
Abstract
This review focuses on the role of Toll-like receptors (TLRs) in lupus and on possibilities to treat lupus using TLR modulating inhibitory oligodeoxynucleotides (INH-ODNs). TLRs bridge innate and adaptive immune responses and may play an important role in the pathogenesis of systemic lupus erythematosus. Of particular interest are TLR3, -7, -8, and -9, which are localized intracellularly. These TLRs recognize single-stranded or double-stranded RNA or hypomethylated CpG-DNA. Exposure to higher order CpG-DNA ligands or to immune complexed self-RNA triggers activation of autoreactive B cells and plasmacytoid dendritic cells. INH-ODNs were recently developed that block all downstream signaling events in TLR9-responsive cells. Some of these INH-ODNs can also target TLR7 signaling pathways. Based on their preferential cell reactivity, we classify INH-ODNs into class B and class R. Class B ('broadly reactive') INH-ODNs target a broad range of TLR-expressing cells. Class R ('restricted') INH-ODNs easily form DNA duplexes or higher order structures, and are preferentially recognized by autoreactive B cells and plasmacytoid dendritic cells, rather than by non-DNA specific follicular B cells. Both classes of INH-ODNs can block animal lupus. Hence, therapeutic application of these novel INH-ODNs in human lupus, particularly class R INH-ODNs, may result in more selective and disease-specific immunosuppression.
Similar content being viewed by others

Introduction
Innate immunity (natural resistance) is recognized as the first echelon in the battle against 'microbial terror'. Microbial recognition is a complex process that depends on the integrity of the complement system and requires specialized receptors on natural killer cells and nucleotide-binding oligomerization domain (NOD) proteins [1].
An important role in innate immunity has been recently ascribed to the Toll-like receptor (TLR) family. TLRs were first identified in Drosophila as receptors that mediate protection against fungal infections [2, 3]. TLRs are surprisingly of very limited heterogeneity [4] but they have potent capacity to sense micro-organisms and alert body defense system about the presence of infectious danger. This is achieved through recognition of conserved microbial patterns such as unmethylated CpG motifs in bacterial DNA [5], single-stranded or double-stranded (ds) viral RNA, lipopolysaccharide, peptidoglycan, and bacterial flagellin (for review, see [6]). However, the role of TLRs extends beyond microbial recognition because recent evidence places them at the interface between innate and adaptive immunity [7]. This function is accomplished through coordinated upregulation of major histocompatibility complex class II and costimulatory molecules (e.g. CD40 and B7 family), resulting in much more efficient antigen presentation [8]. Furthermore, TLR-induced secretion of type I IFNs IL-6, tumor necrosis factor-α, and IL-12 directs maturation and sublineage commitment of immune cells that participate in the adaptive immune response [9, 10]. For example, type I IFNs augment antigen-specific CD4+ and CD8+ T cell responses and, in an autocrine manner, upregulate costimulatory molecule expression on dendritic cells [11–14]. When combined with IL-12, type I IFNs increase natural killer cell mediated cytotoxicity and, together with IL-6, they drive B cell differentiation and immunoglobulin secretion [15–19].
In this review we discuss recent evidence that suggests a role for TLRs in the pathogenesis of systemic autoimmunity.
Do Toll-like receptors contribute to systemic autoimmunity?
Infections frequently precede the occurrence of either organ-specific or systemic autoimmune diseases. Traditionally, this was thought to occur because of the structural cross-reactivity between the pathogenic micro-organisms and self-antigens (theory of molecular mimicry) [20]. Alternatively, microbial products may induce autoimmunity by triggering the bystander activation of the immune system, which, if not regulated, can break the anergic state, leading to the expansion of dormant autoreactive clones.
Because the innate response to microbial products primarily depends on TLRs, it is not surprising that recent studies have implicated these receptors in the pathogenesis of autoimmunity, particularly TLR3, -7, -8, and -9 [21–28]. In contrast to other TLRs, which are primarily localized at the outer cell membrane, this subgroup of TLRs is localized intracellularly. Even more importantly, these TLRs recognize nucleic acid motifs or their synthetic analogs (e.g. ds or single-stranded viral or host RNA [TLR3 and TLR7/8] or hypomethylated bacterial CpG-DNA [TLR9]). Macromolecular complexes of RNA or DNA associated with proteins, such as in chromatin, Ku autoantigen, topoisomerase, Sm/snRNP complexes, ribosomes, or in Ro/La (SS-A/SS-B) antigens, are well known targets of autoimmunity in systemic autoimmune diseases (mixed connective tissue disease, systemic lupus erythematosus [SLE], Sjögren's syndrome, and scleroderma) [29–35]. Another distinctive feature of this subfamily of TLRs is that, in humans, TLR7/8 and TLR9 exhibit a very limited cellular distribution and are detectable only in B cells and plasmacytoid dendritic cells (pDCs) [36]. In mice, in addition to B cells and pDCs, myeloid dendritic cells and macrophages also express TLR7 and TLR9 receptors and can respond to poly I:C – a synthetic ligand for TLR3 [37]. Mouse B cells, but not human B cells, also proliferate and differentiate into antibody-secreting cells when stimulated with lipopolysaccharide. This requires full assembly of the multireceptor signaling complex containing TLR4 and other accessory molecules (for review, see [2, 7]).
The question remains regarding whether TLR activation can bypass tolerance and induce autoimmunity, such that autoimmunity is a natural and recurring consequence of systemic infection. Before attempting to answer this question, one must distinguish between normal transient low-affinity autoimmune responses and self-destructive autoimmune diseases. In a normal individual short exposure to microbial products will induce only a transient activation of TLR-expressing cells, including low-affinity self-reactive B cell clones and pDCs, because these cells are under tight regulation by SOCS (suppressors of cytokine stimulation) proteins [38] and regulatory cytokines [39–41]. However, one can postulate a number a ways in which this normal low-affinity autoimmune response could progress to autoimmune disease, given the right circumstances or host factors. For example, one might suspect that TLR expression or its subcellular distribution is abnormal in mouse strains with autoimmune diseases. Whereas a loss-of-function mutation in TLR results in increased susceptibility to infections [42], at the opposite end of the spectrum, productive mutations in TLR might result in increased receptor avidity for otherwise low-affinity foreign or even unrelated self TLR ligands. Another possibility is that the inducibility or the function of SOCS proteins may be inadequate in autoimmune cells. Finally, the endogenous supply of self TLR ligands may be increased either because of the abnormal apoptotic cell death or because of the inefficient clearance of apoptotic material.
What all these possibilities have in common is the paradigm that TLR can respond to endogenous ligands (e.g. to self-DNA, heat shock proteins, minimally oxidized low density lipoprotein, and saturated fatty acids, to name but a few) [28, 43, 44] (for review, see [45]). Alternatively, presumed endogenous TLR ligands might need to undergo some type of modification before they can gain the capacity to stimulate TLRs. With respect to chromatin complexes, aberrant DNA methylation, oxidative DNA damage, differential cleavage of DNA, and histone phosphorylation are just a few modifications that may result in increased TLR stimulation and 'adjuvanticity'.
Toll-like receptors and interferons in lupus
Although at this time we cannot confirm or rule out any of the above possibilities, we know that some of the downstream events that follow TLR activation (e.g. type I and type II IFN secretion) play a well established role in the pathogenesis of systemic autoimmunity [19, 46, 47]. IFN-γ has long been considered the core cytokine in the pathogenesis of SLE [47]. Even more abundant evidence supports a role for type I IFNs. For example, similarly to IFN-γ, type I IFNs can promote isotype switching to T-helper-1-like isotypes (IgG2a, IgG2b, and IgG3 in mice) that are capable of activating the complement system [48]. This may explain the predominance of these isotypes in animal models of lupus and the well known contribution of complement-mediated activation to the tissue injury in lupus. Increased concentrations of IFN-α correlate directly with disease activity and severity in human SLE [49–52], and there are well documented case reports of patients who developed SLE-like clinical manifestations after treatment with IFN-α [53, 54]. Furthermore, IFN genetic signatures are found in SLE patients [55, 56]. Moreover, IFN-α in sera of lupus patients mediated differentiation of monocytes into potent antigen-presenting cells [13], whereas DNAse-sensitive immune complexes stimulated the FcγR-dependent production of IFN-α by pDCs [57]. A similar requirement for FcγR and TLR9 was seen in murine myeloid dendritic cells stimulated with chromatin immune complexes [58]. Finally, in vivo treatment of NZB mice with type A(D) CpG oligodeoxynucleotides (ODNs) induced abnormally high serum levels of IFN-α [59].
Where is this IFN-α coming from? Accumulating evidence suggests that pDCs (also known as natural IFN-α-producing cells) are the major, but not exclusive, producers of type I IFNs following infections with DNA viruses (e.g. mouse cytomegalovirus, herpes simplex viruses types 2 and 1) [60, 61], and following stimulation with certain types of CpG-ODNs (types A and C [62, 63]). In all of these instances, IFN-α production required TLR9. However, even though the above evidence suggests a primary role for TLR9, this is not the exclusive innate receptor that can trigger high IFN-α production in lupus mice. Indeed, in a study conducted almost 4 decades ago, injection of poly I:C into NZB/W F1 mice accelerated lupus nephritis [64]. Poly I:C closely resembles dsRNA viruses and requires a TLR3 receptor in order to induce high IFN-α secretion. A recent study conducted by Braun and coworkers [65] provided additional evidence that poly I:C can aggravate renal disease in B6lprmice, simultaneously causing polyclonal B cell activation and autoantibody secretion. In concordance with these results, NZB mice lacking the α-chain of the IFN-α/β receptor produced fewer autoantibodies and exhibited reduced renal pathology. More importantly, these mice survived longer [66]. Similar results were observed in B6lprmice, in which type I IFNs appeared to be responsible for both lymphoproliferation and for immune complex deposition in kidneys [65]. However, opposing results were observed in a recent study by Hron and Peng [67], in which MRL-Faslpr/lprmice lacking IFN receptor 1 exhibited worsened lymphoproliferation, autoantibody production and end-organ disease, suggesting that type I IFNs may play a protective role, at least in some animal models of autoimmunity.
Curiously, immature pDCs and human B cells do not express TLR3 [36]. A recent study by Pawar and coworkers [68] showed that treatment with the TLR7/8 agonist R-848 worsened immune complex glomerulonephritis in MRL-Faslpr/lprlupus mice, but it remains to be determined whether such effect required type I IFN.
Diversity of Toll-like receptor 9 ligands and their role in the pathogenesis of lupus
TLR9 is a receptor for microbial CpG-DNA [5, 69]. It recognizes a single-stranded 'CpG motif', consisting of unmethylated CpG dinucleotides flanked by particular bases [70, 71]. The GACGTT hexamer is the most stimulatory CpG motif in rodents, whereas GTCGTT motif works best in primates. However, recent studies have identified additional requirements for optimal TLR9 stimulation (e.g. the need for the 5' T and additional bases 3' to the CpG hexamer) [72–74]. Stimulation with bacterial CpG-DNA can be mimicked both in vitro and in vivo with synthetic CpG-ODNs. Based on cell type preferences and the ability to form duplexes or complex structures, CpG-ODNs can be divided into three major types. Type A(D) CpG-ODNs contain poly-G rich tails and a central palindromic CpG sequence with the natural phosphodiester (PO) backbone. These ODNs easily form secondary structures (e.g. G4 strands), or even larger aggregates [75], and preferentially stimulate pDCs in humans and in mice [62, 63]. Type B(K) CpG-ODNs have linear nonpalindromic CpG-ODN sequences, and are typically made with the nuclease-resistant phosphorothioate (PS) backbone. These ODNs are very good stimulators of both human and mouse B cells [62, 63, 71]. Finally, type C CpG-ODNs contain a 5' TCG motif and CpG-containing palindromes, allowing them to form secondary structures. These ODNs stimulate both B cells and pDCs in the human system. Interestingly, the length of the palindrome correlates well with the ability of these ODNs to activate pDCs but not B cells [76–78].
Even though stimulation with microbial CpG-DNA can induce anti-dsDNA antibodies in animal models of lupus [79, 80], and TLR9-deficient lupus mice fail to produce anti-chromatin (dsDNA) antibodies [81], it is still difficult to establish a direct link between exposure to microbial CpG-DNA and induction of lupus. Rather, microbial DNA may be involved in triggering lupus flares. Endogenous retroviruses were once considered to be etiologic factors in lupus, but strong evidence supporting this possibility is lacking [82]. Interestingly, some mainly circumstantial evidence suggests a role for chronic Epstein–Barr virus infection in the pathogenesis of SLE [83]. However, instead of considering lupus a model for the 'chronic silent infection', one must seek alternative (endogenous) ligands for the TLR9. Clearly, the prime candidate would be mammalian DNA itself. Indeed, unbound and immune-complexed host dsDNA was identified in lupus sera several decades ago [84]. However, in contrast to bacterial DNA, freshly purified unmodified mammalian DNA is not immune stimulatory. There are several possible explanations for this [85]:
-
CpG suppression: mammalian DNA has reduced numbers of stimulatory CpG motifs (one-seventh of the expected frequency) [71, 85].
-
CpG methylation: the majority of CpG motifs in mammalian DNA are methylated; however, even after complete demethylation, mammalian DNA is still poorly stimulatory.
-
Inefficient uptake: uptake of mammalian DNA into immune cells mediated via receptor-mediated endocytosis is saturable but highly inefficient, failing to deliver high enough intracytoplasmic concentrations.
-
Inhibitory DNA motifs: mammalian DNA, in contrast to bacterial DNA, contains a higher frequency of inhibitory DNA motifs, like those found in telomeric DNA (TTAGGGn) [86] or in several other regions of the mammalian genome (e.g. immunoglobulin switch regions). These motifs can block TLR9-induced activation in both a cis and trans manner.
Surprisingly, chromatin-containing immune complexes in lupus sera were capable of inducing proliferation of rheumatoid factor-specific B cells (AM14-transgenic B cells) and DNA-specific B cells (3H9-transgenic B cells) [24, 87]. This proliferation was sensitive to treatment with DNAse and required unmethylated CpG sequences. It also required a synergy between the TLR9/MyD88 pathway and B cell receptor for antigen (BCR)-mediated signaling. For example, blocking the calcineurin pathway with cyclosporine A diminished BCR-dependent proliferation, whereas treatment with either inhibitory ODNs or chloroquine blocked TLR9-mediated signaling. Although CpG-DNA fails to induce extracellular signal-regulated kinase (ERK) phosphorylation in B cells, immune complexes containing chromatin were capable of inducing ERK activation in B cells. They also stimulated IFN-α production by pDCs [58].
Taken together these data suggest that circulating DNA in lupus sera is either enriched in immunostimulatory CpG sequences, or is epigenetically modified to ensure more avid interaction between the DNA and TLR9. Indeed, DNA isolated from serum immune complexes in lupus contains disproportionably more guanosine/cytosine nucleotides than adenine/thymine residues [88]. Some data suggest that such circulating DNA may be derived from activated T cells [89, 90]. CpG islands [91] or sequestered Alu or LINE-1 sequences [92] are particularly good candidates for serving as endogenous TLR9 ligands in lupus. Another possibility is that the ratio between inhibitory DNA sequences and stimulatory CpG motifs is changed in lupus in favor of the later. Abnormal telomerase activity may be responsible for this imbalance. Finally, because BCR-mediated uptake of CpG-DNA is much more efficient than passive uptake, DNA-reactive B cells should have much higher intracellular concentrations of CpG-DNA.
IFN priming may further decrease the threshold for TLR9-mediated (and TLR7-mediated [93]) activation, allowing lower affinity TLR9 ligands to promote downstream signaling (Brummel and coworkers, unpublished data). This may be the case with dsCpG-DNA ligands because they bind to TLR9 with much lower affinity compared with single-stranded CpG-DNA [94]. Thus, complex TLR9 ligands may need prior processing by DNA-specific enzymes (e.g. helicases and/or topoisomerases) in order to bind more avidly to the TLR9 and to initiate downstream signaling. Interestingly, unprimed follicular B cells, in contrast to marginal zone (MZ)-B cells, are very poor responders to bacterial DNA and to other 'natural' TLR9 ligands. However, IFN priming may result in more efficient TLR9 signalosome formation and DNA processing, even in follicular B cells (Brummel and coworkers, unpublished data).
Toll-like receptor 9, B cell ontogeny, and the innate model of lupus
During ontogeny, B cells progress through several developmental stages, during which they appear extremely sensitive to microenvironmental influences and/or self-antigens, resulting in either positive or negative selection of B cells [95–97]. A negative selection of B cells, similarly to T cells, depends on the overall affinity of clonotypic BCR for the self-antigen. Immature B cells are enriched in self-reactive specificities, including those against DNA, and are particularly vulnerable to strong BCR signals [98]. The encounter with self-BCR ligands will typically result in clonal deletion, but a few clones may be rescued by receptor editing or by clonal anergy. A question is whether exposure to self-TLR ligands during development can rescue potentially self-reactive clones from negative selection. Under normal circumstances, the balance between the inhibitory DNA motifs and stimulatory CpG sequences in self-DNA will probably favor apoptosis in immature B cells specific for self-DNA, because the TLR9-mediated cosignal will not be generated. Although single-stranded CpG-ODNs can rescue immature non-DNA-reactive WEHI-231 cells from BCR-induced apoptotic cell death [99], similar studies performed with natural TLR9 ligands (e.g. with ds bacterial DNA) suggest the opposite (Lenert P, unpublished data). However, anti-DNA specificity of at least some B cells in 3H9 mice transgenic for the heavy chain of an anti-DNA antibody [100] suggests that exposure to self-DNA may not always result in clonal deletion, and that some DNA-specific clones may escape tolerance.
The next question is toward which differentiation pathway will surviving self-reactive B cell clones be directed – MZ/B1-B pathway or follicular B cell pathway? Some studies suggest that low-affinity autoreactive B cells will likely be diverted toward the MZ-B cell pathway [101–106]. However, despite the block in MZ-B cell development, male BXSB mice develop a systemic autoimmune disease, suggesting that under some conditions low-affinity autoreactive B cells may be redirected toward the follicular B cell pathway [107]. Although more studies are needed to better understand the role of self-TLR ligands in the rescue of self-reactive immature/transitional B cells, recent studies have revealed substantial differences in responsiveness to natural and complex CpG-DNA ligands at the level of mature B cells. For example, MZ-B cells responded vigorously to bacterial DNA and dsCpG-ODNs, as well as to G4-DNA forming type A(D) CpG-ODNs; however, highly purified follicular B cells failed to respond to any of the above ligands [108] (Brummel and coworkers, unpublished data). Enhanced responsiveness of MZ-B cells was due to the combination of increased numbers of MZ-B cells in lupus mice and their hypersensitivity to bacterial DNA and complex CpG-DNA ligands (Brummel and coworkers, unpublished data). Notably, follicular B cells from lupus mice stimulated with single-stranded type B(K) CpG-ODNs responded similarly to follicular B cells from normal mice, suggesting a normal reactive pattern of TLR9-dependent activation. Although an explanation for this differential TLR responsiveness between follicular and MZ-B cells is missing, it cannot be attributed to TLR9 expression because both cell types express TLR9 similarly [108]. We hypothesize that this lack of responsiveness may be an important safety feature of follicular B cells, protecting them from the bystander activation induced with exogenous or self-derived dsCpG-DNA.
Priming with IFNs, or BCR-mediated delivery of CpG-antigen complexes may allow antigen-specific follicular B cells to become responsive to complex TLR9 ligands, boosting B cell proliferation and promoting immunoglobulin secretion and isotype switching. Interestingly, both IFN priming and CD40-mediated activation, together with autocrine IL-6 (IL-10 in the human system [109]) secretion and BAFF-initiated signaling, are required for CpG-DNA mediated isotype switching toward complement-fixing IgG isotypes (e.g. IgG2a, IgG2b and IgG3 in mice; IgG1 and IgG3 in humans). This suggests that T cells and BAFF-producing antigen-presenting cells may play an indispensable role in lupus pathogenesis. Interestingly, IFN priming induces higher IL-6 and IL-10 secretion from CpG-DNA stimulated MZ-B cells [110]. Although MZ-B cells, at least in murine lupus, may play an important role as self-antigen processing/presenting cells for T cell activation, there is some controversy about the role of these B cells in humans and whether they may have a distinct origin. For example, a recent study showed that human splenic MZ-B cells do not express activation-induced deaminase, which is a key enzyme necessary for isotype switching, thus questioning the relationship between splenic MZ-B cells and circulating hypermutated IgM+CD27+ memory B cells [111]. Although additional studies are needed to resolve this controversy in humans, there is a clear evidence that, in mice, MZ-B cells are a distinct B cell lineage likely derived from nondividing CD21highCD23+ transitional B cells [112], which are capable of undergoing isotype switching [113].
The existence of low-affinity anti-DNA or rheumatoid factor producing B cell clones within the MZ-B cell compartment may be beneficial because these antibodies induce more efficient removal of cellular debris by phagocytes. Transient appearance of low affinity anti-single-stranded or dsDNA anti-bodies, preferentially of IgM and IgG3 isotypes, will probably accompany any systemic exposure to bacteria. At the same time, MZ-B cell derived IL-10 secretion may be important for downregulating the endotoxin-induced inflammatory cytokine storm that characterizes early stages of bacterial sepsis. In addition, IL-10 may also finely modulate the activity of antigen-presenting cells, including pDCs, for example by suppressing the IFN-α production.
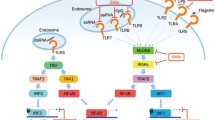
In normal circumstances the TLR9-mediated (and TLR7-mediated) activation of splenic autoreactive B cells will be self-limiting and will probably cause only minimal tissue damage. We propose a different scenario in the pathogenesis of lupus (Fig. 1). Although exogenous TLR9 (and TLR7) ligands (such as microbial DNA) may trigger lupus flares through formation of phlogogenic CpG-DNA (or RNP)/ anti-DNA (anti-RNP) immune complexes and simultaneous activation of pDCs [19], it is the continuous (or repetitive) exposure to modified self-CpG-DNA that probably drives the survival and expansion of self-DNA-reactive B cell clones. Exposure to self-hypomethylated DNA will induce secretion of low-affinity anti-DNA antibodies initially. These antibodies are likely to derive from either MZ or B1 B cells, and not from the follicular B cells. Resulting immune complexes will activate pDCs to induce type I IFN secretion. Through the positive feedback loop, this will further decrease the threshold for B cell activation, promoting survival and expansion of transitional B cell precursors with antichromatin and rheumatoid factor specificity, diverting some of these cells toward the follicular B cell pathway. Help from autoantigen (histone, Ku autoantigen, among others), activated T cells in germinal centers will further promote affinity maturation and isotype switching in autoreactive B cells. Productive rearrangement of immunoglobulin genes will eventually result in the generation of higher affinity anti-dsDNA specific B cell clones. Likewise, continuous exposure to RNA/protein complexes may favor expansion of Sm/RNP, SS-A/SS-B or anti-ribosomal clones, dependent on either TLR3 or TLR7/8. Indeed, recent studies have shown that RNA-associated autoantigens can activate autoreactive lupus B cells through BCR/TLR7 co-engagement [93]. They can also stimulate IFN-α secretion from human pDCs [114].

The innate model of lupus pathogenesis: central role of TLR-activated MZ-B cells and pDCs. Presented is a schematic overview of innate activation of MZ-B cells and pDCs with hypomethylated CpG-DNA or microbial DNA. Exposure to self CpG-DNA derived from apoptotic cells initially engages anti-dsDNA reactive low-affinity MZ-B cells. Endosomal delivery of CpG-DNA leads to TLR9-dependent and TLR9-independent activation of MZ-B cells, resulting in enhanced MHC class II, CD40, and CD86 upregulation and more efficient antigen processing/peptide presentation to histone-specific autoreactive T cells. Maturation of MZ-B cells drives secretion of anti-dsDNA antibodies, which then combine with freely circulating dsDNA to promote FcγR-dependent endogenous IFN-α secretion by pDCs (as well as activation of RF-specific B cells). IFN-α produced by pDCs, through a positive feedback loop, further enhances MZ-B cell activation and autoantibody secretion. IFN-α additionally diverts some autoreactive B cell precursors toward the follicular B cell pathway, activates T cells, and promotes development of myeloid dendritic cells (mDCs). Activated T cells direct isotype switching and affinity maturation in autoreactive B cells dependent on CD40/CD40L interaction and IFN-γ secretion. Independently of T cells, pDC-derived IFN-α can additionally help CpG-DNA activated B cells to express T-bet, a key transcription factor that induces isotype switch to complement fixing IgG2a in mice. Myeloid dendritic cell derived BAFF may further promote survival and differentiation of autoreactive B cells. Higher affinity microbial CpG-DNA released during infections directly triggers MZ-B cells and pDC activation, causing flares of lupus. ds, double stranded; IFN, interferon; MHC, major histocompatibility complex; MZ, marginal zone; pDC, plasmacytoid dendritic cell; TCR, T-cell receptor; TLR, Toll-like receptor.
Exposure to ultraviolet light and hormonal (estrogenic) influences may further increase the availability of self-TLR ligands, generating the characteristic lupus autoantibody profile [115]. Not surprisingly, inappropriate release of self-DNA from damaged tissues, such as skin, has long been suspected to underlie SLE pathogenesis [84]. Nucleosomes released from apoptotic cells in lupus are somehow enriched in self-TLR9-binding DNA sequences, possibly related to decreased DNA methyltransferase activity [89] secondary to cellular activation [116]. Interestingly, medications that can cause drug-induced lupus (e.g. hydralazine and procainamide) are capable of blocking methyltransferase activity [117], and some stimulatory effects of DNA may not require TLR9 receptor at all, as recently noticed in TLR9-deficient cells [118]. Thus, although MZ-B cells may play a crucial role in the initiation of animal lupus, it is the interaction between autoreactive T cells and IFN-primed follicular B cells that is critically important for the amplification of the autoimmune circuit, generating high affinity complement-fixing autoantibodies. Therefore, attempts to block CD40L/CD40 or CD28/B7 interactions, or type I IFNs may be promising therapeutic options for lupus.
Targeting Toll-like receptor activation of autoreactive B cells and plasmacytoid dendritic cells with inhibitory DNA sequences
Halpern and Pisetsky made the original observation that certain poly-G ODNs containing the nuclease-resistant PS backbone, but not those synthesized with the PO backbone, could block the production of IFN-γ induced by mitogens (concanavalin A), bacterial DNA, or PMA/ionomycin. In macrophages, these ODNs also blocked IL-12 secretion induced by bacterial DNA [119, 120]. However, these effects occurred at high micromolar concentrations and were not specific for the TLR9 pathway [121].
Several years later, while studying the role of CpG motifs in the immunogenicity of adenoviral vectors, Krieg and coworkers [122] discovered that certain CpG motifs, particularly those preceded by C and followed by G (e.g. the CCGG motif), were not only nonstimulatory but also could specifically block CpG-induced immune activation. It was subsequently shown that methylated CpG motifs in mammalian DNA, and ODNs containing GC flips were also capable of suppressing bacterial DNA-induced immune activation when used at high-enough concentrations [85, 120, 123, 124]. Interestingly, mammalian DNA, in addition to being heavily methylated, also contains telomeric sequences not found in bacterial DNA. Synthetic ODNs containing repetitive telomeric repeats (TTAGGGn) were capable of blocking CpG-DNA induced activation in vitro [86] and, most importantly, reduced morbidity and mortality when administered to lupus prone NZB/NZW mice [125]. Inhibitory action of telomeric repeats heavily depended on the ability of G repeats to form stable secondary structures [86] but, interestingly, purified G tetrad containing aggregates, in contrast to monomers, failed to inhibit CpG-DNA induced IL-6 by human B cells [126]. Similarly to poly-G sequences, ODNs containing repetitive TTAGGG motifs could directly block murine IFN-γ or IL-12 induced STAT (signal transducer and activator of transcription) phosphorylation, T-bet induction, and T-helper-1 differentiation [121].
Our major contribution to the field was to demonstrate the existence of short (10–15 bases long) single-stranded DNA sequences that could inhibit the action of stimulatory CpG sequences with high potency (50% inhibition at 10–20 nmol/l concentrations) [127–130]. We determined that the exact specificity requirements were located in three regions of the sequence [129, 131]. The optimal sequence contains a 5' CCT, a C-free linker four to five bases long, and a GGG(G) tail, with the order also being critical [129, 131]. Despite the differing sequence preferences for stimulation between cell types and species, the most potent inhibitory sequences for all human and mouse cell types tested were as follows: TCCTGGAGGGGAAGT (2114); TCCTGGCGGGGAAGT (2088); TCCTGGATGGGAAGT (4024); and CCTGGATGGGAAGT (4084). All of these were made with the nuclease-resistant PS backbones. Natural PO versions of the most potent inhibitory oligodeoxynucleotides (INH-ODNs) were still inhibitory, although with about 10- to 100-fold lower potency, in contrast to simple poly-G strings, which were noninhibitory [85]. We now call these inhibitors class B INH-ODNs (B for 'broadly reactive'), because they block TLR9–mediated activation in all TLR9-expressing cells. In B cells, class B INH-ODNs only block TLR9-mediated activation, and do not block proliferation, apoptosis protection, or cytokine secretion induced by anti-CD40, lipopolysaccharide, or anti-IgM+IL-4 [127].
All downstream signaling and gene induction triggered by CpG-DNA tested to date is blocked by INH-ODNs. The inhibition is competitive and reversible, suggesting avidity-driven competition for binding to a common receptor structure, probably TLR9. Indeed, one particular INH-ODN, named 2114, binds to recombinant TLR9-immunoglobulin fusion protein (Ashman RF, personal communication), and similar INH-ODNs block colocalization of CpG-DNA with TLR9 [124]. However, it remains to be determined whether higher affinity for TLR9 translates into higher potency for inhibition of TLR9-mediated signaling. Like telomeric repeats, INH-ODN 2114, when administered twice weekly, successfully prevented renal disease in lupus-prone MRL-Faslpr/lprmice [132]. Interestingly, some INH-ODNs were capable of blocking both TLR7- and TLR9-induced activation of autoreactive B cells [93], whereas the others exhibited preferential specificity for the TLR7 pathway [114].
A new classification
We propose a model classifying cell subsets by the structures they require for CpG stimulation. The first category (type I TLR9-expressing cells) includes myeloid and pDCs, macrophages and MZ-B cells in mice, and pDCs, MZ-B cells and memory B cells in humans, which can respond directly and without priming to both single-stranded and more complex CpG-DNA structures.
In contrast, the second category (type II TLR9-expressing cells) includes primarily follicular B cells in mice (and possibly colon epithelial cells and naïve B cells in humans) that directly respond to single stranded CpG-DNA, whereas they require additional co-signals to gain responsiveness to more complex CpG-DNA. We also propose that the primary abnormality in lupus occurs in type I cells, which develop a lower threshold for responding to self CpG-DNA (or self-RNP), ultimately leading to progression from low-specificity autoimmune reactivity to high-specificity pathogenic autoimmunity, or disease. We next offer the concept of class R INH-ODNs (R for 'restricted'), which specifically and primarily target type I responsive cells but spare type II TLR9-expressing cells. We discovered that when our initial INH-ODNs were modified to include partial or complete palindromic sequences, G-rich ends, 3' or 5' overhangs, or other modifications that result in the formation of more complex secondary and tertiary structures (Table 1), these INH-ODNs were much less potent in follicular B cells, which are type II TLR9-expressing cells. Because all type R INH-ODNs have at least partial dsDNA structure, they will be preferentially recognized by dsDNA-reactive, and therefore lupus-pathogenic, B cells. Furthermore, these class R INH-ODNs will be delivered to TLR9-containing endosomes in B cells, not by less efficient passive uptake but, rather, via highly efficient uptake through the B cell antigen receptors specific for dsDNA. Therefore, we propose that class R INH-ODNs might specifically target autoreactive B cells and thereby also inhibit pDC-mediated IFN-α production. This may be advantageous over the existing protocols that nonselectively suppress all lupus B cells [133, 134]. Eventually, this may result in more lupus friendly therapy, offering new hopes for lupus patients.
Conclusion
TLR signaling plays an important role in the pathogenesis of SLE. Circulating DNA/anti-DNA complexes (or RNP/anti-RNP complexes) are capable of inducing proliferation of autoreactive B cells and IFN-α secretion from pDCs. Lupus mice that lack TLR9 do not produce anti-chromatin (dsDNA) antibodies. We propose herein a model in which initial activation of MZ-B cells and pDCs with self-derived hypomethylated DNA triggers a chain of events ultimately leading to systemic autoimmune disease. TLR9-induced activation can be specifically and potently blocked with INH-ODNs when used at low nanomolar concentrations. Based on the ability of certain INH-ODNs to block selectively TLR9-induced activation of a subset of TLR9-expressing cells (e.g. MZ-B cells and pDCs) but spare activation of other TLR9+ cells (e.g. follicular B cells), we now classify INH-ODN into two categories: class B (broadly reactive) and class R (restricted reactivity). We therefore see a possible therapeutic role for class R INH-ODNs as a means to suppress disease-specific autoimmune B cell responses, while sparing non-autoimmune and protective humoral and T-cell-mediated antimicrobial immune responses. Moreover, because some INH-ODNs share specificity for both TLR7 and TLR9 pathways, whereas others preferentially block TLR7-mediated activation, intelligible application of these small molecular compounds may result in better treatment protocols for different clinical subsets of SLE patients.
Abbreviations
- BCR:
-
B cell receptor for antigen
- ds:
-
double stranded
- ERK:
-
extracellular signal-regulated kinase
- IFN:
-
interferon
- IL:
-
interleukin
- INH-ODN:
-
inhibitory oligodeoxynucleotide
- MZ:
-
marginal zone
- NOD:
-
nucleotide-binding oligomerization domain
- ODN:
-
oligodeoxynucleotide
- pDC:
-
plasmacytoid dendritic cell
- PO:
-
phosphodiester
- PS:
-
phosphorothioate
- SLE:
-
systemic lupus erythematosus
- TLR:
-
Toll-like receptor.
References
Abreu MT, Fukata M, Arditi M: TLR signaling in the gut in health and disease. J Immunol. 2005, 174: 4453-4460.
Takeda K, Kaisho T, Akira S: Toll-like receptors. Annu Rev Immunol. 2003, 21: 335-376.
Barton GM, Medzhitov R: Toll-like receptors and their ligands. Curr Top Microbiol Immunol. 2002, 270: 81-92.
Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, et al: Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA. 2004, 101: 3516-3521.
Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, et al: A toll-like receptor recognizes bacterial DNA. Nature. 2000, 408: 740-745.
Hoebe K, Janseen E, Beutler B: The interface between innate and adaptive immunity. Nat Immunol. 2004, 5: 971-974.
Akira S, Takeda K, Kaisho T: Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001, 2: 675-680.
Chu RS, Askew D, Harding C: CpG DNA switches on Th1 immunity and modulates antigen-presenting cell function. Curr Top Microbiol Immunol. 2000, 247: 199-210.
Klinman DM, Yi AK, Beaucage SL, Conover J, Krieg AM: CpG motifs present in bacterial DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12 and interferon γ. J Immunol. 1996, 93: 2879-2883.
Poeck H, Wagner M, Battiany J, Rothenfusser S, Wellisch D, Hornung V, Jahrsdorfer B, Giese T, Endres S, Hartmann G: Plasmacytoid dendritic cells, antigen, and CpG-C license human B cells for plasma cell differentiation and immunoglobulin production in the absence of T-cell help. Blood. 2004, 103: 3058-3064.
Banchereau J, Pascual V, Palucka AK: Autoimmunity through cytokine-induced dendritic cell activation. Immunity. 2004, 20: 539-550.
Banchereau J, Steinman RM: Dendritic cells and the control of immunity. Nature. 1998, 392: 245-252.
Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J: Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001, 294: 1540-1543.
Balazs M, Martin F, Zhou T, Kearney JF: Blood dendritic cells interact with splenic marginal zone B cells to initiate T-independent immune responses. Immunity. 2002, 17: 341-352.
Dubois B, Vanbervliet B, Fayette J, Massacrier C, Van Kooten C, Briere F, Banchereau J, Caux C: Dendritic cells enhance growth and differentiation of CD40-activated B lymphocytes. J Exp Med. 1997, 185: 941-951.
Dubois B, Massacrier C, Vanbervliet B, Fayette J, Briere F, Banchereau J, Caux C: Critical role of IL-12 in dendritic cell-induced differentiation of naive B lymphocytes. J Immunol. 1998, 161: 2223-2231.
Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J: Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003, 19: 225-234.
Braun D, Caramalho I, Demengeot J: IFN-α/β enhances BCR-dependent B cell responses. Int Immunol. 2002, 14: 411-419.
Ronnblom L, Alm VG: Systemic lupus erythematosus and type I interferon system. Arthritis Res Ther. 2003, 5: 68-75.
Oldstone MB: Molecular mimicry and immune-mediated diseases. FASEB J. 1998, 12: 1255-1265.
Anders HJ: A toll for lupus. Lupus. 2005, 14: 417-422.
Lenert P, Goeken A, Handwerger BS, Ashman RF: Innate immune responses in lupus-prone Palmerston North mice: differential responses to LPS and bacterial DNA/CpG oligonucleotides. J Clin Immunol. 2003, 23: 202-213.
Lenert P: Inhibitory oligodeoxynucleotides: therapeutic promise for systemic autoimmune diseases?. Clin Exp Immunol. 2005, 140: 1-10.
Leadbetter EA, Rifkin AR, Hohlbaum AM, Beaudette BC, Schlomchik MJ, Marshak-Rothstein A: Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002, 416: 603-607.
Vallin H, Perers A, Alm GV, Ronnblom L: Anti-double-stranded DNA antibodies and immunostimulatory plasmid DNA in combination mimic the endogenous IFN-α inducer in systemic lupus erythematosus. J Immunol. 1999, 163: 6306-6313.
Viau M, Zouali M: B-lymphocytes, innate immunity, and autoimmunity. Clin Immunol. 2005, 114: 17-26.
Krieg AM: CpG DNA: a pathogenic factor in systemic lupus erythematosus. J Clin Immunol. 1995, 15: 284-292.
Rifkin IR, Leadbetter EA, Busconi L, Viglianti G, Marshak-Rothstein A: Toll-like receptors, endogenous ligands, and systemic autoimmune disease. Immunol Rev. 2005, 204: 27-42.
Blank M, Shoenfeld Y: Experimental models of systemic lupus erythematosus: anti-dsDNA in murine lupus. Rheumatology. 2005, 44: 1086-1089.
Sherer Y, Gorstein A, Fritzler MJ, Shoenfeld Y: Autoantibody explosition in systemic lupus erythematosus: more than 100 different antibodies found in SLE patients. Semin Arthritis Rheum. 2004, 34: 501-537.
Mohan C, Adams S, Stanik V, Datta SK: Nucleosome: a major immunogen for the pathogenic autoantibody-inducing T cells of lupus. J Exp Med. 1993, 177: 1367-1381.
Mevorach D, Zhou JL, Song X, Elkon KB: Systemic exposure to irradiated apoptotic cells induces autoantibody production. J Exp Med. 1998, 188: 387-392.
Radic MZ, Weigert M: Genetic and structural evidence for antigen selection of anti-DNA antibodies. Annu Rev Immunol. 1994, 12: 487-520.
Tan EM: Antinuclear antibodies: diagnostic markers for autoimmune diseases and probes for cell biology. Adv Immunol. 1989, 44: 93-151.
Casciola-Rosen LA, Anhalt G, Rosen A: Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994, 179: 1317-1330.
Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G: Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002, 168: 4531-4537.
Zarember KA, Godowski PJ: Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products and cytokines. J Immunol. 2002, 168: 554-561.
Naka T, Fujimoto M, Tsutsui H, Yoshimura A: Negative regulation of cytokine and TLR signalings by SOCS and others. Adv Immunol. 2005, 87: 61-122.
Lenert P, Brummel R, Field EH, Ashman RF: TLR-9 activation of marginal zone B cells in lupus mice regulates immunity through increased IL-10 production. J Clin Immunol. 2005, 25: 29-40.
Burke F, Stagg AJ, Bedford PA, English N, Knight SC: IL-10-producing B220+ CD11c- APC in mouse spleen. J Immunol. 2004, 173: 2362-2372.
Fillatreau S, Sweenie CH, McGeachy MJ, Gray D, Anderton SM: B cells regulate autoimmunity by provision of IL-10. Nat Immunol. 2002, 3: 944-950.
Ku CL, Yang K, Bustamante J, Puel A, von Bernuth H, Santos OF, Lawrence T, Chang HH, Al-Mousa H, Picard C, et al: Inherited disorders of human Toll-like receptor signaling: immunological implications. Immunol Rev. 2005, 203: 10-20.
Johnson GB, Brunn GJ, Platt JL: Activation of mammalian Toll-like receptors by endogenous agonists. Crit Rev Immunol. 2003, 23: 15-44.
Seong SY, Matzinger P: Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004, 4: 469-478.
Vabulas RM, Wagner H, Schild H: Heat shock proteins as ligands of toll-like receptors. Curr Top Microbiol Immunol. 2002, 270: 169-184.
Seery JP: IFN-γ transgenic mice: clues to the pathogenesis of systemic lupus erythematosus?. Arthritis Res. 2000, 2: 437-440.
Theofilopoulos AN, Koundouris S, Kono DH, Lawson BR: The role of IFN-γ in systemic lupus erythematosus: a challenge to the Th1/Th2 paradigm in autoimmunity. Arthritis Res Ther. 2001, 3: 136-141.
Cerutti A, Qiao X, He B: Plasmocytoid dendritic cells and the regulation of immunoglobulin heavy chain class switching. Immunol Cell Biol. 2005, 83: 554-562.
Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL: Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med. 1979, 301: 5-8.
Preble OT, Black RJ, Friedman RM, Klippel JH, Vilcek J: Systemic lupus erythematosus: presence in human serum of an unusual acid-labile leukocyte interferon. Science. 1982, 216: 429-431.
Bengtsson A, Sturfelt G, Truedsson L, Blomberg J, Alm G, Vallin H, Ronnblom L: Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not antiretroviral antibodies. Lupus. 2000, 9: 664-671.
Ytterberg SR, Schnitzer TJ: Serum interferon levels in patients with systemic lupus erythematosus. Arthritis Rheum. 1982, 25: 401-406.
Ronnblom LE, Alm GV, Oberg KE: Possible induction of systemic lupus erythematosus by interferon-a treatment in a patient with a malignant carcinoid tumor. J Intern Med. 1990, 227: 207-210.
Ioannou Y, Isenberg DA: Current evidence for the induction of autoimmune rheumatic conditions by cytokine therapy. Arthritis Rheum. 2000, 43: 1431-1442.
Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, et al: Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. 2003, 100: 2610-2615.
Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V: Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003, 197: 711-723.
Ronnblom L, Alm GV: An etiopathogenic role for the type I IFN system in SLE. Trends Immunol. 2001, 22: 427-431.
Boule MW, Broughton C, Mackay F, Akira S, Marshak-Rothstein A, Rifkin IR: Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes. J Exp Med. 2004, 199: 1631-1640.
Lian ZX, Kikuchi K, Yang GX, Ansari AA, Ikehara S, Gershwin ME: Expansion of bone marrow IFN-alpha-producing dendritic cells in New Zealand Black [NZB] mice: high level expression of TLR9 and secretion of IFN-alpha in NZB bone marrow. J Immunol. 2004, 173: 5283-5289.
Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Briere F, Trinchieri G, Biron CA: Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J Exp Med. 2002, 195: 517-528.
Colonna M, Trinchieri G, Liu YJ: Plasmocytoid dendritic cells in immunity. Nat Immunol. 2004, 5: 1219-1226.
Krug A, Rothenfusser S, Hornung V, Jahrsdorfer B, Blackwell S, Ballas ZK, Endres S, Krieg AM, Hartmann G: Identification of CpG oligonucleotide sequences with high induction of IFN-alpha/beta in plasmocytoid dendritic cells. Eur J Immunol. 2001, 31: 2154-2163.
Verthelyi D, Ishii KJ, Gursel M, Takeshita F, Klinman DM: Human peripheral blood cells differentially recognize and respond to two distinct CpG motifs. J Immunol. 2001, 166: 2372-2377.
Carpenter DF, Steinberg AD, Schur PH, Talal N: The pathogenesis of autoimmunity in New Zealand mice. II. Acceleration of glomerulonephritis by polyinosinic-polycytidylic acid. Lab Invest. 1970, 23: 628-634.
Braun D, Geraldes P, Demengeot J: Type I Interferon controls the onset and severity of autoimmune manifestations in lpr mice. J Autoimmun. 2003, 20: 15-25.
Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, Theofilopoulos AN: Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med. 2003, 197: 777-788.
Hron JD, Peng SL: Type I IFN protects against murine lupus. J Immunol. 2004, 173: 2134-2142.
Pawar RD, Patole PS, Zecher D, Segerer S, Kretzler M, Schlondorff D, Anders HJ: Toll-like receptor-7 modulates immune complex glomerulonephritis. J Am Soc Nephrol. 2005,
Bauer S, Kirschning CJ, Hacker H, Redecke V, Hausmann S, Akira S, Wagner H, Lipford GB: Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci USA. 2001, 98: 9237-9242.
Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM: CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995, 374: 546-549.
Krieg AM: CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002, 20: 709-760.
Vollmer J, Weeratna RD, Jurk M, Samulowitz U, McCluskie MJ, Payette P, Davis HL, Schetter C, Krieg AM: Oligodeoxynucleotides lacking CpG dinucleotides mediate Toll-like receptor 9 dependent T helper type 2 biased immune stimulation. Immunology. 2004, 113: 212-223.
Marshall JD, Fearon KL, Higgins D, Hessel EM, Kanzler H, Abbate C, Yee P, Gregorio J, Cruz TD, Lizcano JO, et al: Superior activity of the type C class of ISS in vitro and in vivo across multiple species. DNA Cell Biol. 2005, 24: 63-72.
Lenert P, Goeken JA, Ashman RF: Extended sequence preferences for oligodeoxyribonucleotide activity. Immunology. 2006,
Kerkmann M, Costa LT, Richter C, Rothenfusser S, Battiany J, Hornung V, Johnson J, Englert S, Ketterer T, Heck W, et al: Spontaneous formation of nucleic acid-based nanoparticules is responsible for high interferon-α induction by CpG-A in plasmocytoid dendritic cells. J Biol Chem. 2005, 280: 8086-8093.
Marshall JD, Fearon K, Abbate C, Subramanian S, Yee P, Gregorio J, Coffman RL, Van Nest G: Identification of a novel CpG DNA class and motif that optimally stimulate B cell and plas-macytoid dendritic cell functions. J Leukoc Biol. 2003, 73: 781-792.
Hartmann G, Battiany J, Poeck H, Wagner M, Kerkmann M, Lubenow N, Rothenfusser S, Endres S: Rational design of new CpG oligonucleotides that combine B cell activation with high IFN-alpha induction in plasmacytoid dendritic cells. Eur J Immunol. 2003, 33: 1633-1641.
Vollmer J, Weeratna R, Payette P, Jurk M, Schetter C, Laucht M, Wader T, Tluk S, Liu M, Davis HL, Krieg AM: Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur J Immunol. 2004, 34: 251-262.
Gilkeson GS, Ruiz P, Pippen AM, Alexander AL, Lefkowith JB, Pisetsky DS: Modulation of renal disease in autoimmune NZB/ NZW mice by immunization with bacterial DNA. J Exp Med. 1996, 183: 1389-1397.
Anders HJ, Vielhauer V, Eis V, Linde Y, Kretzler M, de Lema GP, Strutz F, Bauer S, Rutz M, Wagner H, et al: Activation of toll-like receptor-9 induces progression of renal disease in MRL-Fas(lpr) mice. FASEB J. 2004, 18: 534-536.
Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ: Toll-like receptor 9 controls anti-DNA autoanti-body production in murine lupus. J Exp Med. 2005, 202: 321-331.
Krieg AM, Steinberg AD: Analysis of thymic endogenous retro-viral expression in murine lupus. Genetic and immune studies. J Clin Invest. 1990, 86: 809-816.
Pender MP: Infection of autoreactive B cells with EBV, causing chronic autoimmune diseases. Trends Immunol. 2003, 29: 3168-3178.
Tan EM, Schur PH, Carr RI, Kunkel HG: Deoxyribonucleic acid DNA and antibodies to DNA in the serum of patients with systemic lupus erythematosus. J Clin Invest. 1966, 45: 1732-1740.
Stacey K, Young GR, Clark F, Sester DP, Roberts TL, Naik S, Sweet MJ, Hume DA: The molecular basis for the lack of immunostimulatory activity of vertebrate DNA. J Immunol. 2003, 170: 3614-3620.
Gursel I, Gursel M, Yamada H, Ishii KJ, Takeshita F, Klinman DM: Repetitive elements in mammalian telomeres suppress bacterial DNA-induced immune activation. J Immunol. 2003, 171: 1393-1400.
Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A: Activation of autoreactive B cells by CpG dsDNA. Immunity. 2003, 19: 837-847.
Sano H, Morimoto C: DNA isolated from DNA/anti-DNA antibody immune complexes in systemic lupus erythematosus is rich in guanine-cytosine content. J Immunol. 1982, 128: 1341-1345.
Richardson B, Scheinbart L, Strahler J, Gross L, Hanash S, Johnson M: Evidence of impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 1990, 33: 1665-1673.
Richardson B: DNA methylation and autoimmune disease. Clin Immunol. 2003, 109: 72-79.
Bird AP: CpG islands as gene markers in the vertebrate nucleus. Trends Genet. 1987, 3: 342-347.
Brooks WH: Systemic lupus erythematosus and related autoimmune diseases are antigen-driven, epigenetic diseases. Med Hypotheses. 2002, 59: 736-741.
Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, et al: RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005, 202: 1171-1177.
Rutz M, Metzger J, Gellert T, Luppa P, Lipford GB, Wagner H, Bauer S: Toll-like receptor 9 binds single-stranded CpG-DNA in a sequence- and pH-dependent manner. Eur J Immunol. 2004, 34: 2541-2550.
Monroe JG, Bannish G, Fuentes-Panana EM, King LB, Sandel PC, Chung J, Sater R: Positive and negative selection during B lymphocyte development. Immunol Res. 2003, 27: 427-442.
Cancro MP, Kearney JF: B cell positive selection: road map to the primary repertoire?. J Immunol. 2004, 173: 15-19.
Grimaldi CM, Hicks R, Diamond B: B cell selection and susceptibility to autoimmunity. J Immunol. 2005, 174: 1775-1781.
Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC: Predominant autoantibody production by early human B cell precursors. Science. 2003, 301: 1374-1377.
Yi AK, Peckham DW, Ashman RF, Krieg AM: CpG DNA rescues B cells from apoptosis by activating NFkappaB and preventing mitochondrial membrane potential disruption via a chloro-quine-sensitive pathway. Int Immunol. 1999, 11: 2015-2024.
Erikson J, Radic MZ, Camper SA, Hardy RR, Carmack C, Weigert M: Expression of anti-DNA immunoglobulin transgenes in non-autoimmune mice. Nature. 1991, 349: 331-334.
Bendelac A, Bonneville M, Kearney JF: Autoreactivity by design: innate B and T lymphocytes. Nat Rev Immunol. 2001, 1: 177-186.
Martin F, Kearney JF: Marginal-zone B cells. Nat Rev Immunol. 2002, 2: 323-335.
Chen X, Martin F, Forbush KA, Perlmutter RM, Kearney JF: Evidence of selection of a population of multireactive B cells into the splenic marginal zone. Int Immunol. 1997, 9: 27-41.
Kenny JJ, Rezanka LJ, Lustig A, Fischer RT, Yoder J, Marshall S, Longo DL: Autoreactive B cells escape clonal deletion by expressing multiple antigen receptors. J Immunol. 2000, 164: 4111-4119.
Li Y, Li H, Weigert M: Autoreactive B cells in the marginal zones that express dual receptors. J Exp Med. 2002, 195: 181-188.
Grimaldi CM, Michael DJ, Diamond B: Cutting edge: expansion and activation of a population of autoreactive marginal zone B cells in a model of estrogen-induced lupus. J Immunol. 2001, 167: 1886-1890.
Moll T, Martinez-Soria E, Santiago-Raber ML, Amano H, Pihlgren-Bosch M, Marinkovic D, Izui S: Differential activation of anti-erythrocyte and anti-DNA autoreactive B lymphocytes by the Yaa mutation. J Immunol. 2005, 174: 702-709.
Brummel R, Lenert P: Activation of marginal zone B cells from lupus mice with type A(D) CpG-oligodeoxynucleotides. J Immunol. 2005, 174: 2429-2434.
He B, Qiao X, Cerutti A: CpG DNA induces IgG class switch DNA recombination by activating human B cells through an innate pathway that requires TLR9 and cooperates with IL-10. J Immunol. 2004, 173: 4479-4491.
Lenert P, Brummel R, Field EH, Ashman RF: TLR-9 activation of marginal zone B cells in lupus mice regulates immunity through increased IL-10 production. J Clin Immunol. 2005, 25: 29-40.
Willenbrock K, Jungnickel B, Hansmann ML, Kuppers R: Human splenic marginal zone B cells lack expression of activation-induced cytidine deaminase. Eur J Immunol. 2005, 35: 3002-3007.
Srivastava B, Quinn WJ, Hazard K, Erikson J, Allman D: Characterization of marginal zone B cell precursors. J Exp Med. 2005, 202: 1225-1234.
Gatto D, Ruedl C, Odermatt B, Bachmann MF: Rapid response of marginal zone B cells to viral particles. J Immunol. 2004, 173: 4308-4316.
Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, Chang B, Duramad O, Coffman RL: Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med. 2005, 202: 1131-1139.
Peeva E, Zouali M: Spotlight on the role of hormonal factors in the emergence of autoreactive B-lymphocytes. Immunol Lett. 2005, 101: 123-143.
Qiao B, Wu J, Chu YW, Wang Y, Wang DP, Wu HS, Xiong SD: Induction of systemic lupus erythematosus-like syndrome in syngeneic mice by immunization with activated lymphocyte-derived DNA. Rheumatology. 2005, 44: 1108-1114.
Richardson BC: Role of DNA methylation in the regulation of cell function: autoimmunity, aging and cancer. J Nutr. 2002, 2401S-2405S. Suppl
Yasuda K, Yu P, Kirschning CJ, Schlatter B, Schmitz F, Heit A, Bauer S, Hochrein H, Wagner H: Endosomal translocation of vertebrate DNA activates dendritic cells via TLR9-dependent and independent pathways. J Immunol. 2005, 174: 6129-6136.
Halpern MD, Pisetsky DS: In vitro inhibition of murine IFNγ production by phosphorothioate deoxyguanosine oligomers. Immunopharmacology. 1995, 29: 47-52.
Pisetsky DS, Reich CF: Inhibition of murine macrophage IL-12 production by natural and synthetic DNA. Clin Immunol. 2000, 96: 198-204.
Shirota H, Gursel M, Klinman DM: Suppressive oligodeoxynucleotides inhibit Th1 differentiation by blocking IFN-gamma-and IL-12-mediated signaling. J Immunol. 2004, 173: 5002-5007.
Krieg AM, Wu T, Weeratna R, Efler SM, Love-Homan L, Yang L, Yi AK, Short D, Davis HL: Sequence motifs in adenoviral DNA block immune activation by stimulatory CpG motifs. Proc Natl Acad Sci USA. 1998, 95: 12631-12636.
Chen Y, Lenert P, Weeratna R, McCluskie M, Wu T, Davis HL, Krieg AM: Identification of methylated CpG motifs as inhibitors of the immune stimulatory CpG motifs. Gene Ther. 2001, 8: 1024-1032.
Klinman DM, Zeuner R, Yamada H, Gursel M, Currie D, Gursel I: Regulation of CpG-induced immune activation by suppressive oligodeoxynucleotides. Ann NY Acad Sci. 2003, 1002: 112-123.
Dong L, Ito S, Ishii KJ, Klinman DM: Suppressive oligodeoxynucleotides delay the onset of glomerulonephritis and prolong survival in lupus-prone NZB × NZW mice. Arthritis Rheum. 2005, 52: 651-658.
Duramad O, Fearon KL, Chang B, Chan JH, Gregorio J, Coffman RL, Barrat FJ: Inhibitors of TLR-9 act on multiple cell subsets in mouse and man in vitro and prevent death in vivo from systemic inflammation. J Immunol. 2005, 174: 5193-5200.
Stunz LL, Lenert P, Peckham D, Yi AK, Haxhinasto S, Chang M, Krieg AM, Ashman RF: Inhibitory oligonucleotides specifically block effects of stimulatory CpG oligonucleotides in B cells. Eur J Immunol. 2002, 32: 1212-1222.
Lenert P, Rasmussen W, Ashman RF, Ballas ZK: Structural characterization of the inhibitory DNA motif for the type A[D]-CpG-induced cytokine secretion and NK-cell lytic activity in mouse spleen cells. DNA Cell Biol. 2003, 22: 621-631.
Lenert P, Stunz L, Yi AK, Krieg AM, Ashman RF: CpG stimulation of primary mouse B cells is blocked by inhibitory oligodeoxyribonucleotides at a site proximal to NF-κB activation. Antisense Nuc Acid Drug Dev. 2001, 11: 247-256.
Lenert P, Yi AK, Krieg AM, Stunz L, Ashman RF: Inhibitory oligonucleotides block the induction of AP-1 transcription factor by stimulatory CpG oligonucleotides in B cells. Antisense Nucl Acid Drug Dev. 2003, 13: 143-150.
Ashman RF, Goeken JA, Drahos J, Lenert P: Sequence requirements for oligodeoxyribonucleotides inhibitory activity. Int Immunol. 2005, 17: 411-420.
Patole PS, Zecher D, Pawar RD, Grone HJ, Schlondorff D, Anders HJ: G-rich DNA suppresses systemic lupus. J Am Soc Nephrol. 2005, 16: 3273-3280.
Lipsky PE: Systemic lupus erythematosus: an autoimmune disease of B cell hyperactivity. Nat Immunol. 2001, 2: 764-766.
Keystone E: B cell targeted therapies. Arthritis Res Ther. 2005, S13-S18. Suppl 3
Acknowledgements
Special thanks to Rachel Brummel, BS, who performed studies on MZ-B cells; Robert F Ashman, MD, who shared recent data on TLR9/INH-ODN interaction; Teresa Ruggle, who helped with the figures; and Rebecca Tuetken, MD PhD, for her invaluable contribution on revising this manuscript. This study was supported by the NIH-RO1 grant AI 047374-01A2.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Lenert, P.S. Targeting Toll-like receptor signaling in plasmacytoid dendritic cells and autoreactive B cells as a therapy for lupus. Arthritis Res Ther 8, 203 (2006). https://doi.org/10.1186/ar1888
Published:
DOI: https://doi.org/10.1186/ar1888