
Abstract
Microtubule-associated protein tau encoded by the MAPT gene binds to microtubules and is important for maintaining neuronal morphology and function. Alternative splicing of MAPT pre-mRNA generates six major tau isoforms in the adult central nervous system resulting in tau proteins with three or four microtubule-binding repeat domains. In a group of neurodegenerative disorders called tauopathies, tau becomes aberrantly hyperphosphorylated and dissociates from microtubules, resulting in a progressive accumulation of intracellular tau aggregates. The spectrum of sporadic frontotemporal lobar degeneration associated with tau pathology includes progressive supranuclear palsy, corticobasal degeneration, and Pick’s disease. Alzheimer’s disease is considered the most prevalent tauopathy. This review is divided into two broad sections. In the first section we discuss the molecular classification of sporadic tauopathies, with a focus on describing clinicopathologic relationships. In the second section we discuss the neuroimaging methodologies that are available for measuring tau pathology (directly using tau positron emission tomography ligands) and tau-mediated neuronal injury (magnetic resonance imaging and fluorodeoxyglucose positron emission tomography). Both sections have detailed descriptions of the following neurodegenerative dementias – Alzheimer’s disease, progressive supranuclear palsy, corticobasal degeneration and Pick’s disease.
Similar content being viewed by others

Background
Molecular classification of tauopathies
Neurodegeneration is the progressive loss of selective populations of vulnerable neurons. Research efforts investigating sporadic and familial neurodegenerative diseases have identified distinct clinicopathologic relationships related to the accumulation of hallmark brain lesions found in selectively vulnerable neuroanatomical distributions. Neurodegeneration can thus be classified by clinical presentation, anatomic distribution, or molecular mechanisms (that is, specific proteinopathies). Strong evidence supports a pathogenic role of altered microtubule-associated protein tau (MAPT) as the shared molecular mechanism of disease amongst the collectively termed tauopathies. Although tauopathies share a common molecular mechanism, the selective vulnerability of anatomic systems and clinical presentations significantly varies across disease types. Moreover, the inclusions and cell types involved can range from neuronal cells to glial cells. Based on the predominance of disorders involving tau neuropathology, it is recognized as the most commonly associated misfolded protein in human neurodegenerative diseases (Table 1).
Hyperphosphorylation of tau is thought to destabilize the microtubule-associated proteins, which act as stabilizers of microtubule networks. The degree of phosphorylation regulates the physiological functions of tau, thus effecting microtubule interaction and intracellular trafficking [1]. Abnormal accumulation of hyperphosphorylated tau that makes up neurofibrillary tangles (NFTs), composed of paired helical filaments (PHFs) and straight filaments, is found in Alzheimer’s disease (AD) brains (Table 2). In addition, there are several non-AD tauopathies with focal cortical neuronal loss and gliosis that fit into the spectrum of sporadic frontotemporal lobar degeneration with tau pathology (FTLD-tau), including progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and Pick’s disease (PiD). Table 1 provides a more extensive list of neurodegenerative diseases with tau inclusions – including, but not limited to, argyrophilic grains disease [2], Parkinsonism–dementia complex of Guam [3], and white matter tauopathy with globular glial inclusions [4–9].
This brief review summarizes the clinicopathologic patterns and neuroimaging signatures of sporadic AD and FTLD-tau. Over the past 15 years, knowledge about the genetics of familial FTLD research has exploded – yielding the discoveries of mutations in the gene for MAPT[10–12], mutations in the gene encoding progranulin (GRN) [13, 14], and recently the abnormal hexanucleotide repeat expansion in the gene C9ORF72[15, 16]. Readers are referred to recent reviews that cover the breadth of genetic forms of AD [17] and FTLD [18].
Clinicopathologic patterns of sporadic Alzheimer’s disease and FTLD-tau
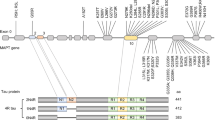
Heterogeneity of tau neuropathology is the consequence of alternative splice forms and post-translational modifications (for example, phosphorylation, ubiquitination, and acetylation) [19]. Six isoforms of the tau protein are expressed in the human brain, which results from alternatively spliced pre-mRNA [20, 21]. Alternative splicing of exon 2, exon 3, and exon 10 of MAPT affects the number of microtubule-binding repeats. Dependent upon the alternative splicing of exon 10, the tau species will contain three or four repeat domains (3R and 4R, respectively). Preferential accumulation of 3R or 4R tau can be found in various tauopathies, revealing a nonuniform biochemical pattern (Table 2) [22–25]. PSP and CBD brains have predominantly 4R tau pathology and are considered 4R tauopathies (4R > 3R), whereas PiD is considered a 3R tauopathy (3R > 4R). In AD the 3R:4R tau ratio is close to one and is thus not referred as a 3R or 4R tauopathy. The recent revision of FTLD neuropathologic diagnostic criteria takes into account molecular genetics, biochemistry characteristics, and current immunohistochemical techniques [26].
AD is a progressive neurodegenerative disorder and is the most common form of dementia in the aging population. Intracellular tau NFTs and extracellular amyloid-beta (Aβ) plaques are the histopathologic hallmarks of AD (Figure 1a,b,c) [27]. AD patients typically present initially with memory impairment, correlating with tau NFTs in medial temporal lobe structures including the entorhinal cortex, amygdala, and cornu ammonis field 1/subiculum of the hippocampus early in the disease process [28, 29]. The stereotypic progression from medial temporal lobe structures to association cortices and eventual involvement of primary cortices was originally described by Braak and Braak [28]. Dementia associated with AD pathology has an insidious onset with progressive worsening of cognition. Patients can have an amnestic presentation or can have nonamnestic presentations including language, visuospatial, and executive dysfunction that are probably due to atypical patterns of AD neuropathology [30–32]. The recently updated clinical diagnostic criterion for dementia associated with AD pathology incorporates imaging and cerebrospinal fluid biomarkers in efforts to improve earlier detection and tracking of disease progression [33, 34].

Neuropathologic inclusions seen in tauopathies range from intracellular to extracellular and from neuron to glia. Alzheimer’s disease neuropathologic inclusions used to classify severity based on an ABC scoring scheme include (a) extracellular amyloid-beta (Aβ) plaque (33.1.1 antibody), (b) neurofibrillary tangle (NFT) composed of abnormal tau fibrils (paired helical filament phosphorylated tau antibody), and (c) Aβ deposits surrounded by dystrophic neurites produce neuritic plaques (observed with Bielschowsky silver stain). Tau immunohistochemistry in progressive supranuclear palsy shows abnormal tau aggregates in (d) astrocytes called tufts or tufted astrocytes, (e) neurons called globose NFTs, and (f) oligodendrocytes termed coiled bodies. Tau-immunoreactivity in corticobasal degeneration (CBD) shows abnormal tau aggregates in (g) astrocytes called astrocytic plaques and tau-immunoreactive threads in the gray and white matter in neocortical and subcortical regions and (h) swollen, achromatic or ballooned neurons (hematoxylin and eosin). (i) Tau-immunoreactive, dense spherical neuronal cytoplasmic inclusions called Pick bodies are observed in granular neurons of the dentate fascia in Pick’s disease. (a), (b), (c) Medial temporal cortex. (d), (e), (f),(g), (i) Phospho-tau antibody CP13. (d), (f) Red nucleus at the level of the oculomotor nerve. (e) Substantia nigra. (g), (h) Mid-frontal cortex.
In AD, hyperphosphorylated, insoluble aggregates composed of 3R and 4R tau develop into NFTs and neuritic plaques (Aβ extracellular lesions surrounded by tau neuropil threads and dystrophic neurites) [20, 35, 36]. Updated AD neuropathologic diagnostic criteria implement an ABC standardized scoring scheme [37] that includes modified versions of Thal phasing for Aβ plaques (A) [38], the Braak and Braak NFT stage (B) [28, 39], and a neuritic plaque score defined by the Consortium to Establish a Registry for Alzheimer’s Disease (C) [40]. These criteria have the advantage of ensuring uniformity in neuropathologic assessment of AD across research institutions to improve clinicopathologic studies, and in particular highlight the occurrence of AD pathology in the absence of cognitive impairment, which may represent a preclinical phase of AD [41].
PSP is a sporadic neurodegenerative disorder with prominent hyperphosphorylated tau aggregates in the brain accompanied by neuronal loss and gliosis. In general, the anatomical distribution of tau pathology correlates with the clinical presentation of PSP patients, with the basal ganglia, substantia nigra, and subthalamic nucleus being the most affected brain regions [42]. PSP can often be diagnosed on macroscopic examination by the presence of midbrain atrophy with dilation of the cerebral aqueduct, superior cerebellar peduncle and subthalamic nucleus atrophy [43], and variable cortical involvement of the peri-Rolandic cortex [44]. Microscopically, PSP neuropathology is characterized by neuronal inclusions called globose NFTs, tufted astrocytes [45], and tau immunoreactive inclusions in oligodendrocytes, termed coiled bodies (Figure 1d,e,f). There is often marked neuronal loss and gliosis in the subthalamic nucleus, globus pallidus, ventral thalamus, and cerebellar dentate nucleus. Ultrastructural characterization of tau pathology in PSP reveals mostly straight filaments, with only rare twisted filaments having been observed (Figure 2 and Table 2).

Ultrastructural characterization of tau filaments in Alzheimer’s disease and frontotemporal lobar degeneration tau. (Top, left) Alzheimer’s disease (AD) tau fibrils form paired helical filaments typically observed in flame-shaped cytoplasmic inclusion. (Top, middle) Progressive supranuclear palsy (PSP) tau fibrils typically form straight filaments with rare twisted filaments, similar to corticobasal degeneration (CBD), that aggregate in less compact bundles associated with dense granular material. (Top, right) Pick’s disease (PiD) tau fibrils are mainly straight filaments with some loosely twisted wide filaments that aggregate in close proximity and can be associated with dense granular material. (Bottom, left) Electron micrograph of tau filaments from AD showing paired helical filaments (PHF) and straight filament (SF). Bar, 50 nm. (Bottom, right) Twisted filaments in PSP, CBD and PiD have longer periodicity. Bar, 100 nm. Arrows point at twists of filaments.
The large majority of PSP patients present with Richardson syndrome, also known as PSP syndrome, characterized by postural instability leading to unexplained backward falls within the first year of symptom onset, axial rigidity, dysarthria, dysphagia, progressive vertical ophthalmoplegia, personality changes, and bradykinesia that is unresponsive to levodopa. Although this description comprises the typical PSP cases, there is a great deal of pathologic heterogeneity that causes patients to present with various clinical syndromes. Atypical variants of PSP include frontotemporal dementia (FTD) [46], nonfluent/agrammatic primary progressive aphasia and apraxia of speech [47], and pure akinesia with gait freezing syndrome due to severe pallido-nigro-luysial degeneration [48, 49]. The cause of this extensive variability associated with PSP is currently unknown, but underlying genetic variation is expected to play a role.
Although there are rare familial cases, CBD and PSP are considered sporadic disorders. Yet, despite their sporadic nature, genetic variants on the H1 major haplotype harboring the MAPT locus that spans ~1.8 Mb of DNA on chromosome 17q21 are a strong genetic risk factor for CBD and PSP [50–55]. Recent progress in our understanding of PSP genetics is credited to the completion of the first, of its kind, PSP genome-wide association study [56], and future studies aim to use common genetic variation within PSP to determine whether they associate with and influence variability in tau neuropathology.
CBD is a rare neurodegenerative disorder classified as a 4R tauopathy due to neuronal and glial aggregates of hyperphosphorylated tau in both gray and white matter of the neocortex, basal ganglia, thalamus, and, to a lesser extent, the brainstem of these patients [57]. The hallmark glial lesion in CBD is the astrocytic plaque (Figure 1g), which is not observed in other disorders [58, 59]. Microscopic inspection of the affected cortices often shows cortical thinning with neuronal loss, gliosis and many ballooned neurons (Figure 1h). Ultrastructural characterization of tau pathology in CBD reveals mostly straight filaments with some wide twisted filaments having been observed (Figure 2 and Table 2). CBD was first described as a distinct clinicopathologic entity in the 1960s by Rebeiz and coworkers [60] and has some overlapping clinical and pathologic features with PSP, yet is considered a distinct disease entity [61, 62]. CBD is associated with focal cortical atrophy and, because of this, patients can present with a wide range of clinical syndromes depending on the location of the highest tau burden pathology and marked cortical atrophy that can be observed on imaging using voxel-based morphometric analysis (reviewed in [63]). Because CBD pathology can cause multiple different neurologic syndromes, defining clinical diagnostic criteria for CBD has been extremely challenging [64–67]. CBD patients can present with corticobasal syndrome [68–70], PSP syndrome [70–73], FTD [71, 74–76], or nonfluent/agrammatic primary progressive aphasia [77, 78]. CBD patients presenting with corticobasal syndrome often have asymmetric atrophy of the superior frontal cortex, whereas those patients presenting with PSP syndrome have symmetric atrophy slightly more anterior than corticobasal syndrome patients and have greater hindbrain involvement (that is, brainstem and cerebellum) [72, 73, 79].
PiD is a rare form of FTLD-tau that is associated with severe circumscribed cortical atrophy of the frontal and temporal lobes, described as knife-edge atrophy of cortical gyri. Patients suffering from PiD will have clinical syndromes corresponding to the location of the most affected cortical regions, most often presenting with behavioral variant FTD [80], nonfluent/agrammatic primary progressive aphasia with peri-Sylvian atrophy [81, 82], an amnestic syndrome [80], upper motor neuron signs due to pyramidal tract degeneration [83], or progressive limb apraxia due to frontoparietal atrophy [84, 85]. Familial forms of PiD are extremely rare and are due to MAPT mutations p.G272V [86] and p.G389R [87, 88]. The histopathologic inclusions observed in PiD, termed Pick bodies, are round intraneuronal inclusions composed of hyperphosphorylated 3R tau [89, 90] and are argyrophilic on Bielschowsky but are Gallyas-negative (PiD neuropathology reviewed in [91]). Hippocampal pyramidal neurons and granular neurons of the dentate fascia are particularly susceptible to Pick bodies (Figure 1i). There is diffuse spongiosis in affected cortical regions and ballooned achromatic neurons (Pick cells) in middle and lower cortical layers, and variable tau-immunoreactive glial inclusions [26]. Ultrastructural characterization of tau pathology in PiD also reveals mostly straight filaments, with some wide twisted filaments having been observed (Figure 2 and Table 2).
Imaging tauopathies in neurodegenerative diseases
In vivo imaging of molecular processes and pathologies has evolved significantly in the last two decades. Imaging surrogates of pathology are especially useful in the neurodegenerative dementias where there is no clear one-to-one correspondence between the neuropathologic findings at autopsy and the clinical expression of the disease in each subject. There are two noninvasive imaging technologies that are used widely to measure tau pathology and/or tau-mediated injury in the brain – positron emission tomography (PET) and magnetic resonance imaging (MRI).
PET imaging involves injecting a radioactive tracer into a subject intravenously. After the tracer is chemically incorporated into a biologically active molecule of interest, the tracer decays and annihilates to produce gamma rays that are measured using the PET cameras. The typical radioactive tracers use carbon-11, oxygen-15 and fluorine-18 (18 F) isotopes. The most commonly used PET tracer is fluorodeoxyglucose (FDG; 18 F agent), which is a glucose analog used to measure glucose uptake in the organ of interest. On the other hand, MRI is based on the principles of nuclear magnetic resonance of the atomic nuclei. The following section discusses both the tau tracers/ligands that are available for direct measurement of tau using PET imaging as well as MRI and PET imaging methods that indirectly measure tau-mediated neuronal injury. We will also specifically discuss the expected patterns of neurodegeneration seen in different tauopathies in MRI.
Tau ligands in positron emission tomography
In the recent past after the invention of excellent amyloid tracers (such as carbon-11-labeled Pittsburgh compound B and [18F]florbetapir), the search for a tau binding ligand has intensified. The search properties include nontoxicity, ability to cross the blood–brain barrier (that is, low molecular weight lipophilic molecules), rapid clearance from the bloodstream and selective binding to specific targets (that is, tau) in a reversible fashion [92]. Due to the longer half-life of 18 F (110 minutes) and a temporal advantage favorable for commercialization and distribution, most of the tau ligands are 18 F-based. Below, we summarize the three tau ligands that have shown the most promise and describe their selective potential in AD and FTLD-tau. For a more in-depth review on the pharmacokinetic requirements of tau imaging ligands, the readers are directed to a recent review by Jensen and colleagues [93].
The radiotracer 2-(1-(6-((2-[18F]fluoroethyl) (methyl)amino)-2-naphthyl)ethylidene) malononitrile ([18F]FDDNP) was the first reported PET contrast agent to successfully detect both Aβ deposits and NFTs in brains of AD patients [94, 95]. [18F]FDDNP was identified through direct visual inspection of tissue fluorescence in postmortem brain tissue. The putative protein target is the aggregate conformation of β-pleated sheets, thus prompting investigations into the specificity of the radiotracer to AD neuropathology. After observing [18F]FDDNP-labeled prion plaques found in Creutzfeldt–Jakob disease and Gerstmann–Sträussler–Scheinker disease, follow-up studies sought to further characterize the protein deposits potentially labeled by the radiotracer [96]. A subsequent immunofluorescent study demonstrated binding of [18F]FDDNP to similarly labeled Congo red protein deposits, including prion plaques, cerebral amyloid angiopathy, amyloid plaques, NFTs, and Lewy bodies [97]. Pick bodies, globose NFTs, and glial cytoplasmic inclusions were not visualized with [18F]FDDNP, implying the absence of structural conditions necessary to bind the molecule [97]. Contradictory to the lack of fluorescent staining in PSP postmortem tissue described in the aforementioned study, a more recent [18F]FDDNP PET study convincingly demonstrates a higher signal in areas known to be vulnerable to tau pathology in PSP – subcortical gray matter and brainstem structures [98]. Representative images from this study [98] are shown in Figure 3. There is contention, however, whether the tracer concentration used on patients is sufficient to adequately label NFTs and/or other pathologies. A major drawback of [18F]FDDNP is its nonspecific binding to other proteins in addition to tau.

Tau ligand binding patterns in progressive supranuclear palsy. (Left) Typical 2-(1-(6-((2-[18F]fluoroethyl) (methyl)amino)-2-naphthyl)ethylidene) malononitrile ([18F]-FDDNP) binding patterns seen in advanced progressive supranuclear palsy (PSP), early PSP and Parkinson’s disease. [18F]-FDDNP signal due to tau binding seen in the basal ganglia, midbrain and pons in PSP subjects but not in Parkinson’s disease. (Right) Sagittal magnetic resonance imaging (MRI) scan of a PSP patient with characteristic midbrain atrophy. DVR, distribution volume ratio, a scaled measure that indicates the linear function of radioligand binding. Reprinted with permission from [98].
A second group from the Tohoku University in Japan employed a screen of organic compounds targeting β-sheet structures (for example, quinolone, benzoxazole, and benzimidazole) in brain tissue [99]. One of these derivatives was found to bind tau with a higher affinity over Aβ,2-(4-aminophenyl)-6-(2-([18F]fluoroethoxy))quinolone ([18F]THK523) [100, 101]. To investigate the binding properties of [18F]THK523, an in vitro binding assay using recombinant tau and Aβ1–42 fibrils was performed. The overall number of binding sites was ~5-fold higher for tau compared with Aβ1–42[100]. Follow-up immunofluorescence and autoradiography studies in postmortem brain tissue demonstrated specificity to tau tangles in the cortex and hippocampus [100, 101]. Although there appears to be white matter retention visible in the autoradiography photomicrographs, the signal relative to the grey matter pathology appeared to remain distinguishable. Further supportive evidence for the selectivity of [18F]THK523 as a tau ligand was demonstrated by microPET assessment of the Alzheimer-like tau pathology in the Tg4510 line, which expresses the P301L MAPT mutation. Higher binding was observed compared with that seen in APP/PS1 mice, which expresses the Swedish APP and presinilin-1 transgene (Alzheimer-like amyloid pathology model). Despite evidence of higher cortical retention in AD, a study comparing AD, semantic dementia, and healthy control patients showed no distinct pattern of [18F]THK523 radiotracer retention [102]. More work demonstrating in vivo PET images of human tauopathies will be of interest to future clinical use of [18F]THK523 as a tau-directed imaging agent, although preliminary work has been quite promising.
The most recently described tau ligand came from the Siemens’ Molecular Imaging Group (recently acquired by Avid/Lily) screening over 900 compounds to determine which had both higher binding affinity and selectivity for tau tangles compared with Aβ plaques [103]. Two compounds, [18F]T807 and [18F]T808, met optimum pharmacokinetic characteristics for tau ligands with >27-fold higher affinity for PHF-tau compared with Aβ, as well as low white matter binding. [18F]T808 reportedly underwent slow defluorination, compared with the metabolically stable [18F]T807 compound. The follow-up study investigating the efficacy of these imaging agents thus focused on [18F]T807 [104]. Autoradiographic evidence of tau selectivity was evident in Aβ-positive/tau-negative brain tissue when compared with Aβ-negative/tau-positive brain tissue [104, 105]. Various brain regions were analyzed for the uptake of [18F]T807 across healthy controls, mild cognitive impairment, and AD patients [104]. Healthy controls showed low binding, whereas medial temporal and association cortices demonstrated stereotypic severity expected in AD [28]. The mild cognitive impairment patient was found centered between healthy controls and the AD patients – except in the occipital cortex, which would be expected.
The favorable pharmacokinetics, low white matter binding, and apparent association with cognitive status in AD make [18F]T807 a promising tau ligand for future clinical studies in AD. Given the initial screen for PHF-tau in AD, it will be of interest to observe the efficacy of [18F]T807 as a tau ligand in FTLD tauopathies because they are primarily composed of straight filaments. Twisted filaments found in CBD and PiD have a wider periodicity (~160 nm) compared with AD (~80 nm), which may interfere with tau ligand binding (Table 2). The PHFs in AD are less compact and more of a pure filamentous bundle compared with PiD, which have a compartmentalized combination of straight and twisted filaments mixed with other material – possibly masking the tau epitope. Labeling PSP and CBD may be easier given the more diffuse, shorter filamentous nature of the tau. Past studies evaluating tau epitopes identified in AD and their specificity in PSP [106], CBD [107], and PiD [108] have shown immunopositive labeling despite differences in periodicity.
Another challenge of tau imaging is the abundance of tau aggregates in the white matter of many tauopathies, as discussed by Villemagne and colleagues [109]. Amyloid imaging has faced the issue of high nonspecific binding of amyloid ligands in white matter [110], but binding of tau to white matter may have a biologic or pathologic mechanism of explanation. Tau has been shown to localize to the axon in white matter, with some evidence of localization to the somatodendritic compartment [111, 112]. Although tau imaging in AD would favor low white matter binding, specific binding in the white matter would probably benefit the differential diagnosis of CBD and PSP [73] or identify cases of white matter tauopathy with globular glial inclusions [4, 5]. In comparison with high specific-to-nonspecific tau binding in the gray matter, the white matter may have an equal ratio or a higher nonspecific-to-specific binding ratio given the reduced blood flow compared with gray matter.
Imaging tau-mediated neuronal injury
Both structural MRI and FDG-PET are used for measuring tau-mediated neuronal injury. Structural MRI measures brain morphometry. MRI captures structural changes that occur on a microscopic level in neurodegenerative disorders: gray matter atrophy related to the loss of neurons, synapses, and dendritic dearborization; white matter atrophy related to loss of structural integrity of white matter tracts, presumably resulting from demyelination and dying back of axonal processes; and ex vacuo expansion of cerebrospinal fluid spaces. Strong correlations have been shown between the volume measured on MRI and histology-based neuronal numbers in the hippocampus [113]. Since there is a significant negative correlation between NFT density and neuronal counts [114], MRI has been considered a sensitive marker of tau pathology – although more work is needed to establish the contribution of coexisting neuropathologies (for example, neuritic plaques, TDP-43, ubiquitin). Pathology studies in AD have shown high correlations between structural changes on MRI and Braak NFT stages [28], validating structural MRI as a biomarker for measuring neuron loss associated with NFT burden [115–117]. Emerging MRI modalities such as diffusion tensor imaging and resting-state functional MRI have also shown significant promise in capturing changes due to tau pathology [118, 119]. FDG-PET, on the other hand, is used to measure net brain metabolism, although including many neural and glial functions, largely indicating synaptic activity [120, 121]. Brain glucose metabolism measured with FDG-PET is highly correlated with postmortem measures of the synaptic structural protein synaptophysin [122].
We now discuss the typical patterns of atrophy seen on MRI and metabolic deficits seen on FDG-PET for each of the major tauopathies – AD, PSP, CBD, and PiD. In AD, atrophy patterns seen on MRI are similar to the progression of NFT pathology discussed earlier. Typical AD begins and is ultimately most severe in the medial temporal lobe, particularly the entorhinal cortex and hippocampus. Later the atrophy is seen in the basal temporal lobe and posterior cingulate gyrus and precuneus. The visual assessment [123] or the quantification of the hippocampus [124] is the most commonly used biomarker for measuring tau-mediated injury in AD and has been validated using several autopsy studies [125]. FDG-PET patterns in AD show significant hypometabolism in the bilateral posterior cingulate gyri and the parietotemporal area in AD [126].
PSP is characterized by significant atrophy and metabolic changes in the brainstem with additional involvement of cortical regions, specifically the medial frontal regions [127]. Atrophy of the midbrain on mid-sagittal MRI, described as the hummingbird sign, is a useful predictor of PSP [128]. Visual assessment or quantification of atrophy in the superior cerebellar peduncle on MRI significantly increases accuracy of the clinical diagnosis [129].
CBD is characterized by significant focal atrophy and metabolic changes that are typically asymmetric and are observed in the frontoparietal regions with involvement of subcortical structures [130, 131]. Additionally, the rates of global atrophy observed in CBD are significantly higher than those in other neurodegenerative disorders [132].
PiD is associated with widespread metabolic abnormality and atrophy in the frontal regions and to a lesser extent in the temporal lobe regions [133, 134]. Imaging examples of cognitive normal subjects, FTD subjects, and AD subjects with an amyloid tracer, FDG-PET, and MRI are shown in Figure 4. The use of neuroimaging for identifying neuroanatomical patterns underlying different FTLD clinical syndromes as well as differential prediction of tau pathology from other pathologies underlying FTLD (ubiquitin, progranulin) has been an area of active research [118, 134–137].

Amyloid imaging of Alzheimer’s disease and frontotemporal dementia. Typical amyloid positron emission tomography (Pittsburgh Compound B-PET), fluorodeoxyglucose (FDG)-PET and magnetic resonance imaging (MRI) images seen in a cognitively normal individual (CN), an Alzheimer’s disease (AD) patient and a frontotemporal dementia (FTD) patient. The CN individual shows no evidence of amyloid deposition, normal metabolic uptake and normal structural MRI scan. The AD patient shows significant amyloid uptake throughout the brain, significant low parietal lobe FDG uptake and significant ventricular expansion on the MRI scan. The FTD patient shows no significant amyloid deposition, significant frontal and temporal lobe deficits and atrophy, which are both highly asymmetric.
Conclusions and future directions
The vast heterogeneity of both clinical presentations and molecular neuropathology across the major tauopathies underlies the importance of biomarker development. Given that there is no one-to-one match between the neuropathologic findings at autopsy and the clinical expression of the disease in each subject, in vivo MRI and PET imaging that measures tau either directly or indirectly will be extremely useful for identifying the pathologic substrate of the disease. In addition to aiding the early detection and differential diagnosis of the tauopathies in neurodegenerative disorders, in vivo imaging measures can play several important roles – predicting the risk of progression in at-risk populations, evaluating disease progression, measuring efficacy of therapeutics, screening for clinical trials, as well as making mechanistic inferences into the disease process. FDG and MRI are currently excellent surrogates for measuring neuronal injury due to tau, but tau imaging will provide clinicians with a direct in vivo tool to measure tau pathology. Thorough validation using antemortem autopsy studies, however, is still needed in future analyses.
Note
This article is part of a series on Tau-based therapeutic strategies, edited by Leonard Petrucelli. Other articles in this series can be found at http://alzres.com/series/tau_therapeutics.
Abbreviations
- [18F]FDDNP:
-
2-(1-(6-((2-[18F]fluoroethyl) (methyl)amino)-2-naphthyl)ethylidene) malononitrile
- [18F]THK523:
-
Aβ,2-(4-aminophenyl)-6-(2-([18F]fluoroethoxy))quinolone
- 18F:
-
Fluorine-18
- 3R:
-
Three repeat domain
- 4R:
-
Four repeat domain
- AD:
-
Alzheimer’s disease
- Aβ:
-
Amyloid-beta
- CBD:
-
Corticobasal degeneration
- FDG:
-
Fluorodeoxyglucose
- FTD:
-
Frontotemporal dementia
- FTLD:
-
Frontotemporal lobar degeneration
- MAPT:
-
Microtubule-associated protein tau
- MRI:
-
Magnetic resonance imaging
- NFT:
-
Neurofibrillary tangle
- PET:
-
Positron emission tomography
- PHF:
-
Paired helical filament
- PiD:
-
Pick’s disease
- PSP:
-
Progressive supranuclear palsy.
References
Billingsley ML, Kincaid RL: Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J. 1997, 323: 577-591.
Braak H, Braak E: Cortical and subcortical argyrophilic grains characterize a disease associated with adult onset dementia. Neuropathol Appl Neurobiol. 1989, 15: 13-26. 10.1111/j.1365-2990.1989.tb01146.x.
Steele JC: Parkinsonism–dementia complex of Guam. Mov Disord. 2005, 20: S99-S107. 10.1002/mds.20547.
Kovacs GG, Majtenyi K, Spina S, Murrell JR, Gelpi E, Hoftberger R, Fraser G, Crowther RA, Goedert M, Budka H, Ghetti B: White matter tauopathy with globular glial inclusions: a distinct sporadic frontotemporal lobar degeneration. J Neuropathol Exp Neurol. 2008, 67: 963-975. 10.1097/NEN.0b013e318187a80f.
Bigio EH, Lipton AM, Yen SH, Hutton ML, Baker M, Nacharaju P, White CL, Davies P, Lin W, Dickson DW: Frontal lobe dementia with novel tauopathy: sporadic multiple system tauopathy with dementia. J Neuropathol Exp Neurol. 2001, 60: 328-341.
Wischik CM, Novak M, Edwards PC, Klug A, Tichelaar W, Crowther RA: Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci USA. 1988, 85: 4884-4888. 10.1073/pnas.85.13.4884.
Takauchi S, Mizuhara T, Miyoshi K: Unusual paired helical filaments in progressive supranuclear palsy. Acta Neuropathol. 1983, 59: 225-228. 10.1007/BF00703207.
Ksiezak-Reding H, Tracz E, Yang LS, Dickson DW, Simon M, Wall JS: Ultrastructural instability of paired helical filaments from corticobasal degeneration as examined by scanning transmission electron microscopy. Am J Pathol. 1996, 149: 639-651.
Kato S, Nakamura H: Presence of two different fibril subtypes in the Pick body: an immunoelectron microscopic study. Acta Neuropathol. 1990, 81: 125-129. 10.1007/BF00334500.
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, et al: Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998, 393: 702-705. 10.1038/31508.
Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD: Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998, 43: 815-825. 10.1002/ana.410430617.
Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B: Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998, 95: 7737-7741. 10.1073/pnas.95.13.7737.
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M: Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006, 442: 916-919. 10.1038/nature05016.
Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C: Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006, 442: 920-924. 10.1038/nature05017.
Dejesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R: Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-Linked FTD and ALS. Neuron. 2011, 72: 245-256. 10.1016/j.neuron.2011.09.011.
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, et al: A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011, 72: 257-268. 10.1016/j.neuron.2011.09.010.
Schellenberg GD, Montine TJ: The genetics and neuropathology of Alzheimer’s disease. Acta Neuropathol. 2012, 124: 305-323. 10.1007/s00401-012-0996-2.
Rademakers R, Neumann M, Mackenzie IR: Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. 2012, 8: 423-434.
Iqbal K, Grundke-Iqbal I: Ubiquitination and abnormal phosphorylation of paired helical filaments in Alzheimer’s disease. Mol Neurobiol. 1991, 5: 399-410. 10.1007/BF02935561.
Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA: Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 1989, 8: 393-399.
Andreadis A, Brown WM, Kosik KS: Structure and novel exons of the human tau gene. Biochemistry. 1992, 31: 10626-10633. 10.1021/bi00158a027.
Greenberg SG, Davies P: A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA. 1990, 87: 5827-5831. 10.1073/pnas.87.15.5827.
Flament S, Delacourte A, Verny M, Hauw JJ, Javoy-Agid F: Abnormal Tau proteins in progressive supranuclear palsy. Similarities and differences with the neurofibrillary degeneration of the Alzheimer type. Acta Neuropathol. 1991, 81: 591-596. 10.1007/BF00296367.
Ksiezak-Reding H, Morgan K, Mattiace LA, Davies P, Liu WK, Yen SH, Weidenheim K, Dickson DW: Ultrastructure and biochemical composition of paired helical filaments in corticobasal degeneration. Am J Pathol. 1994, 145: 1496-1508.
Delacourte A, Robitaille Y, Sergeant N, Buee L, Hof PR, Wattez A, Laroche-Cholette A, Mathieu J, Chagnon P, Gauvreau D: Specific pathological Tau protein variants characterize Pick’s disease. J Neuropathol Exp Neurol. 1996, 55: 159-168. 10.1097/00005072-199602000-00004.
Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL, Schneider JA, Grinberg LT, Halliday G, Duyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DM: Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007, 114: 5-22. 10.1007/s00401-007-0237-2.
Hyman BT, Trojanowski JQ: Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol. 1997, 56: 1095-1097. 10.1097/00005072-199710000-00002.
Braak H, Braak E: Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82: 239-259. 10.1007/BF00308809.
Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT: Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992, 42: 631-639. 10.1212/WNL.42.3.631.
Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW: Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011, 10: 785-796. 10.1016/S1474-4422(11)70156-9.
Alladi S, Xuereb J, Bak T, Nestor P, Knibb J, Patterson K, Hodges JR: Focal cortical presentations of Alzheimer’s disease. Brain. 2007, 130: 2636-2645. 10.1093/brain/awm213.
Galton CJ, Patterson K, Xuereb JH, Hodges JR: Atypical and typical presentations of Alzheimer’s disease: a clinical, neuropsychological, neuroimaging and pathological study of 13 cases. Brain. 2000, 123: 484-498. 10.1093/brain/123.3.484.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH: The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging–Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7: 263-269. 10.1016/j.jalz.2011.03.005.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH: The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging–Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7: 270-279. 10.1016/j.jalz.2011.03.008.
Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM: Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986, 261: 6084-6089.
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI: Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986, 83: 4913-4917. 10.1073/pnas.83.13.4913.
Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ: National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012, 8: 1-13. 10.1016/j.jalz.2011.10.007.
Thal DR, Rüb U, Orantes M, Braak H: Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002, 58: 1791-1800. 10.1212/WNL.58.12.1791.
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K: Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112: 389-404. 10.1007/s00401-006-0127-z.
Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L: The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991, 41: 479-486. 10.1212/WNL.41.4.479.
Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH: Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging–Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7: 280-292. 10.1016/j.jalz.2011.03.003.
Hauw JJ, Daniel SE, Dickson D, Horoupian DS, Jellinger K, Lantos PL, McKee A, Tabaton M, Litvan I: Preliminary NINDS neuropathologic criteria for Steele–Richardson–Olszewski syndrome (progressive supranuclear palsy). Neurology. 1994, 44: 2015-2019. 10.1212/WNL.44.11.2015.
Tsuboi Y, Slowinski J, Josephs KA, Honer WG, Wszolek ZK, Dickson DW: Atrophy of superior cerebellar peduncle in progressive supranuclear palsy. Neurology. 2003, 60: 1766-1769. 10.1212/01.WNL.0000068011.21396.F4.
Josephs KA, Whitwell JL, Dickson DW, Boeve BF, Knopman DS, Petersen RC, Parisi JE, Jack CR: Voxel-based morphometry in autopsy proven PSP and CBD. Neurobiol Aging. 2008, 29: 280-289. 10.1016/j.neurobiolaging.2006.09.019.
Yamada T, McGeer PL, McGeer EG: Appearance of paired nucleated, Tau-positive glia in patients with progressive supranuclear palsy brain tissue. Neurosci Lett. 1992, 135: 99-102. 10.1016/0304-3940(92)90145-W.
Bigio EH, Brown DF, White CL: Progressive supranuclear palsy with dementia: cortical pathology. J Neuropathol Exp Neurol. 1999, 58: 359-364. 10.1097/00005072-199904000-00006.
Josephs KA, Boeve BF, Duffy JR, Smith GE, Knopman DS, Parisi JE, Petersen RC, Dickson DW: Atypical progressive supranuclear palsy underlying progressive apraxia of speech and nonfluent aphasia. Neurocase. 2005, 11: 283-296. 10.1080/13554790590963004.
Ahmed Z, Josephs KA, Gonzalez J, DelleDonne A, Dickson DW: Clinical and neuropathologic features of progressive supranuclear palsy with severe pallido-nigro-luysial degeneration and axonal dystrophy. Brain. 2008, 131: 460-472. 10.1093/brain/awm301.
Williams DR, Holton JL, Strand K, Revesz T, Lees AJ: Pure akinesia with gait freezing: a third clinical phenotype of progressive supranuclear palsy. Mov Disord. 2007, 22: 2235-2241. 10.1002/mds.21698.
Baker M, Litvan I, Houlden H, Adamson J, Dickson D, Perez-Tur J, Hardy J, Lynch T, Bigio E, Hutton M: Association of an extended haplotype in the tau gene with progressive supranuclear palsy. Hum Mol Genet. 1999, 8: 711-715. 10.1093/hmg/8.4.711.
Conrad C, Andreadis A, Trojanowski JQ, Dickson DW, Kang D, Chen X, Wiederholt W, Hansen L, Masliah E, Thal LJ, Katzman R, Xia Y, Saitoh T: Genetic evidence for the involvement of tau in progressive supranuclear palsy. Ann Neurol. 1997, 41: 277-281. 10.1002/ana.410410222.
Ezquerra M, Pastor P, Valldeoriola F, Molinuevo JL, Blesa R, Tolosa E, Oliva R: Identification of a novel polymorphism in the promoter region of the tau gene highly associated to progressive supranuclear palsy in humans. Neurosci Lett. 1999, 275: 183-186. 10.1016/S0304-3940(99)00738-7.
Houlden H, Baker M, Morris HR, MacDonald N, Pickering-Brown S, Adamson J, Lees AJ, Rossor MN, Quinn NP, Kertesz A, Khan MN, Hardy J, Lantos PL, St George-Hyslop P, Munoz DG, Mann D, Lang AE, Bergeron C, Bigio EH, Litvan I, Bhatia KP, Dickson D, Wood NW, Hutton M: Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology. 2001, 56: 1702-1706. 10.1212/WNL.56.12.1702.
Pastor P, Ezquerra M, Perez JC, Chakraverty S, Norton J, Racette BA, McKeel D, Perlmutter JS, Tolosa E, Goate AM: Novel haplotypes in 17q21 are associated with progressive supranuclear palsy. Ann Neurol. 2004, 56: 249-258. 10.1002/ana.20178.
Rademakers R, Melquist S, Cruts M, Theuns J, Del-Favero J, Poorkaj P, Baker M, Sleegers K, Crook R, De Pooter T, Bel Kacem S, Adamson J, Van den Bossche D, Van den Broeck M, Gass J, Corsmit E, De Rijk P, Thomas N, Engelborghs S, Heckman M, Litvan I, Crook J, De Deyn PP, Dickson D, Schellenberg GD, Van Broeckhoven C, Hutton ML: High-density SNP haplotyping suggests altered regulation of tau gene expression in progressive supranuclear palsy. Hum Mol Genet. 2005, 14: 3281-3292. 10.1093/hmg/ddi361.
Höglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, van Swieten JC, Heutink P, Wszolek ZK, Uitti RJ, Vandrovcova J, Hurtig HI, Gross RG, Maetzler W, Goldwurm S, Tolosa E, Borroni B, Pastor P, Cantwell LB, Han MR, Dillman A, van der Brug MP, Gibbs JR, Cookson MR, Hernandez DG, PSP Genetics Study Group: Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011, 43: 699-705. 10.1038/ng.859.
Dickson DW, Bergeron C, Chin SS, Duyckaerts C, Horoupian D, Ikeda K, Jellinger K, Lantos PL, Lippa CF, Mirra SS, Tabaton M, Vonsattel JP, Wakabayashi K, Litvan I, Office of Rare Diseases of the National Institutes of Health: Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol. 2002, 61: 935-946.
Feany MB, Dickson DW: Widespread cytoskeletal pathology characterizes corticobasal degeneration. Am J Pathol. 1995, 146: 1388-1396.
Komori T, Arai N, Oda M, Nakayama H, Mori H, Yagishita S, Takahashi T, Amano N, Murayama S, Murakami S, Shibata N, Kobayashi M, Sasaki S, Iwata M: Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. 1998, 96: 401-408. 10.1007/s004010050911.
Rebeiz JJ, Kolodny EH, Richardson EP: Corticodentatonigral degeneration with neuronal achromasia: a progressive disorder of late adult life. Trans Am Neurol Assoc. 1967, 92: 23-26.
Litvan I, Grimes DA, Lang AE, Jankovic J, McKee A, Verny M, Jellinger K, Chaudhuri KR, Pearce RK: Clinical features differentiating patients with postmortem confirmed progressive supranuclear palsy and corticobasal degeneration. J Neurol. 1999, 246: II/1-II/5.
Dickson DW: Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol. 1999, 246: II/6-II/15.
Kouri N, Whitwell JL, Josephs KA, Rademakers R, Dickson DW: Corticobasal degeneration: a pathologically distinct 4R tauopathy. Nat Rev Neurol. 2011, 7: 263-272. 10.1038/nrneurol.2011.43.
Watts RL, Mirra SS: Corticobasal ganglionic degeneration. Movement Disorders. Edited by: Marsden CD, Fahn S. 1994, London: Butterworth, 282-299.
Litvan I, Agid Y, Goetz C, Jankovic J, Wenning GK, Brandel JP, Lai EC, Verny M, Ray-Chaudhuri K, McKee A, Jellinger K, Pearce RK, Bartko JJ: Accuracy of the clinical diagnosis of corticobasal degeneration: a clinicopathologic study. Neurology. 1997, 48: 119-125. 10.1212/WNL.48.1.119.
Boeve BF, Lang AE, Litvan I: Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol. 2003, 54: S15-S19. 10.1002/ana.10570.
Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, Boxer AL, Dickson DW, Grossman M, Hallett M, Josephs KA, Kertesz A, Lee SE, Miller BL, Reich SG, Riley DE, Tolosa E, Troster AI, Vidailhet M, Weiner WJ: Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013, 80: 496-503. 10.1212/WNL.0b013e31827f0fd1.
Riley DE, Lang AE, Lewis A, Resch L, Ashby P, Hornykiewicz O, Black S: Cortical-basal ganglionic degeneration. Neurology. 1990, 40: 1203-1212. 10.1212/WNL.40.8.1203.
Gibb WR, Luthert PJ, Marsden CD: Clinical and pathological features of corticobasal degeneration. Adv Neurol. 1990, 53: 51-54.
Litvan I, Grimes DA, Lang AE: Phenotypes and prognosis: clinicopathologic studies of corticobasal degeneration. Adv Neurol. 2000, 82: 183-196.
Murray R, Neumann M, Forman MS, Farmer J, Massimo L, Rice A, Miller BL, Johnson JK, Clark CM, Hurtig HI, Gorno-Tempini ML, Lee VM, Trojanowski JQ, Grossman M: Cognitive and motor assessment in autopsy-proven corticobasal degeneration. Neurology. 2007, 68: 1274-1283. 10.1212/01.wnl.0000259519.78480.c3.
Ling H, O’Sullivan SS, Holton JL, Revesz T, Massey LA, Williams DR, Paviour DC, Lees AJ: Does corticobasal degeneration exist? A clinicopathological re-evaluation. Brain. 2010, 133: 2045-2057. 10.1093/brain/awq123.
Kouri N, Murray ME, Hassan A, Rademakers R, Uitti RJ, Boeve BF, Graff-Radford NR, Wszolek ZK, Litvan I, Josephs KA, Dickson DW: Neuropathological features of corticobasal degeneration presenting as corticobasal syndrome or Richardson syndrome. Brain. 2011, 134: 3264-3275. 10.1093/brain/awr234.
Bergeron C, Davis A, Lang AE: Corticobasal ganglionic degeneration and progressive supranuclear palsy presenting with cognitive decline. Brain Pathol. 1998, 8: 355-365.
Grimes DA, Lang AE, Bergeron CB: Dementia as the most common presentation of cortical-basal ganglionic degeneration. Neurology. 1999, 53: 1969-1974. 10.1212/WNL.53.9.1969.
Lee SE, Rabinovici GD, Mayo MC, Wilson SM, Seeley WW, DeArmond SJ, Huang EJ, Trojanowski JQ, Growdon ME, Jang JY, Sidhu M, See TM, Karydas AM, Gorno-Tempini ML, Boxer AL, Weiner MW, Geschwind MD, Rankin KP, Miller BL: Clinicopathological correlations in corticobasal degeneration. Ann Neurol. 2011, 70: 327-340. 10.1002/ana.22424.
Kertesz A, Martinez-Lage P, Davidson W, Munoz DG: The corticobasal degeneration syndrome overlaps progressive aphasia and frontotemporal dementia. Neurology. 2000, 55: 1368-1375. 10.1212/WNL.55.9.1368.
Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR, Parisi JE, Dickson DW: Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology. 2006, 66: 41-48. 10.1212/01.wnl.0000191307.69661.c3.
Hassan A, Whitwell JL, Boeve BF, Jack CR, Parisi JE, Dickson DW, Josephs KA: Symmetric corticobasal degeneration (S-CBD). Parkinsonism Relat Disord. 2010, 16: 208-214. 10.1016/j.parkreldis.2009.11.013.
Kovacs GG, Rozemuller AJ, van Swieten JC, Gelpi E, Majtenyi K, Al-Sarraj S, Troakes C, Bódi I, King A, Hortobágyi T, Esiri MM, Ansorge O, Giaccone G, Ferrer I, Arzberger T, Bogdanovic N, Nilsson T, Leisser I, Alafuzoff I, Ironside JW, Kretzschmar H, Budka H: Neuropathology of the hippocampus in FTLD-Tau with Pick bodies: a study of the BrainNet Europe Consortium. Neuropathol Appl Neurobiol. 2013, 39: 166-178. 10.1111/j.1365-2990.2012.01272.x.
Graff-Radford NR, Damasio AR, Hyman BT, Hart MN, Tranel D, Damasio H, Van Hoesen GW, Rezai K: Progressive aphasia in a patient with Pick’s disease: a neuropsychological, radiologic, and anatomic study. Neurology. 1990, 40: 620-626. 10.1212/WNL.40.4.620.
Caso F, Gesierich B, Henry M, Sidhu M, LaMarre A, Babiak M, Miller BL, Rabinovici GD, Huang EJ, Magnani G, Filippi M, Comi G, Seeley WW, Gorno-Tempini ML: Nonfluent/agrammatic PPA with in-vivo cortical amyloidosis and Pick’s disease pathology. Behav Neurol. 2013, 26: 95-106.
Tsuchiya K, Piao YS, Oda T, Mochizuki A, Arima K, Hasegawa K, Haga C, Kakita A, Hori K, Tominaga I, Yagishita S, Akiyama H, Takahashi H: Pathological heterogeneity of the precentral gyrus in Pick’s disease: a study of 16 autopsy cases. Acta Neuropathol. 2006, 112: 29-42. 10.1007/s00401-005-0028-6.
Lang AE, Bergeron C, Pollanen MS, Ashby P: Parietal Picks-disease mimicking cortical-basal ganglionic degeneration. Neurology. 1994, 44: 1436-1440. 10.1212/WNL.44.8.1436.
Fukui T, Sugita K, Kawamura M, Shiota J, Nakano I: Primary progressive apraxia in Pick’s disease: a clinicopathologic study. Neurology. 1996, 47: 467-473. 10.1212/WNL.47.2.467.
Bronner IF, ter Meulen BC, Azmani A, Severijnen LA, Willemsen R, Kamphorst W, Ravid R, Heutink P, van Swieten JC: Hereditary Pick’s disease with the G272V tau mutation shows predominant three-repeat tau pathology. Brain. 2005, 128: 2645-2653. 10.1093/brain/awh591.
Murrell JR, Spillantini MG, Zolo P, Guazzelli M, Smith MJ, Hasegawa M, Redi F, Crowther RA, Pietrini P, Ghetti B, Goedert M: Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J Neuropathol Exp Neurol. 1999, 58: 1207-1226. 10.1097/00005072-199912000-00002.
Pickering-Brown S, Baker M, Yen SH, Liu WK, Hasegawa M, Cairns N, Lantos PL, Rossor M, Iwatsubo T, Davies Y, Allsop D, Furlong R, Owen F, Hardy J, Mann D, Hutton M: Pick’s disease is associated with mutations in the tau gene. Ann Neurol. 2000, 48: 859-867. 10.1002/1531-8249(200012)48:6<859::AID-ANA6>3.0.CO;2-1.
Probst A, Tolnay M, Langui D, Goedert M, Spillantini MG: Pick’s disease: hyperphosphorylated tau protein segregates to the somatoaxonal compartment. Acta Neuropathol. 1996, 92: 588-596. 10.1007/s004010050565.
de Silva R, Lashley T, Strand C, Shiarli AM, Shi J, Tian J, Bailey KL, Davies P, Bigio EH, Arima K, Iseki E, Murayama S, Kretzschmar H, Neumann M, Lippa C, Halliday G, MacKenzie J, Ravid R, Dickson D, Wszolek Z, Iwatsubo T, Pickering-Brown SM, Holton J, Lees A, Revesz T, Mann DM: An immunohistochemical study of cases of sporadic and inherited frontotemporal lobar degeneration using 3R- and 4R-specific tau monoclonal antibodies. Acta Neuropathol. 2006, 111: 329-340. 10.1007/s00401-006-0048-x.
Dickson DW: Neuropathology of Pick’s disease. Neurology. 2001, 56: S16-S20. 10.1212/WNL.56.suppl_4.S16.
Laruelle M, Slifstein M, Huang Y: Relationships between radiotracer properties and image quality in molecular imaging of the brain with positron emission tomography. Mol Imaging Biol. 2003, 5: 363-375. 10.1016/j.mibio.2003.09.009.
Jensen JR, Cisek K, Funk KE, Naphade S, Schafer KN, Kuret J: Research towards tau imaging. J Alzheimers Dis. 2011, 26: 147-157.
Agdeppa ED, Kepe V, Liu J, Flores-Torres S, Satyamurthy N, Petric A, Cole GM, Small GW, Huang SC, Barrio JR: Binding characteristics of radiofluorinated 6-dialkylamino-2-naphthylethylidene derivatives as positron emission tomography imaging probes for beta-amyloid plaques in Alzheimer’s disease. J Neurosci. 2001, 21: RC189-
Shoghi-Jadid K, Small GW, Agdeppa ED, Kepe V, Ercoli LM, Siddarth P, Read S, Satyamurthy N, Petric A, Huang SC, Barrio JR: Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry. 2002, 10: 24-35.
Bresjanac M, Smid LM, Vovko TD, Petric A, Barrio JR, Popovic M: Molecular-imaging probe 2-(1-[6-[(2-fluoroethyl)(methyl) amino]-2-naphthyl]ethylidene) malononitrile labels prion plaques in vitro. J Neurosci. 2003, 23: 8029-8033.
Smid LM, Vovko TD, Popovic M, Petric A, Kepe V, Barrio JR, Vidmar G, Bresjanac M: The 2,6-disubstituted naphthalene derivative FDDNP labeling reliably predicts Congo red birefringence of protein deposits in brain sections of selected human neurodegenerative diseases. Brain Pathol. 2006, 16: 124-130. 10.1111/j.1750-3639.2006.00006.x.
Kepe V, Bordelon Y, Boxer A, Huang SC, Liu J, Thiede FC, Mazziotta JC, Mendez MF, Donoghue N, Small GW, Barrio JR: PET imaging of neuropathology in tauopathies: progressive supranuclear palsy. J Alzheimers Dis. 2013, 36: 145-153.
Okamura N, Suemoto T, Furumoto S, Suzuki M, Shimadzu H, Akatsu H, Yamamoto T, Fujiwara H, Nemoto M, Maruyama M, Arai H, Yanai K, Sawada T, Kudo Y: Quinoline and benzimidazole derivatives: candidate probes for in vivo imaging of tau pathology in Alzheimer’s disease. J Neurosci. 2005, 25: 10857-10862. 10.1523/JNEUROSCI.1738-05.2005.
Fodero-Tavoletti MT, Okamura N, Furumoto S, Mulligan RS, Connor AR, McLean CA, Cao D, Rigopoulos A, Cartwright GA, O’Keefe G, Gong S, Adlard PA, Barnham KJ, Rowe CC, Masters CL, Kudo Y, Cappai R, Yanai K, Villemagne VL: 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer’s disease. Brain. 2011, 134: 1089-1100. 10.1093/brain/awr038.
Harada R, Okamura N, Furumoto S, Tago T, Maruyama M, Higuchi M, Yoshikawa T, Arai H, Iwata R, Kudo Y, Yanai K: Comparison of the binding characteristics of [18F]THK-523 and other amyloid imaging tracers to Alzheimer’s disease pathology. Eur J Nucl Med Mol Imaging. 2013, 40: 125-132. 10.1007/s00259-012-2261-2.
Villemagne V, Furumoto S, Fodero-Tavoletti M, Mulligan R, Hodges J, Kudo Y, Masters C, Yanai K, Rowe C, Okamura N: In vivo tau imaging in Alzheimer’s disease. J Nucl Med Meeting Abstracts. 2012, 53: 36-
Zhang W, Arteaga J, Cashion DK, Chen G, Gangadharmath U, Gomez LF, Kasi D, Lam C, Liang Q, Liu C, Mocharla VP, Mu F, Sinha A, Szardenings AK, Wang E, Walsh JC, Xia C, Yu C, Zhao T, Kolb HC: A highly selective and specific PET tracer for imaging of tau pathologies. J Alzheimers Dis. 2012, 31: 601-612.
Chien DT, Szardenings AK, Bahri S, Walsh JC, Mu F, Xia C, Shankle WR, Lerner AJ, Su MY, Elizarov A, Kolb HC: Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis. 2013, 34: 457-468.
Xia CF, Arteaga J, Chen G, Gangadharmath U, Gomez LF, Kasi D, Lam C, Liang Q, Liu C, Mocharla VP, Mu F, Sinha A, Su H, Szardenings AK, Walsh JC, Wang E, Yu C, Zhang W, Zhao T, Kolb HC: [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimers Dement. 2013, [Epub ahead of print]
Schmidt ML, Huang R, Martin JA, Henley J, Mawal-Dewan M, Hurtig HI, Lee VM, Trojanowski JQ: Neurofibrillary tangles in progressive supranuclear palsy contain the same tau epitopes identified in Alzheimer’s disease PHFtau. J Neuropathol Exp Neurol. 1996, 55: 534-539. 10.1097/00005072-199605000-00006.
Mori H, Nishimura M, Namba Y, Oda M: Corticobasal degeneration: a disease with widespread appearance of abnormal tau and neurofibrillary tangles, and its relation to progressive supranuclear palsy. Acta Neuropathol. 1994, 88: 113-121. 10.1007/BF00294503.
Perry G, Stewart D, Friedman R, Manetto V, Autilio-Gambetti L, Gambetti P: Filaments of Pick’s bodies contain altered cytoskeletal elements. Am J Pathol. 1987, 127: 559-568.
Villemagne VL, Furumoto S, Fodero-Tavoletti MT, Harada R, Mulligan RS, Kudo Y, Masters CL, Yanai K, Rowe CC, Okamura N: The challenges of tau imaging. Future Neurol. 2012, 7: 409-421. 10.2217/fnl.12.34.
Fodero-Tavoletti MT, Rowe CC, McLean CA, Leone L, Li QX, Masters CL, Cappai R, Villemagne VL: Characterization of PiB binding to white matter in Alzheimer disease and other dementias. J Nucl Med. 2009, 50: 198-204. 10.2967/jnumed.108.057984.
Khatoon S, Grundke-Iqbal I, Iqbal K: Levels of normal and abnormally phosphorylated tau in different cellular and regional compartments of Alzheimer disease and control brains. FEBS Lett. 1994, 351: 80-84. 10.1016/0014-5793(94)00829-9.
Binder LI, Frankfurter A, Rebhun LI: The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985, 101: 1371-1378. 10.1083/jcb.101.4.1371.
Bobinski M, de Leon MJ, Wegiel J, Desanti S, Convit A, Saint Louis LA, Rusinek H, Wisniewski HM: The histological validation of post mortem magnetic resonance imaging-determined hippocampal volume in Alzheimer’s disease. Neuroscience. 2000, 95: 721-725.
Gómez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT: Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997, 41: 17-24. 10.1002/ana.410410106.
Gosche KM, Mortimer JA, Smith CD, Markesbery WR, Snowdon DA: Hippocampal volume as an index of Alzheimer neuropathology: findings from the Nun Study. Neurology. 2002, 58: 1476-1482. 10.1212/WNL.58.10.1476.
Jack CR, Dickson DW, Parisi JE, Xu YC, Cha RH, O’Brien PC, Edland SD, Smith GE, Boeve BF, Tangalos EG, Kokmen E, Petersen RC: Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology. 2002, 58: 750-757. 10.1212/WNL.58.5.750.
Silbert LC, Quinn JF, Moore MM, Corbridge E, Ball MJ, Murdoch G, Sexton G, Kaye JA: Changes in premorbid brain volume predict Alzheimer’s disease pathology. Neurology. 2003, 61: 487-492. 10.1212/01.WNL.0000079053.77227.14.
McMillan CT, Irwin DJ, Avants BB, Powers J, Cook PA, Toledo JB, McCarty Wood E, Van Deerlin VM, Lee VM, Trojanowski JQ, Grossman M: White matter imaging helps dissociate tau from TDP-43 in frontotemporal lobar degeneration. J Neurol Neurosurg Psychiatry. 2013, 84: 949-955. 10.1136/jnnp-2012-304418.
Xiao-Chuan W, Zheng-Hui H, Zheng-Yu F, Yue F, Yun-Huang Y, Qun W, Xiao-Wei T, Yi-Gen W, Jian-Zhi W: Correlation of Alzheimer-like tau hyperphosphorylation and fMRI bold intensity. Curr Alzheimer Res. 2004, 1: 143-148. 10.2174/1567205043332216.
Schwartz WJ, Smith CB, Davidsen L, Savaki H, Sokoloff L, Mata M, Fink DJ, Gainer H: Metabolic mapping of functional activity in the hypothalamo-neurohypophysial system of the rat. Science. 1979, 205: 723-725. 10.1126/science.462184.
Attwell D, Laughlin SB: An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001, 21: 1133-1145.
Rocher AB, Chapon F, Blaizot X, Baron JC, Chavoix C: Resting-state brain glucose utilization as measured by PET is directly related to regional synaptophysin levels: a study in baboons. Neuroimage. 2003, 20: 1894-1898. 10.1016/j.neuroimage.2003.07.002.
Scheltens P, Leys D, Barkhof F, Huglo D, Weinstein HC, Vermersch P, Kuiper M, Steinling M, Wolters EC, Valk J: Atrophy of medial temporal lobes on MRI in 'probable’ Alzheimer’s disease and normal ageing: diagnostic value and neuropsychological correlates. J Neurol Neurosurg Psychiatry. 1992, 55: 967-972. 10.1136/jnnp.55.10.967.
Jack CR, Petersen RC, O’Brien PC, Tangalos EG: MR-based hippocampal volumetry in the diagnosis of Alzheimer’s disease. Neurology. 1992, 42: 183-188. 10.1212/WNL.42.1.183.
Whitwell JL, Josephs KA, Murray ME, Kantarci K, Przybelski SA, Weigand SD, Vemuri P, Senjem ML, Parisi JE, Knopman DS, Boeve BF, Petersen RC, Dickson DW, Jack CR: MRI correlates of neurofibrillary tangle pathology at autopsy: a voxel-based morphometry study. Neurology. 2008, 71: 743-749. 10.1212/01.wnl.0000324924.91351.7d.
Jagust WJ, Haan MN, Eberling JL, Wolfe N, Reed BR: Functional imaging predicts cognitive decline in Alzheimer’s disease. J Neuroimaging. 1996, 6: 156-160.
Eckert T, Tang C, Ma Y, Brown N, Lin T, Frucht S, Feigin A, Eidelberg D: Abnormal metabolic networks in atypical parkinsonism. Mov Disord. 2008, 23: 727-733. 10.1002/mds.21933.
Kato N, Arai K, Hattori T: Study of the rostral midbrain atrophy in progressive supranuclear palsy. J Neurol Sci. 2003, 210: 57-60. 10.1016/S0022-510X(03)00014-5.
Paviour DC, Price SL, Stevens JM, Lees AJ, Fox NC: Quantitative MRI measurement of superior cerebellar peduncle in progressive supranuclear palsy. Neurology. 2005, 64: 675-679. 10.1212/01.WNL.0000151854.85743.C7.
Hosaka K, Ishii K, Sakamoto S, Mori T, Sasaki M, Hirono N, Mori E: Voxel-based comparison of regional cerebral glucose metabolism between PSP and corticobasal degeneration. J Neurol Sci. 2002, 199: 67-71. 10.1016/S0022-510X(02)00102-8.
Josephs KA, Whitwell JL, Boeve BF, Shiung MM, Gunter JL, Parisi JE, Dickson DW, Jack CR: Rates of cerebral atrophy in autopsy-confirmed progressive supranuclear palsy. Ann Neurol. 2006, 59: 200-203. 10.1002/ana.20707.
Whitwell JL, Jack CR, Parisi JE, Knopman DS, Boeve BF, Petersen RC, Ferman TJ, Dickson DW, Josephs KA: Rates of cerebral atrophy differ in different degenerative pathologies. Brain. 2007, 130: 1148-1158.
Ishii K, Sakamoto S, Sasaki M, Kitagaki H, Yamaji S, Hashimoto M, Imamura T, Shimomura T, Hirono N, Mori E: Cerebral glucose metabolism in patients with frontotemporal dementia. J Nucl Med. 1998, 39: 1875-1878.
Whitwell JL, Josephs KA: Neuroimaging in frontotemporal lobar degeneration - predicting molecular pathology. Nat Rev Neurol. 2011, 8: 131-142.
Pereira JM, Williams GB, Acosta-Cabronero J, Pengas G, Spillantini MG, Xuereb JH, Hodges JR, Nestor PJ: Atrophy patterns in histologic vs clinical groupings of frontotemporal lobar degeneration. Neurology. 2009, 72: 1653-1660. 10.1212/WNL.0b013e3181a55fa2.
Rohrer JD, Lashley T, Schott JM, Warren JE, Mead S, Isaacs AM, Beck J, Hardy J, de Silva R, Warrington E, Troakes C, Al-Sarraj S, King A, Borroni B, Clarkson MJ, Ourselin S, Holton JL, Fox NC, Revesz T, Rossor MN, Warren JD: Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain. 2011, 134: 2565-2581. 10.1093/brain/awr198.
Rohrer JD, Ridgway GR, Modat M, Ourselin S, Mead S, Fox NC, Rossor MN, Warren JD: Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage. 2010, 53: 1070-1076. 10.1016/j.neuroimage.2009.12.088.
Acknowledgements
The authors would like to thank Farzan Fatemi for help with creating the imaging figure. The neuropathology figure would not be possible without the technical expertise of Virginia Philips, Linda Rousseau, and Monica Castanedes-Casey and the gifts of CP13 and PHF-1 antibodies from Dr Peter Davies at The Feinstein Institute for Medical Research, Manhasset, NY, USA. This review was supported by National Institutes of Health grants: Mayo Alzheimer Disease Research grant (P50 AG016574), Mayo Clinic Study on Aging (Alzheimer Disease Patient Registry; U01 AG006786), NIH/NINDS Morris K. Udall Center (P50-NS072187), K99-AG37573, R01-AG011378 and R01-AG041851. The authors would also like to thank the CurePSP|Society for Progressive Supranuclear Palsy, The Alexander Family Alzheimer’s Disease Research Professorship of the Mayo Foundation, The Robert E. Jacoby Professorship for Alzheimer’s Research, and the Mayo Foundation for Education and Research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
CRJ has no financial activities related to the present article, but financial activities not related to the present article include consultation fees from Pfizer, Johnson & Johnson, Janssen, Eisai, and Elan for consultancies. The remaining authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Murray, M.E., Kouri, N., Lin, WL. et al. Clinicopathologic assessment and imaging of tauopathies in neurodegenerative dementias. Alz Res Therapy 6, 1 (2014). https://doi.org/10.1186/alzrt231
Published:
DOI: https://doi.org/10.1186/alzrt231