage are generally regarded as professional antigen-presenting cells, and as such key players in the induction of immune responses, they can negatively regulate immune functions when exposed to particular environments [58]. Furthermore, specific subsets are intrinsically capable of being suppressive. The ligation of CD80/CD86 co-stimulatory molecules on certain subsets of dendritic cells induces the expression of functional indoleamine 2,3-dioxygenase (IDO-competent dendritic cells). IDO is a haeme-containing enzyme that catabolizes compounds containing indole rings, such as the essential amino acid tryptophan. IDO-competent dendritic cells can function as potent suppressors of T-cell responses both in vivo and in vitro [59].
Another monocyte subset with immunosuppressive properties has recently been identified in the tumour setting and is characterized by the expression of CD11b and Gr-1. Their accrual has been correlated with the induction of T-lymphocyte unresponsiveness to antigenic stimulation both in vitro and in vivo. CD11b+Gr-1+ cells inhibit antigen-activated T cells through a cognate-independent mechanism that involves arginase and nitric oxide synthase as the main effector pathways [60]. These cells are named myeloid suppressor cells and include a heterogeneous population ranging from immature myelomonocytic cells to terminally differentiated monocytes and granulocytes [61]. Tumours release soluble factors (that is, the cytokines granulocyte–macrophage colony-stimulating factor, granulocyte colony-stimulating factor, and IL-3) that contribute to myeloid suppressor cell recruitment [62], thus accounting in some cases for the poor outcome of tumour vaccination strategies.
Mesenchymal stem cells
Mesenchymal stem cells (MSC) are cells of stromal origin that display a variety of features of paramount relevance in the field of chronic inflammatory diseases. Several reports have shown that MSC not only differentiate into limb-bud mesodermal tissues [63], but can also acquire characteristics of cell lineages outside the limb-bud, such as endothelial cells [64], neural cells [65], and cells of the endoderm [66]. Whereas in some cases the ability of MSC to provide newly generated tissues may be ascribed to 'reprogramming' of gene expression in MSC [66], in other situations it appears that MSC act through differentiation-independent mechanisms probably mediated by soluble factors [67]. Despite the efforts to adopt a consensus definition [68], the identification of MSC based on the isolation method and the use of specific markers remains rather loose. A generally accepted profile includes their ability to differentiate in vitro into multiple lineages and the expression of CD73, CD105, and CD90 as well as the absence of haematopoietic markers [69–71]. The most well studied and accessible source of MSC is bone marrow, although even in this tissue the cells are present in a very low frequency. As well as being present in bone marrow, MSC have also been isolated from peripheral blood, fat, and synovial tissue [72].
Much interest has recently been generated by the observation that MSC may also exert a profound immunosuppressive and anti-inflammatory effect in vitro and in vivo. Such an effect is dose dependent and is exerted on T-cell responses to polyclonal stimuli [73, 74] or to their cognate peptide [75]. The inhibition does not appear to be antigen specific [73] and targets both primary and secondary T-cell responses [75]. MSC-induced T-cell suppression is not cognate dependent because it can be observed using class I-negative MSC [75] and can be exerted by MSC of different MHC origin from the target T cells [76]. The inhibitory effect of MSC is directed mainly at the level of cell proliferation as a result of cyclin D2 downregulation and p27 upregulation [77, 78], and it affects other cells of the immune system [77, 79, 80] as well as tumour cells of nonhaematopoieic origin [81].
The mechanisms underlying the immunosuppressive effect of MSC remain to be clarified, but they involve mechanisms mediated by both soluble factors [74, 82–84] and cell contact [75, 79, 82–85]. Candidate molecules are similar to those identified in other immunosuppressive cells and include IDO [84], hepatocyte growth factor, transforming growth factor beta [74], prostaglandins [86], or nitric oxide [87]. IL-10 secretion by MSC has also been attributed to play a major role in the immunosuppressive effect by determining a T-helper 1–T-helper 2 shift [79].
Such immunosuppressive activity does not seem to be spontaneous but requires MSC to be 'licensed' in an appropriate environment. It has been shown that IFNγ is a powerful inducer of such activity [88], probably via the upregulation of IDO [84]. On the contrary, TNFα can reverse the immunosuppressive activity of MSC in a collagen-induced arthritis model [89].
MSC have great potential to become a new tool in the list of cellular therapies for AD. The initial observation that MSC can exert an immunosuppressive activity in vivo by prolonging allogeneic skin grafts [73] has been confirmed in animal models of AD [90], but other workers have reported opposing results [89]. A common finding is the poor engraftment of the infused MSC, which could be attributed either to a natural contraction in their numbers or the use of allogeneic MSC. There is in fact emerging evidence that the immunosuppressive activity of MSC does not eventually avoid their rejection [91, 92]. Nevertheless, MSC have been tested in the clinical setting of HSCT whereby a patient with severe GVHD of the gut transiently benefited from the infusion of a third-party MSC from a haplo-identical donor [93]. More encouraging results are being reported [94].
Mesenchymal stem cells and autoimmune diseases
AD could be the ideal scenario in which to test the therapeutic potentials of MSC for their anti-inflammatory properties. It is still unclear, however, whether MSC derived from patients with AD display altered functions. Bone-marrow-derived MSC from RA patients, SLE patients, and SSc patients were shown to be deficient in their ability to support haematopoiesis and to exhibit features of early senescence, possibly as a result of TNFα [95]. Furthermore, the differentiation potential of MSC into adipogeneic or osteogenic lineages was reported as impaired in SSc patients [96]. Recent data similarly suggest that the MSC in these patients have a defective ability to differentiate into endothelial precursor cells (R Giacomelli, personal communication). Despite these faults, MSC derived from the bone marrow of AD patients have consistently been shown to retain their immunosuppressive activity [97]. In these experiments MSC were derived from a variety of AD, including SSc, RA, and primary Sjoegren's syndrome. The possibility of using autologous MSC for therapeutic application has become important following the demonstration in nonmyeloablated mice that allogeneic MSC are immunogenic and can be rejected [91, 92].
As already mentioned, some animal models of AD have been successfully treated by the intravenous infusion of syngeneic MSC [90, 98], as has acute GvHD (Tisato V, et al., submitted). In addition, other models of tissue damage such as ischemia-reperfusion of the kidney [67], bleomycin-induce lung fibrosis [99], and carbon tetrachloride-induced liver damage [100] appear to benefit from the early administration but not late administration of bone marrow MSC. Very limited data exist regarding the use of MSC in humans, most being derived from patients treated for acute GvHD [94] and those receiving MSC post myocardial infarct [101].
Although little is known about the long-term fate of infused MSC, a common theme is emerging that they may localize in inflamed and damaged tissue, where they might exert a protective effect [67], after which they are difficult to detect.
Most knowledge currently comes from limited animal experiments. Engraftment was estimated to be from 2.7% in the gastrointestinal tract to 0.1% in a broad range of other tissues [102]. Some MSC may transdifferentiate in situ, but probably not in sufficient numbers to be of clinical significance. One recent study of children and adults who had received either bone marrow or cord-blood transplants for various disorders looked at the origin of MSC in the bone marrow up to 192 months following transplant. Donor MSC were detected from 3 to 17 months in around 30% of the children, but never in the adults. All children had mixed chimerism and most had received a fully myeloablative regimen [103].
Acute toxicity in humans and animals appears minimal. Long-term toxicity is entirely unknown but may be negligible in view of the very low level of engraftment. There is evidence, however, that extensive in vitro passages could expose MSC to mutations, and in principle the possibility that MSC could produce tumours when transferred in vivo, as demonstrated in mice [104]. In the long term MSC might promote tumour growth either by impairing immune surveillance [83] or by facilitating tumour survival [81].
Following the preliminary successes of MSC in acute GvHD, several groups are planning similar studies for the treatment of AD that have some similarities with GvHD, whereby an underlying inflammatory, multisystem disorder compromises the function of vital organs. Unlike acute GvHD patients, AD patients are not as severely immunosuppressed and the use of autologous MSC should be considered as the first option. In vitro data suggest that, at least as far as their immuno-suppressive activity is concerned, MSC from AD patients are fully functional. The use of allogeneic, third-party MSC would probably merely resolve into a short period of 'salvage and respite', as in the case of acute GvHD. These and other issues such as optimal expansion media (for example, animal protein free, platelet lysate, autologous serum) and the source of MSC (bone marrow, cord blood, adipose tissue) will only be answered by proper randomized studies.
Conclusion
Cell therapies for AD have seen a dramatic development during the past 10 years, especially with the successful use of HSCT for otherwise untreatable forms of AD. The recent identification of cell populations of immune and nonimmune origin capable of producing profound immunosuppression is providing new strategies to narrow the specificity of the immune modulation and, as in the case of MSC, also to facilitate tissue repair.
Abbreviations
- AD:
-
= autoimmune diseases
- ASTIS:
-
= Autologous Stem cell Transplantation International Scleroderma
- EBMT/EULAR:
-
= European Blood and Marrow Transplant/European League Against Rheumatism
- GvHD:
-
= graft-versus-host disease
- HSCT:
-
= haemopoietic stem cell transplantation
- IDO:
-
= indoleamine 2,3-dioxygenase
- IFN:
-
= interferon
- IL:
-
= interleukin
- MSC:
-
= mesenchymal stem cells
- RA:
-
= rheumatoid arthritis
- SLE:
-
= systemic lupus erythematosus
- SSc:
-
= systemic sclerosis
- TNF:
-
= tumour necrosis factor
- Treg:
-
= regulatory T.
References
Feldmann M, Maini RN: Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned?. Annu Rev Immunol. 2001, 19: 163-196. 10.1146/annurev.immunol.19.1.163.
Hyrich KL, Symmons DP, Watson KD, Silman AJ: Comparison of the response to infliximab or etanercept monotherapy with the response to cotherapy with methotrexate or another disease-modifying antirheumatic drug in patients with rheumatoid arthritis: results from the British Society for Rheumatology Biologics Register. Arthritis Rheum. 2006, 54: 1786-1794. 10.1002/art.21830.
Listing J, Strangfeld A, Rau R, Kekow J, Gromnica-Ihle E, Klopsch T, Demary W, Burmester GR, Zink A: Clinical and functional remission: even though biologics are superior to conventional DMARDs overall success rates remain low – results from RABBIT, the German biologics register. Arthritis Res Ther. 2006, 8: R66-10.1186/ar1933.
Moreland LW, Bucy RP, Tilden A, Pratt PW, LoBuglio AF, Khazaeli M, Everson MP, Daddona P, Ghrayeb J, Kilgarriff C, et al: Use of a chimeric monoclonal anti-CD4 antibody in patients with refractory rheumatoid arthritis. Arthritis Rheum. 1993, 36: 307-318. 10.1002/art.1780360304.
van der Lubbe PA, Dijkmans BA, Markusse HM, Nassander U, Breedveld FC: A randomized, double-blind, placebo-controlled study of CD4 monoclonal antibody therapy in early rheumatoid arthritis. Arthritis Rheum. 1995, 38: 1097-1106. 10.1002/art.1780380812.
Leandro MJ, Edwards JC, Cambridge G, Ehrenstein MR, Isenberg DA: An open study of B lymphocyte depletion in systemic lupus erythematosus. Arthritis Rheum. 2002, 46: 2673-2677. 10.1002/art.10541.
Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T: Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004, 350: 2572-2581. 10.1056/NEJMoa032534.
Edwards JC, Cambridge G: B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat Rev Immunol. 2006, 6: 394-403. 10.1038/nri1838.
Ansel KM, Lee DU, Rao A: An epigenetic view of helper T cell differentiation. Nat Immunol. 2003, 4: 616-623. 10.1038/ni0703-616.
Koetz K, Bryl E, Spickschen K, O'Fallon WM, Goronzy JJ, Weyand CM: T cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci USA. 2000, 97: 9203-9208. 10.1073/pnas.97.16.9203.
Grigor C, Capell H, Stirling A, McMahon AD, Lock P, Vallance R, Kincaid W, Porter D: Effect of a treatment strategy of tight control for rheumatoid arthritis (the TICORA study): a single-blind randomised controlled trial. Lancet. 2004, 364: 263-269. 10.1016/S0140-6736(04)16676-2.
Goekoop-Ruiterman YP, de Vries-Bouwstra JK, Allaart CF, van Zeben D, Kerstens PJ, Hazes JM, Zwinderman AH, Ronday HK, Han KH, Westedt ML, et al: Clinical and radiographic outcomes of four different treatment strategies in patients with early rheumatoid arthritis (the BeSt study): a randomized, controlled trial. Arthritis Rheum. 2005, 52: 3381-3390. 10.1002/art.21405.
Sykes M, Nikolic B: Treatment of severe autoimmune disease by stem-cell transplantation. Nature. 2005, 435: 620-627. 10.1038/nature03728.
Roux E, Dumont-Girard F, Starobinski M, Siegrist CA, Helg C, Chapuis B, Roosnek E: Recovery of immune reactivity after T-cell-depleted bone marrow transplantation depends on thymic activity. Blood. 2000, 96: 2299-2303.
van Bekkum DW: Stem cell transplantation for autoimmune disorders. Preclinical experiments. Best Pract Res Clin Haematol. 2004, 17: 201-222. 10.1016/j.beha.2004.04.003.
Snowden JA, Kearney P, Kearney A, Cooley HM, Grigg A, Jacobs P, Bergman J, Brooks PM, Biggs JC: Long-term outcome of autoimmune disease following allogeneic bone marrow transplantation. Arthritis Rheum. 1998, 41: 453-459. 10.1002/1529-0131(199803)41:3<453::AID-ART11>3.0.CO;2-#.
Burt RK, Fassas A, Snowden J, van Laar JM, Kozak T, Wulffraat NM, Nash RA, Dunbar CE, Arnold R, Prentice G, et al: Collection of hematopoietic stem cells from patients with autoimmune diseases. Bone Marrow Transplant. 2001, 28: 1-12. 10.1038/sj.bmt.1703081.
Tyndall A, Saccardi R: Haematopoietic stem cell transplantation in the treatment of severe autoimmune disease: results from phase I/II studies, prospective randomized trials and future directions. Clin Exp Immunol. 2005, 141: 1-9. 10.1111/j.1365-2249.2005.02806.x.
Binks M, Passweg JR, Furst D, McSweeney P, Sullivan K, Besenthal C, Finke J, Peter HH, van Laar J, Breedveld FC, et al: Phase I/II trial of autologous stem cell transplantation in systemic sclerosis: procedure related mortality and impact on skin disease. Ann Rheum Dis. 2001, 60: 577-584. 10.1136/ard.60.6.577.
Farge D, Passweg J, van Laar JM, Marjanovic Z, Besenthal C, Finke J, Peter HH, Breedveld FC, Fibbe WE, Black C, et al: Autologous stem cell transplantation in the treatment of systemic sclerosis: report from the EBMT/EULAR Registry. Ann Rheum Dis. 2004, 63: 974-981. 10.1136/ard.2003.011205.
van Laar JM, Farge D, Tyndall A: Autologous Stem cell Transplantation International Scleroderma (ASTIS) trial: hope on the horizon for patients with severe systemic sclerosis [letter]. Ann Rheum Dis. 2005, 64: 1515-10.1136/ard.2005.043240.
McSweeney PA, Nash RA, Sullivan KM, Storek J, Crofford LJ, Dansey R, Mayes MD, McDonagh KT, Nelson JL, Gooley TA, et al: High-dose immunosuppressive therapy for severe systemic sclerosis: initial outcomes. Blood. 2002, 100: 1602-1610.
De Kleer IM, Brinkman DM, Ferster A, Abinun M, Quartier P, Van Der Net J, Ten Cate R, Wedderburn LR, Horneff G, Oppermann J, et al: Autologous stem cell transplantation for refractory juvenile idiopathic arthritis: analysis of clinical effects, mortality, and transplant related morbidity. Ann Rheum Dis. 2004, 63: 1318-1326. 10.1136/ard.2003.017798.
Gratwohl A, Passweg J, Bocelli-Tyndall C, Fassas A, van Laar JM, Farge D, Andolina M, Arnold R, Carreras E, Finke J, et al: Autologous hematopoietic stem cell transplantation for autoimmune diseases. Bone Marrow Transplant. 2005, 35: 869-879. 10.1038/sj.bmt.1704892.
Wulffraat NM, de Kleer IM, Prakken BJ, Kuis W: Stem cell transplantation for autoimmune disorders. Refractory juvenile idiopathic arthritis. Best Pract Res Clin Haematol. 2004, 17: 277-289. 10.1016/j.beha.2004.05.003.
Lisukov IA, Sizikova SA, Kulagin AD, Kruchkova IV, Gilevich AV, Konenkova LP, Zonova EV, Chernykh ER, Leplina OY, Sentyakova TN, et al: High-dose immunosuppression with autologous stem cell transplantation in severe refractory systemic lupus erythematosus. Lupus. 2004, 13: 89-94. 10.1191/0961203304lu491oa.
Burt RK, Traynor A, Statkute L, Barr WG, Rosa R, Schroeder J, Verda L, Krosnjar N, Quigley K, Yaung K, et al: Nonmyeloablative hematopoietic stem cell transplantation for systemic lupus erythematosus. JAMA. 2006, 295: 527-535. 10.1001/jama.295.5.527.
Traynor AE, Corbridge TC, Eagan AE, Barr WG, Liu Q, Oyama Y, Burt RK: Prevalence and reversibility of pulmonary dysfunction in refractory systemic lupus: improvement correlates with disease remission following hematopoietic stem cell transplantation. Chest. 2005, 127: 1680-1689. 10.1378/chest.127.5.1680.
Teng YK, Verburg RJ, Sont JK, van den Hout WB, Breedveld FC, van Laar JM: Long-term followup of health status in patients with severe rheumatoid arthritis after high-dose chemotherapy followed by autologous hematopoietic stem cell transplantation. Arthritis Rheum. 2005, 52: 2272-2276. 10.1002/art.21219.
Snowden JA, Passweg J, Moore JJ, Milliken S, Cannell P, Van Laar J, Verburg R, Szer J, Taylor K, Joske D, et al: Autologous hemopoietic stem cell transplantation in severe rheumatoid arthritis: a report from the EBMT and ABMTR. J Rheumatol. 2004, 31: 482-488.
McColl GJ, Szer J, Wicks IP: Sustained remission, possibly cure, of seronegative arthritis after high-dose chemotherapy and syngeneic hematopoietic stem cell transplantation [letter]. Arthritis Rheum. 2005, 52: 3322-10.1002/art.21380.
van Oosterhout M, Verburg RJ, Levarht EW, Moolenburgh JD, Barge RM, Fibbe WE, van Laar JM: High dose chemotherapy and syngeneic stem cell transplantation in a patient with refractory rheumatoid arthritis: poor response associated with persistence of host autoantibodies and synovial abnormalities. Ann Rheum Dis. 2005, 64: 1783-1785. 10.1136/ard.2004.034793.
Burt RK, Oyama Y, Verda L, Quigley K, Brush M, Yaung K, Statkute L, Traynor A, Barr WG: Induction of remission of severe and refractory rheumatoid arthritis by allogeneic mixed chimerism. Arthritis Rheum. 2004, 50: 2466-2470. 10.1002/art.20451.
Flierman R, Witteveen HJ, van der Voort EI, Huizinga TW, de Vries RR, Fibbe WE, Toes RE, van Laar JM: Control of systemic B cell-mediated autoimmune disease by nonmyeloablative conditioning and major histocompatibility complex-mismatched allogeneic bone marrow transplantation. Blood. 2005, 105: 2991-2994. 10.1182/blood-2004-09-3715.
Griffith LM, Pavletic SZ, Tyndall A, Bredeson CN, Bowen JD, Childs RW, Gratwohl A, van Laar JM, Mayes MD, Martin R, et al: Feasibility of allogeneic hematopoietic stem cell transplantation for autoimmune disease: position statement from a National Institute of Allergy and Infectious Diseases and National Cancer Institute-Sponsored International Workshop, Bethesda, MD, March 12 and 13, 2005. Biol Blood Marrow Transplant. 2005, 11: 862-870. 10.1016/j.bbmt.2005.07.009.
van Laar JM, McSweeney PA: High-dose immunosuppressive therapy and autologous progenitor cell transplantation for systemic sclerosis. Best Pract Res Clin Haematol. 2004, 17: 233-245. 10.1016/j.beha.2004.05.005.
Muraro PA, Douek DC, Packer A, Chung K, Guenaga FJ, Cassiani-Ingoni R, Campbell C, Memon S, Nagle JW, Hakim FT, et al: Thymic output generates a new and diverse TCR repertoire after autologous stem cell transplantation in multiple sclerosis patients. J Exp Med. 2005, 201: 805-816. 10.1084/jem.20041679.
Kappler JW, Roehm N, Marrack P: T cell tolerance by clonal elimination in the thymus. Cell. 1987, 49: 273-280. 10.1016/0092-8674(87)90568-X.
Webb S, Morris C, Sprent J: Extrathymic tolerance of mature T cells: clonal elimination as a consequence of immunity. Cell. 1990, 63: 1249-1256. 10.1016/0092-8674(90)90420-J.
Burkly LC, Lo D, Kanagawa O, Brinster RL, Flavell RA: T-cell tolerance by clonal anergy in transgenic mice with nonlymphoid expression of MHC class II I-E. Nature. 1989, 342: 564-566. 10.1038/342564a0.
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M: Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995, 155: 1151-1164.
Hori S, Nomura T, Sakaguchi S: Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003, 299: 1057-1061. 10.1126/science.1079490.
Shimizu J, Yamazaki S, Sakaguchi S: Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. 1999, 163: 5211-5218.
Waldmann H, Chen TC, Graca L, Adams E, Daley S, Cobbold S, Fairchild PJ: Regulatory T cells in transplantation. Semin Immunol. 2006, 18: 111-119. 10.1016/j.smim.2006.01.010.
Vukmanovic-Stejic M, Zhang Y, Cook JE, Fletcher JM, McQuaid A, Masters JE, Rustin MH, Taams LS, Beverley PC, Macallan DC, et al: Human CD4+ CD25hi Foxp3+ regulatory T cells are derived by rapid turnover of memory populations in vivo. J Clin Invest. 2006, 116: 2423-2433.
Ehrenstein MR, Evans JG, Singh A, Moore S, Warnes G, Isenberg DA, Mauri C: Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti-TNFα therapy. J Exp Med. 2004, 200: 277-285. 10.1084/jem.20040165.
Cohen JL, Trenado A, Vasey D, Klatzmann D, Salomon BL: CD4(+)CD25(+) immunoregulatory T cells: new therapeutics for graft-versus-host disease. J Exp Med. 2002, 196: 401-406. 10.1084/jem.20020090.
Trenado A, Charlotte F, Fisson S, Yagello M, Klatzmann D, Salomon BL, Cohen JL: Recipient-type specific CD4+CD25+ regulatory T cells favor immune reconstitution and control graft-versus-host disease while maintaining graft-versus-leukemia. J Clin Invest. 2003, 112: 1688-1696. 10.1172/JCI200317702.
Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, Negrin RS: CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. 2003, 9: 1144-1150. 10.1038/nm915.
Joffre O, Gorsse N, Romagnoli P, Hudrisier D, van Meerwijk JP: Induction of antigen-specific tolerance to bone marrow allo-grafts with CD4+CD25+ T lymphocytes. Blood. 2004, 103: 4216-4221. 10.1182/blood-2004-01-0005.
Gross DA, Chappert P, Leboeuf M, Monteilhet V, Van Wittenberghe L, Danos O, Davoust J: Simple conditioning with mono-specific CD4+CD25+ regulatory T cells for bone marrow engraftment and tolerance to multiple gene products. Blood. 2006, 108: 1841-1848. 10.1182/blood-2006-02-011981.
Barthlott T, Kassiotis G, Stockinger B: T cell regulation as a side effect of homeostasis and competition. J Exp Med. 2003, 197: 451-460. 10.1084/jem.20021387.
de Kleer I, Vastert B, Klein M, Teklenburg G, Arkesteijn G, Yung GP, Albani S, Kuis W, Wulffraat N, Prakken B: Autologous stem cell transplantation for autoimmunity induces immunologic self-tolerance by reprogramming autoreactive T cells and restoring the CD4+CD25+ immune regulatory network. Blood. 2006, 107: 1696-1702. 10.1182/blood-2005-07-2800.
Herrmann MM, Gaertner S, Stadelmann C, van den Brandt J, Boscke R, Budach W, Reichardt HM, Weissert R: Tolerance induction by bone marrow transplantation in a multiple sclerosis model. Blood. 2005, 106: 1875-1883. 10.1182/blood-2004-12-4607.
Godfrey DI, Kronenberg M: Going both ways: immune regulation via CD1d-dependent NKT cells. J Clin Invest. 2004, 114: 1379-1388. 10.1172/JCI200423594.
van der Vliet HJ, Molling JW, von Blomberg BM, Nishi N, Kolgen W, van den Eertwegh AJ, Pinedo HM, Giaccone G, Scheper RJ: The immunoregulatory role of CD1d-restricted natural killer T cells in disease. Clin Immunol. 2004, 112: 8-23. 10.1016/j.clim.2004.03.003.
Van Kaer L: α-Galactosylceramide therapy for autoimmune diseases: prospects and obstacles. Nat Rev Immunol. 2005, 5: 31-42. 10.1038/nri1531.
Rutella S, Danese S, Leone G: Tolerogenic dendritic cells: cytokine modulation comes of age. Blood. 2006, 108: 1435-1440. 10.1182/blood-2006-03-006403.
Mellor AL, Munn DH: IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. 2004, 4: 762-774. 10.1038/nri1457.
Zea AH, Rodriguez PC, Atkins MB, Hernandez C, Signoretti S, Zabaleta J, McDermott D, Quiceno D, Youmans A, O'Neill A, et al: Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res. 2005, 65: 3044-3048.
Serafini P, Borrello I, Bronte V: Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol. 2006, 16: 53-65. 10.1016/j.semcancer.2005.07.005.
Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, Basso G, Brombacher F, Borrello I, Zanovello P, et al: Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. 2006, 116: 2777-2790. 10.1172/JCI28828.
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR: Multilineage potential of adult human mesenchymal stem cells. Science. 1999, 284: 143-147. 10.1126/science.284.5411.143.
Reyes M, Dudek A, Jahagirdar B, Koodie L, Marker PH, Verfaillie CM: Origin of endothelial progenitors in human postnatal bone marrow. J Clin Invest. 2002, 109: 337-346. 10.1172/JCI200214327.
Woodbury D, Schwarz EJ, Prockop DJ, Black IB: Adult rat and human bone marrow stromal cells differentiate into neurons. J Neurosci Res. 2000, 61: 364-370. 10.1002/1097-4547(20000815)61:4<364::AID-JNR2>3.0.CO;2-C.
Sato Y, Araki H, Kato J, Nakamura K, Kawano Y, Kobune M, Sato T, Miyanishi K, Takayama T, Takahashi M, et al: Human mesenchymal stem cells xenografted directly to rat liver differentiated into human hepatocytes without fusion. Blood. 2005, 106: 756-763. 10.1182/blood-2005-02-0572.
Togel F, Hu Z, Weiss K, Isaac J, Lange C, Westenfelder C, Stasko T, Brown MD, Carucci JA, Euvrard S, et al: Amelioration of acute renal failure by stem cell therapy – paracrine secretion versus transdifferentiation into resident cells: administered mesenchymal stem cells protect against ischemic acute renal failure through differentiation-independent mechanisms. Am J Physiol Renal Physiol [E-pub 15 February 2005]. J Am Soc Nephrol. 2005, 16: 1153-1163. 10.1681/ASN.2005030294.
Horwitz E, Le Blanc K, Dominici M, Mueller I, Slaper-Cortenbach I, Marini F, Deans R, Krause D, Keating A: Clarification of the nomenclature for MSC: the International Society for Cellular Therapy position statement. Cytotherapy. 2005, 7: 393-395. 10.1080/14653240500319234.
Colter DC, Sekiya I, Prockop DJ: Identification of a subpopulation of rapidly self-renewing and multipotential adult stem cells in colonies of human marrow stromal cells. Proc Natl Acad Sci USA. 2001, 98: 7841-7845. 10.1073/pnas.141221698.
Barry FP, Boynton RE, Haynesworth S, Murphy JM, Zaia J: The monoclonal antibody SH-2, raised against human mesenchymal stem cells, recognizes an epitope on endoglin (CD105). Biochem Biophys Res Commun. 1999, 265: 134-139. 10.1006/bbrc.1999.1620.
Barry F, Boynton R, Murphy M, Haynesworth S, Zaia J: The SH-3 and SH-4 antibodies recognize distinct epitopes on CD73 from human mesenchymal stem cells. Biochem Biophys Res Commun. 2001, 289: 519-524. 10.1006/bbrc.2001.6013.
Jones EA, English A, Henshaw K, Kinsey SE, Markham AF, Emery P, McGonagle D: Enumeration and phenotypic characterization of synovial fluid multipotential mesenchymal progenitor cells in inflammatory and degenerative arthritis. Arthritis Rheum. 2004, 50: 817-827. 10.1002/art.20203.
Bartholomew A, Sturgeon C, Siatskas M, Ferrer K, McIntosh K, Patil S, Hardy W, Devine S, Ucker D, Deans R, et al: Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp Hematol. 2002, 30: 42-48. 10.1016/S0301-472X(01)00769-X.
Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P, Grisanti S, Gianni AM: Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002, 99: 3838-3843. 10.1182/blood.V99.10.3838.
Krampera M, Glennie S, Dyson J, Scott D, Laylor R, Simpson E, Dazzi F: Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood. 2003, 101: 3722-3729. 10.1182/blood-2002-07-2104.
Le Blanc K, Tammik L, Sundberg B, Haynesworth SE, Ringden O: Mesenchymal stem cells inhibit and stimulate mixed lymphocyte cultures and mitogenic responses independently of the major histocompatibility complex. Scand J Immunol. 2003, 57: 11-20. 10.1046/j.1365-3083.2003.01176.x.
Glennie S, Soeiro I, Dyson PJ, Lam EW, Dazzi F: Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood. 2005, 105: 2821-2827. 10.1182/blood-2004-09-3696.
Ramasamy R, Fazekasova H, Lam EW-P, Soeiro I, Lombardi G, Dazzi F: Mesenchymal stem cells inhibit dendritic cell differentiation and function by preventing entry into the cell cycle. Transplantation. 2007, 83: 71-76. 10.1097/01.tp.0000244572.24780.54.
Beyth S, Borovsky Z, Mevorach D, Liebergall M, Gazit Z, Aslan H, Galun E, Rachmilewitz J: Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unresponsiveness. Blood. 2005, 105: 2214-2219. 10.1182/blood-2004-07-2921.
Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F, Risso M, Gualandi F, Mancardi GL, Pistoia V, et al: Human mesenchymal stem cells modulate B cell functions. Blood. 2006, 107: 367-372. 10.1182/blood-2005-07-2657.
Ramasamy RL, Lam EW, Soeiro I, Tisato V, Bonnet D, Dazzi F: Mesenchymal stem cells inhibit proliferation and apoptosis of tumor cells: impact on in vivo tumor growth. Leukemia. 2007, 21: 304-310. 10.1038/sj.leu.2404489.
Tse WT, Pendleton JD, Beyer WM, Egalka MC, Guinan EC: Suppression of allogeneic T-cell proliferation by human marrow stromal cells: implications in transplantation. Transplantation. 2003, 75: 389-397. 10.1097/01.TP.0000045055.63901.A9.
Djouad F, Plence P, Bony C, Tropel P, Apparailly F, Sany J, Noel D, Jorgensen C: Immunosuppressive effect of mesenchymal stem cells favors tumor growth in allogeneic animals. Blood. 2003, 102: 3837-3844. 10.1182/blood-2003-04-1193.
Meisel R, Zibert A, Laryea M, Gobel U, Daubener W, Dilloo D: Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004, 103: 4619-4621. 10.1182/blood-2003-11-3909.
Potian JA, Aviv H, Ponzio NM, Harrison JS, Rameshwar P: Veto-like activity of mesenchymal stem cells: functional discrimination between cellular responses to alloantigens and recall antigens. J Immunol. 2003, 171: 3426-3434.
Aggarwal S, Pittenger MF: Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005, 105: 1815-1822. 10.1182/blood-2004-04-1559.
Sato K, Ozaki K, Oh I, Meguro A, Hatanaka K, Nagai T, Muroi K, Ozawa K: Nitric oxide plays a critical role in suppression of T cell proliferation by mesenchymal stem cells. Blood. 2007, 109: 228-234. 10.1182/blood-2006-02-002246.
Krampera M, Cosmi L, Angeli R, Pasini A, Liotta F, Andreini A, Santarlasci V, Mazzinghi B, Pizzolo G, Vinante F, et al: Role for interferon-gamma in the immunomodulatory activity of human bone marrow mesenchymal stem cells. Stem Cells. 2006, 24: 386-398. 10.1634/stemcells.2005-0008.
Djouad F, Fritz V, Apparailly F, Louis-Plence P, Bony C, Sany J, Jorgensen C, Noel D: Reversal of the immunosuppressive properties of mesenchymal stem cells by tumor necrosis factor alpha in collagen-induced arthritis. Arthritis Rheum. 2005, 52: 1595-1603. 10.1002/art.21012.
Zappia E, Casazza S, Pedemonte E, Benvenuto F, Bonanni I, Gerdoni E, Giunti D, Ceravolo A, Cazzanti F, Frassoni F, et al: Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood. 2005, 106: 1755-1761. 10.1182/blood-2005-04-1496.
Eliopoulos N, Stagg J, Lejeune L, Pommey S, Galipeau J: Allogeneic marrow stromal cells are immune rejected by MHC class I- and class II-mismatched recipient mice. Blood. 2005, 106: 4057-4065. 10.1182/blood-2005-03-1004.
Nauta AJ, Westerhuis G, Kruisselbrink AB, Lurvink EG, Willemze R, Fibbe WE: Donor-derived mesenchymal stem cells are immunogenic in an allogeneic host and stimulate donor graft rejection in a nonmyeloablative setting. Blood. 2006, 108: 2114-2120. 10.1182/blood-2005-11-011650.
Le Blanc K, Rasmusson I, Sundberg B, Gotherstrom C, Hassan M, Uzunel M, Ringden O: Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004, 363: 1439-1441. 10.1016/S0140-6736(04)16104-7.
Ringden O, Uzunel M, Rasmusson I, Remberger M, Sundberg B, Lonnies H, Marschall HU, Dlugosz A, Szakos A, Hassan Z, et al: Mesenchymal stem cells for treatment of therapy-resistant graft-versus-host disease. Transplantation. 2006, 81: 1390-1397. 10.1097/01.tp.0000214462.63943.14.
Papadaki HA, Marsh JC, Eliopoulos GD: Bone marrow stem cells and stromal cells in autoimmune cytopenias. Leuk Lymphoma. 2002, 43: 753-760. 10.1080/10428190290016854.
Del Papa N, Quirici N, Soligo D, Scavullo C, Cortiana M, Borsotti C, Maglione W, Comina DP, Vitali C, Fraticelli P, et al: Bone marrow endothelial progenitors are defective in systemic sclerosis. Arthritis Rheum. 2006, 54: 2605-2615. 10.1002/art.22035.
Bocelli-Tyndall C, Bracci L, Spagnoli G, Braccini A, Bouchenaki M, Ceredig R, Pistoia V, Martin I, Tyndall A: Bone marrow mesenchymal stromal cells (BM-MSCs) from healthy donors and auto-immune disease patients reduce the proliferation of autologous- and allogeneic-stimulated lymphocytes in vitro. Rheumatology (Oxford). 2007, 46: 403-408. 10.1093/rheumatology/kel267.
Zhang J, Li Y, Chen J, Cui Y, Lu M, Elias SB, Mitchell JB, Hammill L, Vanguri P, Chopp M: Human bone marrow stromal cell treatment improves neurological functional recovery in EAE mice. Exp Neurol. 2005, 195: 16-26. 10.1016/j.expneurol.2005.03.018.
Ortiz LA, Gambelli F, McBride C, Gaupp D, Baddoo M, Kaminski N, Phinney DG: Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates its fibrotic effects. Proc Natl Acad Sci USA. 2003, 100: 8407-8411. 10.1073/pnas.1432929100.
Fang B, Shi M, Liao L, Yang S, Liu Y, Zhao RC: Systemic infusion of FLK1(+) mesenchymal stem cells ameliorate carbon tetrachloride-induced liver fibrosis in mice. Transplantation. 2004, 78: 83-88. 10.1097/01.TP.0000128326.95294.14.
Rosenzweig A: Cardiac cell therapy – mixed results from mixed cells. N Engl J Med. 2006, 355: 1274-1277. 10.1056/NEJMe068172.
Devine SM, Cobbs C, Jennings M, Bartholomew A, Hoffman R: Mesenchymal stem cells distribute to a wide range of tissues following systemic infusion into nonhuman primates. Blood. 2003, 101: 2999-3001. 10.1182/blood-2002-06-1830.
Pozzi S, Lisini D, Podesta M, Bernardo ME, Sessarego N, Piaggio G, Cometa A, Giorgiani G, Mina T, Buldini B, et al: Donor multi-potent mesenchymal stromal cells may engraft in pediatric patients given either cord blood or bone marrow transplantation. Exp Hematol. 2006, 34: 934-942. 10.1016/j.exphem.2006.03.007.
Wang Y, Huso DL, Harrington J, Kellner J, Jeong DK, Turney J, McNiece IK: Outgrowth of a transformed cell population derived from normal human BM mesenchymal stem cell culture. Cytotherapy. 2005, 7: 509-519. 10.1080/14653240500363216.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
About this article
Cite this article
Dazzi, F., van Laar, J.M., Cope, A. et al. Cell therapy for autoimmune diseases . Arthritis Res Ther 9, 206 (2007). https://doi.org/10.1186/ar2128
Published:
DOI: https://doi.org/10.1186/ar2128