
Abstract
Osteoclasts are multinucleated cells of hematopoietic origin and are the primary bone resorbing cells. Numerous osteoclasts are found within the synovial tissue at sites adjacent to bone, creating resorption pits and local bone destruction. They are equipped with specific enzymes and a proton pump that enable them to degrade bone matrix and solubilize calcium, respectively. The synovial tissue of inflamed joints has a particularly high potential to accumulate osteoclasts because it harbors monocytes/macrophages, which function as osteoclast precursors, as well as cells that provide the specific molecular signals that drive osteoclast formation. Osteoclasts thus represent a link between joint inflammation and structural damage since they resorb mineralized tissue adjacent to the joint and destroy the joint architecture.
Similar content being viewed by others
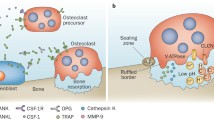


Introduction
Practically all disciplines in medicine are exposed to trends, which focus on a certain aspect of a disease while other aspects attract less interest. Rheumatology is not spared from such gradients in scientific interest. When reviewing rheumatology, it appears that research interests time-dependently switch from one topic to another, as if they represent television programs selected by the remote control of the scientists of the field. B cells comprise one example; these had been of particular interest after the detection of rheumatoid factor as an autoantibody in rheumatoid arthritis (RA) decades ago, before entering a sleep mode during the phases of intensive T cell and cytokine research. Later, B cells were rediscovered as a potential target for B-cell depleting antibodies to treat RA and have regained scientific interest. Osteoclasts have shared a similar fate, but the lag time for the rediscovery of osteoclasts in the synovial tissue took more than 100 years.
Theodor Billroth gained his honor and reputation by introducing new operating techniques that enabled the effective treatment of serious ulcers of the stomach and the rescue of patients from lethal gastrointestinal bleeding. As a typical feature of doctors during these times, Billroth was not addicted to surgery but was also interested in other fields in medicine, especially anatomy and pathology. When reading the slides of tissue sections derived from joint surgery of patients with inflammatory arthritis, he observed giant cells at the interphase between inflammatory tissue and bone. He termed these cells "bone breakers" based on the appearance of microscopic sites of bone resorption (lacunae) adjacent to these cells [1]. His contemporary chairman of pathology, Anton Weichselbaum, first described the appearance of local bone erosions in RA (at this time termed fungous synovitis because of the fungous-like appearance of the synovial inflammatory tissue) and characterized these lesions as caries of the joint ends [2]. These two findings did actually represent a very detailed and informative description of structural damage in RA: a special giant-like cell type populates chronically inflamed joints, appears to resorb the bone and creates localized skeletal defects within the inflamed joint. This finding was basically the 'end of the show' for the osteoclast in RA until its rediscovery and comeback in the late 1980s and much more detailed studies in the late 1990s. Until then, osteoclasts were not attractive enough to compete with the rise of immunology, the discovery of antibodies, the insights in cellular immunity and the rise of molecular biology in the field of immunology.
A short introduction to the osteoclasts
Osteoclasts are the primary bone resorbing cells and are essential for the remodeling of bone throughout life [3]. These giant cells are a fusion product of up to 20 single cells, also called a syncytium. Osteoclasts enable the shaping of bone architecture in early life, remodel the skeleton during adulthood and pave the way to bone loss during old age. Osteoclasts have two pivotal molecular machineries that allow them to resorb bone (Figure 1). One of these is a proton/protein pump, which is molecularly characterized as a vacuolar ATPase. This pump creates an acidic milieu between the metabolically active part of the plasma membrane of the osteoclast, the ruffled border, and the bone surface. This acidification allows the cell to solubilize calcium from the bone matrix. The second includes special matrix degrading enzymes, such as matrix metalloproteinases and cathepsins, which cleave matrix molecules such as collagen type-1 and thus remove non-mineralized substances from bone. These two specificities allow osteoclasts to invade bone and create a resorption pit, which can latter be filled up by osteoblasts synthesizing new bone matrix. Based on these attributes (polykaryons, proton pump and high enzymatic activity), osteoclasts are highly specialized cells that are particularly designed to degrade bone, a job that cannot be done by other cell types in a similar manner. Osteoclasts are not found at places where no mineralized tissue is present. Generation of these cells occurs only in the vicinity of bone, suggesting that mineralized tissue provides key differentiation signals. Osteoclasts are hematopoietic cells stemming from the monocytic lineage that undergo a series of differentiation steps until they ultimately end up as activated osteoclasts, which stick to bone and start resorbing it.

Osteoclast invading bone. Osteoclasts are multinucleated cells that resorb mineralized tissue. This image shows osteoclasts that have created a resorption lacuna. The cells are stained for tartrate-acid phosphatase (TRAP; top) and for the calcitonin receptor (CT-R; bottom).
Osteoclasts in the synovial tissue of rheumatoid arthritis
Normally, osteoclasts are found at the surface of the trabeculae of cancellous bone, where they create resorption pits. These pits are then repopulated by osteoblasts refilling these sites with new bone matrix. Osteoclasts are also active in cortical bone, which is remodeled on the basis of thin bone channels that harbor osteoclasts and osteoblasts. Besides this physiological situation, osteoclast-mediated bone resorption can be enhanced systemically, leading to increased bone resorption and bone loss as found in postmenopausal osteoporosis. Aside from these systemic changes, local accumulations of osteoclasts also trigger bone erosions. Two clinical conditions are typical examples of this form of local bone loss: skeletal metastasis of tumors and arthritis. Thus, malignant plasma cells in multiple myeloma, transformed mammary gland epithelial cells in breast cancer and inflammatory tissue in RA all induce the local formation of osteoclasts, which then triggers local bone erosion (Figure 2).

Early structural damage in arthritis. Osteoclasts are part of the synovial inflammatory tissue (arrow), which invades mineralized cartilage (double asterisk) and bone (hash symbol). The single asterisk indicates unmineralized cartilage. Arrowheads mark the bone erosion.
Synovial inflammatory tissue is the source of osteoclasts in RA. In the 1980s, Bromley and Woolley identified cells with multiple nuclei, a ruffled membrane, positive acid phosphatase and ATPase in the majority of samples of knee joints derived from patients with RA [4]. All these features are typical characteristics of osteoclasts and the authors concluded from their findings that osteoclasts populate the inflammatory synovial infiltrate. Based on their localization, Bromley and Woolley termed them 'chondroclasts' when attached to the articular cartilage rather than to subchondral bone. Final identification of these cells as osteoclasts was done in the late 1990s, when Gravallese and Goldring from Harvard Medical School molecularly characterized these cells as osteoclasts [5]. Importantly, multinucleated cells in the synovial tissue express the calcitonin receptor, which is specific to osteoclasts and only expressed in later stages of osteoclast differentiation. Expression of the calcitonin receptor was thus only found at sites where the inflammatory synovial tissue was in direct contact with the bone surface, suggesting that final differentiation to osteoclasts depends on direct contact with mineralized tissue. Apart from this late-differentiation marker, early-differentiation markers for osteoclastogenesis, such as cathepsin K and tartrate-resistant acid phosphatase, are also expressed in the synovium of RA. These markers indicate the formation of osteoclast precursors, which are mononuclear cells that have entered osteoclast differentiation and are to undergo fusion with polykaryons. These cells also accumulate at sites close to the bone surface, although they are not strictly dependent on direct contact with the bone surface. Notably, cells of the osteoclast lineage carry monocytic differentiation markers, such as CD68, identifying them as hematopoietic cells and distinguishing them from mesenchymal cells such as synovial fibroblasts. This is important since synovial fibroblasts have some characteristics that are known to be typical features of osteoclasts, for example, the expression of molecules such as cathepsin K or the vacuolar ATPase [6]. Whether this 'aberrant' expression of osteoclast differentiation markers on synovial fibroblasts allows them to resorb bone to some extent is unclear and is not supported by data from animal models with defective osteoclastogenesis [7–9]. However, these feature may contribute to the invasive properties of these cells towards articular cartilage, which is a well-described feature of synovial fibroblasts derived from the joints of patients with RA [10].
Promotion of osteoclast formation in the inflamed joint
As a typical feature of inflammatory tissue, the synovial membrane in RA contains many monocytes/macrophages, which can undergo osteoclast differentiation upon contact with the appropriate signals. It is so far unclear whether the osteoclasts develop from monocytes being trafficked to the inflammatory tissue, or whether there is a certain commitment to the osteoclast lineage before entering the joint. Monocytes entering the inflamed joint space receive signals that allow further differentiation into osteoclasts. Synovial fibroblast-like cells and activated T cells appear as the most important cell types in the synovial membrane, providing the necessary signals for monocytes to finally differentiate into osteoclasts. Synovial fibroblast-like cells are part of the so-called pannus tissue, which invades cartilage and bone and is located close to osteoclasts at sites of bone erosion. Moreover, these cells express receptor activator of nuclear factor (NF)κB ligand (RANKL) and can thus drive osteoclast formation [11, 12]. A second source of pro-osteoclastogenic factors are activated T lymphocytes, which do not only express RANKL but also produce IL-17, which supports osteoclast formation. IL-17-producing T cells (Th17 cells) have recently been described as potent stimulators of osteoclast formation [13]
Key molecules for osteoclast differentiation are macrophage colony stimulating factor and RANKL, which are both expressed locally in the synovial tissue of RA patients, enabling full differentiation of osteoclasts [11–14]. These essential molecules receive additional support from pro-inflammatory cytokines, such as tumor necrosis factor (TNF), IL-17 and IL-1, which themselves drive osteoclast formation [15–17]. RANKL is a molecule with structural homologies to TNFα, but it engages a receptor on the surface of monocytes (RANK), which drives them into osteoclastogenesis. Importantly, expression of RANKL is regulated by pro-inflammatory cytokines such as TNFα, IL-1, IL-6 and IL-17, which are abundant in the synovial membrane of RA patients and increase RANKL expression. In fact, RANKL is upregulated in experimental models of arthritis as well as human RA and psoriatic arthritis [11, 12, 18, 19], suggesting that RANKL is a key driving force of osteoclast formation in the joint. Expression of RANKL is found on mesenchymal cells such as synovial fibroblasts but also on activated T cells, which constitute a considerable proportion of inflammatory cells in the synovial membrane. Thus, there appears to be a tight interplay between inflammatory cytokines, RANKL expression and osteoclast formation in the joint.
Another key mediator for osteoclast formation is TNFα. It is not only an inducer of RANKL expression and, thus, indirectly promotes osteoclast formation but also directly binds to osteoclasts through TNFα-receptor type 1 [15, 20]. The concomitant presence of TNF thus potentiates the effect of RANKL and boosts osteoclast formation. This dual role of TNFα on osteoclast formation is an attractive explanation for the influence of TNFα on bone structure and the high efficacy of TNFα-blocking agents in the protection of bone structure in patients with RA. Signaling through TNFα-receptor type 1 involves mitogen-activated protein kinases (MAPKs) and NFkB, which then activate key transcription factors for osteoclast formation, such as c-fos of the activator protein-1 family or NFATc1. Activation of p38MAPK, for instance, is highly important for the differentiation of osteoclasts [21]. In vivo activation of p38MAPK has been observed in the inflamed synovial membrane of arthritis and deregulation of p38MAPK increases osteoclast formation and promotes a more severe destructive phenotype of arthritis [22]. In line with these molecular interactions, systemic overexpression of TNF leads to enhanced formation of osteoclasts, severe osteoporosis and erosive arthritis in mice [23]. Moreover, TNF influences the trafficking of osteoclast precursors within the body, allowing an accumulation of Cd11b-positive monocytes within lymphoid organs, such as the spleen, which then can home to the inflammatory sites [9].
The impact of osteoclast formation in inflamed joints
Since osteoclasts are found in the synovial membrane of all relevant RA animal models, such as collagen-induced arthritis, adjuvant-induced arthritis, the serum transfer model of arthritis as well as mice transgenic for human TNF, the effects of targeting these cells using genetic as well as pharmacological approaches have been intensively studied during the past years. From these models it is evident that osteoclast formation is an early and rapidly occurring process that starts right from the onset of arthritis and leads to a fast resorption of the juxta-articular bone (Figure 3) [24]. Experiments that have induced arthritis in osteoclast-free models, such as c-fos knockout mice [7] or mice deficient in either rankl or rank, have shown that osteoclasts are essential for joint destruction [8, 9]. In these models, no osteoclasts can be built up, which not only results in osteopetrosis but also in a complete protection of the joint from bone damage. Inflammatory signs of arthritis are not affected by the removal of osteoclasts, suggesting that osteoclasts are strictly linked to bone damage but not to the inflammatory features of arthritis. Very similar results were also obtained with therapeutic administration of potent bisphosphonates like zolendronic acid and osteoprotegerin, a decoy receptor and thus negative regulator of RANKL [16, 25–29]. In all models, administration of osteoprotegerin results in an almost complete protection of the articular bone and disappearance of osteoclasts from the inflamed synovium [16, 26–29]. In contrast, inflammation is not affected by the inhibition of RANKL. Thus, inhibition of osteoclasts in arthritis appears to particularly affect the onset and progression of structural damage in the joint.

Accumulation of osteoclast precursors upon induction of arthritis. Osteoclast precursors are rapidly built upon induction of arthritis in mice. This shows the junction zone as well as Haversian channels in cortical bone one day after onset of arthritis. Osteoclast precursors are stained brown for cathepsin K expression as shown in the right panels. The left panels show corresponding hematoxylin eosin stained sections. Arrows indicate bone erosion.
The role of structural damage in rheumatoid arthritis
Virtually all clinical studies on anti-inflammatory and immunomodulatory drugs for the treatment of RA have not only used clinical endpoints as efficacy measures but also radiological endpoints to define their effect on structural damage. This is attributable to the current concept that the clinical picture of RA as a debilitating joint disease is composed of chronic inflammation as well as accumulation of structural damage. This concept is reflected by the fact that bone erosion is part of the diagnostic criteria of RA and has become a valuable tool for monitoring the disease [30–34]. It soon became evident that bone erosion starts early in disease and progresses most rapidly during the first year [35]. These findings have fostered the concept that retardation, arrest or even repair of structural damage are central goals in the treatment of RA. It is also driven by the strong association between increased radiographic damage and poor functional outcome in patients with RA [33–35].
Osteoclasts and the cartilage
Structural damage in RA results from a complex process that involves bone erosion, cartilage degradation and the inflammation of the tendons close to joints. Cartilage also includes unmineralized cartilage, which builds the surface of the joint. This structure is not a target of osteoclast-mediated joint damage because osteoclasts do not affect non-mineralized tissue. In fact, investigation of samples from joint replacement surgery has revealed that osteoclasts do not invade unmineralized cartilage, suggesting that other mechanisms lead to its degradation (Figure 4). Although the molecular mechanism of degradation of the surface cartilage of the inflamed joint is not fully understood, a combination of the invasive properties of the synovial tissue and the expression of degrading enzymes such as matrix metalloproteinases are likely to be the key players in cartilage damage [10]. Underneath the surface cartilage, however, is a layer of mineralized cartilage, which connects it to the subchondral bone.

Erosion of mineralized cartilage by osteoclasts. The image shows a section through a metocarpophalangeal joint of a patient with rheumatoid arthritis. A deep invasion into mineralized cartilage (double asterisks) by synovial inflammatory tissue (arrow) harbors osteoclasts at the front of the erosion. Unmineralized surface cartilage (single asterisk) appears intact, whereas subchondral bone (hash symbol) shows resorption lacunae.
Mineralized cartilage is usually as thick as unmineralized cartilage and is particularly sensitive to osteoclast-mediated bone resorption. This is quite conceivable since the most abundant pathway of ossification, enchondral ossification, is based on the removal of mineralized cartilage and its remodeling into bone. Thus, the mineralized cartilage is actually a weak point in the joint, which allows osteoclasts to invade properly and to undermine the surface cartilage. These tunnels are then filled by inflammatory tissue, the pannus, which allows the inflammatory tissue to build up a forceps-like structure around the remaining surface cartilage, which then faces rapid degradation due to direct exposure to high levels of cytokines and matrix-degrading enzymes. Invasion into mineralized cartilage also paves the way for breaking the subchondral bone barrier, which is only a thin barrier, allowing synovial tissue to gain access to the bone marrow.
Conclusion
Osteoclasts populate the synovial membrane of patients with RA and psoriatic arthritis. As these cells are specialized to destroy mineralized tissue, osteoclasts are of central importance in the structural damage of chronic inflammatory joint disease. The unique functions of osteoclasts rely on special molecular properties that allow selective targeting of these cells by specific drugs. Osteoclasts are dependent on the presence of RANKL, which is an essential signal for osteoclast differentiation. Whether RANKL inhibition is effective at protecting human joints from inflammatory damage remains to be elucidated. Currently, the best-studied drug interfering with RANKL is a neutralizing human antibody termed denosumab (formerly AMG162), which is highly effective at suppressing bone resorption within days of administration [36, 37]. Other molecular targets of osteoclasts are cathepsin-K, a matrix degrading enzyme, the matrix-binding molecule αvβ3 integrin and the vacuolar ATPase that creates an acidic milieu to remove calcium from bone [38–40]. Whether targeting these with potential drugs would be effective in stopping structural damage in inflammatory arthritis remains to be elucidated. A recent clinical study on the structural effects of new potent bisphosphonates in RA suggests a good rationale for osteoclast inhibition in RA [41]. However, intensive therapy with very potent bisphosphonates may be necessary since osteoclast formation itself is not affected by these agents, which primarily target the resorptive properties of these cells [42].
It is important to state that therapies currently in use for the treatment of RA, such as TNF and IL-1 blockers, interfere with osteoclast formation. Particularly, TNF blockers show profound bone sparing effects in arthritis, which suggests that these agents interfere with osteoclast formation in addition to inhibiting synovial inflammation. This is in line with the observation that TNF blockers can even slow bone erosion in the absence of a major clinical response [43]. Whether other targeted therapies such as rituximab or abatacept similarly affect osteoclast formation is unknown. Both agents reduce the signs and symptoms of RA and they also show effects on joint structure. The latter effect may either be an indirect one through lowering joint inflammation or is based on a direct inhibition of the osteoclast. Current and future concepts of treatment of chronic arthritis will thus combine an optimal inhibition of inflammation as well as structural protection. Interference with osteoclasts could thus be an important tool to optimize the structural protection of joints and may allow maintaining long-term protection of joint structure during inflammatory disease.
Note
This review is part of a series on Cells of the synovium in rheumatoid arthritis edited by Gary Firestein.
Other articles in this series can be found at http://arthritis-research.com/articles/review-series.asp?series=ar_Cells
Abbreviations
- IL:
-
interleukin
- MAPK:
-
mitogen-activated protein kinase
- NF:
-
nuclear factor
- RA:
-
rheumatoid arthritis
- RANK:
-
receptor activator of NFκB
- RANKL:
-
receptor activator of NFκB ligand
- TNF:
-
tumor necrosis factor.
References
Billroth T, Von Winiwarter Al: Die allgemeine chirurgische Pathologie und Therapie in fünfzig Vorlesungen: Ein Handbuch f. Studierende u. Ärzte. 1882, Reimer, Berlin, 10
Weichselbaum A: Die feineren Veränderungen des Gelenkknorpels bei fungöser Synovitis und Karies der Gelenkenden. Archiv Pathol Anat Physiol Klin Med. 1878, 73: 461-475. 10.1007/BF01995720.
Teitelbaum SL: Bone resorption by osteoclasts. Science. 2000, 289: 1504-1508. 10.1126/science.289.5484.1504.
Bromley M, Woolley DE: Chondroclasts and osteoclasts at subchondral sites of erosion in the rheumatoid joint. Arthritis Rheum. 1984, 27: 968-975. 10.1002/art.1780270902.
Gravallese EM, Harada Y, Wang JT, Gorn AH, Thornhill TS, Goldring SR: Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am J Pathol. 1998, 152: 943-951.
Hummel KM, Petrow PK, Franz JK, Muller-Ladner U, Aicher WK, Gay RE, Bromme D, Gay S: Cysteine proteinase cathepsin K mRNA is expressed in synovium of patients with rheumatoid arthritis and is detected at sites of synovial bone destruction. J Rheumatol. 1998, 25: 1887-1894.
Redlich K, Hayer S, Ricci R, David JP, Tohidast-Akrad M, Kollias G, Steiner G, Smolen JS, Wagner EF, Schett G: Osteoclasts are essential for TNF-alpha-mediated joint destruction. J Clin Invest. 2002, 110: 1419-1427. 10.1172/JCI200215582.
Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR, Choi Y, Benoist C, Gravallese EM: TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol. 2001, 159: 1689-1699.
Li P, Schwarz EM, O'Keefe ML, Boyce BF, Xing L: RANK signaling is not required for TNF-mediated increase in CD11(hi) osteoclast precursors but is essential for mature osteoclast formation in TNFalpha-mediated inflammatory arthritis. J Bone Miner Res. 2004, 19: 207-213. 10.1359/JBMR.0301233.
Pap T, Aupperle KR, Gay S, Firestein GS, Gay RE: Invasiveness of synovial fibroblasts is regulated by p53 in the SCID mouse in vivo model of cartilage invasion. Arthritis Rheum. 2001, 44: 676-681. 10.1002/1529-0131(200103)44:3<676::AID-ANR117>3.0.CO;2-6.
Gravallese EM, Manning C, Tsay A, Naito A, Pan C, Amento E, Goldring SR: Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 2000, 43: 250-258. 10.1002/1529-0131(200002)43:2<250::AID-ANR3>3.0.CO;2-P.
Shigeyama Y, Pap T, Kunzler P, Simmen BR, Gay RE, Gay S: Expression of osteoclast differentiation factor in rheumatoid arthritis. Arthritis Rheum. 2000, 43: 2523-2530. 10.1002/1529-0131(200011)43:11<2523::AID-ANR20>3.0.CO;2-Z.
Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, Tanaka S, Kodama T, Akira S, Iwakura Y, et al: Th17 functions as an osteoclastogenic T helper cell subset that links T cell activation and bone destruction. J Exp Med. 2006, 203: 2673-2682. 10.1084/jem.20061775.
Seitz M, Loetscher P, Fey MF, Tobler A: Constitutive mRNA and protein production of macrophage colony-stimulating factor but not of other cytokines by synovial fibroblasts from rheumatoid arthritis and osteoarthritis patients. Br J Rheumatol. 1994, 33: 613-619. 10.1093/rheumatology/33.7.613.
Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL: TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000, 106: 1481-1488.
Lubberts E, van den Bersselaar L, Oppers-Walgreen B, Schwarzenberger P, Coenen-de Roo CJ, Kolls JK, Joosten LA, van den Berg WB: IL-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of NF-kappa B ligand/osteoprotegerin balance. J Immunol. 2003, 170: 2655-2662.
Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL: IL-1 mediates TNF-induced osteoclastogenesis. J Clin Invest. 2005, 115: 282-290. 10.1172/JCI200523394.
Stolina M, Adamu S, Ominsky M, Dwyer D, Asuncion F, Geng Z, Middleton S, Brown H, Pretorius J, Schett G, et al: RANKL is a marker and mediator of local and systemic bone loss in two rat models of inflammatory arthritis. J Bone Miner Res. 2005, 20: 1756-1765. 10.1359/JBMR.050601.
Ritchlin CT, Haas-Smith SA, Li P, Hicks DG, Schwarz EM: Mechanisms of TNF-alpha- and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. J Clin Invest. 2003, 111: 821-831. 10.1172/JCI200316069.
Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A: Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J Biol Chem. 2000, 275: 4858-4864. 10.1074/jbc.275.7.4858.
Matsumoto M, Sudo T, Maruyama M, Osada H, Tsujimoto M: Activation of p38 mitogen-activated protein kinase is crucial in osteoclastogenesis induced by tumor necrosis factor. FEBS Lett. 2000, 486: 23-28. 10.1016/S0014-5793(00)02231-6.
Hayer S, Steiner G, Gortz B, Reiter E, Tohidast-Akrad M, Amling M, Hoffmann O, Redlich K, Zwerina J, Skriner K, et al: CD44 is a determinant of inflammatory bone loss. J Exp Med. 2005, 201: 903-914. 10.1084/jem.20040852.
Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G: Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991, 10: 4025-4031.
Schett G, Stolina M, Bolon B, Middleton S, Adlam M, Brown H, Zhu L, Feige U, Zack DJ: Analysis of the kinetics of osteoclastogenesis in arthritic rats. Arthritis Rheum. 2005, 52: 3192-3201. 10.1002/art.21343.
Herrak P, Gortz B, Hayer S, Redlich K, Reiter E, Gasser J, Bergmeister H, Kollias G, Smolen JS, Schett G: Zoledronic acid protects against local and systemic bone loss in tumor necrosis factor-mediated arthritis. Arthritis Rheum. 2004, 50: 2327-2337. 10.1002/art.20384.
Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, et al: OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999, 397: 315-323. 10.1038/16852.
Redlich K, Hayer S, Maier A, Dunstan CR, Tohidast-Akrad M, Lang S, Turk B, Pietschmann P, Woloszczuk W, Haralambous S, et al: Tumor necrosis factor a-mediated joint destruction is inhibited by targeting osteoclasts with osteoprotegerin. Arthritis Rheum. 2002, 46: 785-792. 10.1002/art.10097.
Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, et al: Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999, 402: 304-309. 10.1038/46303.
Romas E, Gillespie MT, Martin TJ: Involvement of receptor activator of NFκB ligand and tumor necrosis factor-α in bone destruction in rheumatoid arthritis. Bone. 2002, 30: 340-346. 10.1016/S8756-3282(01)00682-2.
Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, et al: The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988, 31: 315-324. 10.1002/art.1780310302.
Sharp JT, Young DY, Bluhm GB, Brook A, Brower AC, Corbett M, Decker JL, Genant HK, Gofton JP, Goodman N, et al: How many joints in the hands and wrists should be included in a score of radiologic abnormalities used to assess rheumatoid arthritis?. Arthritis Rheum. 1985, 28: 1326-1335. 10.1002/art.1780281203.
Van der Heijde DM: Joint erosions and patients with early rheumatoid arthritis. Br J Rheumatol. 1995, 34: 74-78.
Scott DL, Pugner K, Kaarela K, Doyle DV, Woolf A, Holmes J, Hieke K: The links between joint damage and disability in rheumatoid arthritis. Rheumatology (Oxford). 2000, 39: 122-132. 10.1093/rheumatology/39.2.122.
Pincus T: Rheumatoid arthritis: disappointing long-term outcomes despite successful short-trem clinical trials. J Clin Epidemiol. 1988, 41: 1037-1041. 10.1016/0895-4356(88)90072-8.
Welsing PM, van Gestel AM, Swinkels HL, Kiemeney LA, van Riel PL: The relationship between disease activity, joint destruction, and functional capacity over the course of rheumatoid arthritis. Arthritis Rheum. 2001, 44: 2009-2017. 10.1002/1529-0131(200109)44:9<2009::AID-ART349>3.0.CO;2-L.
Bekker PJ, Holloway DL, Rasmussen AS, Murphy R, Martin SW, Leese PT, Holmes GB, Dunstan CR, DePaoli AM: A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res. 2004, 19: 1059-1066. 10.1359/JBMR.040305.
McClung MR, Lewiecki EM, Cohen SB, Bolognese MA, Woodson GC, Moffett AH, Peacock M, Miller PD, Lederman SN, Chesnut CH, et al: AMG 162 Bone Loss Study Group. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med. 2006, 354: 821-831. 10.1056/NEJMoa044459.
Yasuda Y, Kaleta J, Bromme D: The role of cathepsins in osteoporosis and arthritis: rationale for the design of new therapeutics. Adv Drug Deliv Rev. 2005, 57: 973-993. 10.1016/j.addr.2004.12.013.
Wilder RL: Integrin alpha V beta 3 as a target for treatment of rheumatoid arthritis and related rheumatic diseases. Ann Rheum Dis. 2002, ii96-99. Suppl 2
Farina C, Gagliardi S: Selective inhibition of osteoclast vacuolar H(+)-ATPase. Curr Pharm Des. 2002, 8: 2033-2048. 10.2174/1381612023393369.
Jarrett SJ, Conaghan PG, Sloan VS, Papanastasiou P, Ortmann CE, O'Connor PJ, Grainger AJ, Emery P: Preliminary evidence for a structural benefit of the new bisphosphonate zoledronic acid in early rheumatoid arthritis. Arthritis Rheum. 2006, 54: 1410-1414. 10.1002/art.21824.
Goldring SR, Gravallese EM: Bisphosphonates: environmental protection for the joint?. Arthritis Rheum. 2004, 50: 2044-2047. 10.1002/art.20383.
Lipsky PE, van der Heijde DM, St Clair EW, Furst DE, Breedveld FC, Kalden JR, Smolen JS, Weisman M, Emery P, Feldmann M, et al: Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med. 2000, 343: 1594-1602. 10.1056/NEJM200011303432202.
Acknowledgements
This work was supported by the START prize of the Austrian Research Fund and the Interdisziplinäres Zentrum für Klinische Forschungproject Erlangen (project C5) of the Deutsche Forschungsgemeinschaft (DFG).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Schett, G. Cells of the synovium in rheumatoid arthritis. Osteoclasts . Arthritis Res Ther 9, 203 (2007). https://doi.org/10.1186/ar2110
Published:
DOI: https://doi.org/10.1186/ar2110