
Abstract
Neuritic plaques in the brain are a major neuropathological hallmark of Alzheimer’s disease. They are formed by the deposition and aggregation of extracellular amyloid-β protein (Aβ). Aβ is derived from the sequential cleavage of amyloid-β precursor protein (APP) by β-secretase and γ-secretase. β-Site APP cleaving enzyme 1 (BACE1) functions as the primary, if not sole, β-secretase in vivo and is essential for Aβ production. Regulation of APP processing is a major focus of research into AD pathogenesis. The trafficking systems of APP and its cleavage enzymes are complex. Transporting APP and secretases into the same subcellular organelles facilitates their interaction and favors APP processing. The role of APP and BACE1 trafficking in the amyloidgenic pathway and the underlying mechanisms for Aβ production are discussed in this review. In addition, the distinct mechanisms of amino- and carboxy-terminal Aβ generation are reviewed.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is the most common type of dementia. The most common initial symptom is gradual memory loss followed by impairment of other intellectual abilities. According to the report of the Alzheimer’s Association in 2013, every 68 seconds one American will develop AD. AD is the sixth leading cause of death in the United States and the only cause of death among the top ten that cannot be prevented, cured or even slowed [1], and it is now at the forefront of biomedical research.
AD patients tend to develop far more neurofibrillary tangles and neuritic plaques in hippocampus and cortex than age-matched healthy people [2, 3]. A small peptide now called the amyloid-β protein (Aβ) was isolated from brains with amyloid depositon and it was subsequently identified as the core protein of neuritic plaques [4, 5]. Aβ is derived from amyloid-β precursor protein (APP) by sequential proteolytic cleavage [6–9]; BACE1 (β-site APP cleaving enzyme 1) and γ-secretase are important enzymes in this process. The spatial proximity of APP and enzymes is essential for APP processing and Aβ production but several important questions regarding these have major implications for our understanding of AD pathogenesis and in AD drug development: how are APP and BACE1 trafficked through subcellular organelles? How do the cleavage enzymes become mature and active? Do these enzymes exhibit preferential activities in certain subcellular locations? And how do substrates encounter these enzymes?
Here, we review current studies that advance our understanding of trafficking of APP and its cleavage enzymes, and emphasize that the subcellular co-localization of these facilitates APP processing. We discuss the preferential β-cleavage sites on APP in different subcellular compartments. Furthermore, we show that amino- and carboxy-terminal Aβ generation occur in different subcellular organelles due to distinct locations for active BACE1 and γ-secretase.
An overview of APP processing and amyloid-β production
APP is first cleaved by β-secretase at the amino terminus of Aβ, producing a secreted form of APP (sAPPβ) and membrane-bound C99. C99 is subsequently cleaved by γ-secretase to generate Aβ and intracellular carboxy-terminal fragment (CTF)γ. β-Secretase could also cleave APP within the Aβ region to produce C89 and truncated amyloid species; however, most of the APP undergoes a non-amyloidogenic cleavage process. APP is cleaved by α-secretase within the Aβ domain to produce a secreted form of APP (sAPPα) and membrane-bound C83. C83 is further cleaved by γ-secretase, producing extracellular fragment p3 and intracellular CTFγ (Figure 1). BACE1 is the β-secretase in vivo, and the components of the γ-secretase complex are presenilin (PS), nicastrin (NCT), presenilin enhance 2 (Pen-2) and anterior pharynx-defective 1 (Aph-1). The activity of α-secretase is associated with several members of the ADAM (a distintergrin and metalloproteinase) family, ADAM9, ADAM10 and tumour necrosis factor-α convertase (also named ADAM17), though other proteases may also contribute.

β-Amyloid precursor protein processing. β-Amyloid precursor protein (APP) can be cleaved via two pathways, the nonamyloidogenic pathway (left, green) or the amyloidogenic pathway (right, red). Under normal conditions, the majority of APP is cleaved within the amyloid-β (Aβ) domain by α-secretase to produce secreted APP (sAPP)α and membrane-bound C83. C83 can be further cleaved by γ-secretase, producing extracellular fragment p3 and intracellular carboxy-terminal fragment (CTF)γ. In the amyloidogenic pathway, APP is first cleaved by β-secretase to produce sAPPβ and membrane-bound C99. Cleavage of C99 by γ-secretase yields Aβ and intracellular CTFγ. γ-Secretase cleaves APP at multiple sites close to the inner membrane leaflet to produce variants of Aβ peptide with different lengths. The 42 amino acid Aβ peptide, Aβ42 (after γ-cleavage indicated in the figure), is considered the major toxic Aβ in Alzheimer’s disease. Insoluble Aβ is deposited and aggregates to form the core of neuritic plaques in the brain, the pathological hallmark of Alzheimer’s disease.
The role of intracellular trafficking of APP and BACE1 in amyloid-β protein production
Both nascent APP and BACE1 molecules mature through the constitutive secretory pathway from endoplasmic reticulum (ER) to the plasma membrane (PM) [10]. The majority of APP localizes to the Golgi complex [11]. Only a small proportion of APP is detected at the cell surface and over 50% is internalized within 10 minutes [12, 13] and sorted into early endosomes [14–16], where one fraction of APP is recycled back to the PM and another fraction is targeted to the lysosome for degradation [17, 18]. α-Secretase is particularly enriched at the cell surface and competes with BACE1 for APP processing [19]. α-Secretase also competes with β-secretase in the trans-Golgi network (TGN), whereas protein kinase C stimulates α-secretase activity to relatively decrease β-cleavage [20]. However, BACE1 is predominantly localized in the TGN and endosomes [21]. These acidic endosomal compartments provide a low pH environment, which is more favorable for BACE1 activity [22]. Moreover, BACE1 is rapidly internalized from the cell surface [23, 24]. It is degraded by the ubiquitin-proteasome pathway [25] and accelerating BACE1 degradation by ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) reduces C99 and Aβ production [26]. Therefore, the majority of cell surface APP is processed through the non-amyloidogenic pathway, whereas intracellular APP processing predominantly involves the amyloidogenic pathway [14, 16]. Only a small fraction of γ-secretase complex components are located on the cell surface, the rest mainly localized at the ER, Golgi/TGN and endosome [27, 28].
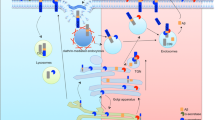
Since 1992, multiple lines of evidence have been used to demonstrate that Aβ is mainly produced in the endosome/lysosome system. Impairing APP trafficking to the cell surface or enhancing APP internalization increased β-secretase-mediated processing of it [16, 29], while enhancing APP routing to, or reducing its internalization from, the cell surface facilitated α-secretase-mediated processing [30]. The endocytosis motif located at the carboxyl terminus of APP (YENPTY) is responsible for the efficient internalization of APP, in clathrin-coated vesicles, to early endosomes [15]. Deletion or mutation of this motif led to APP endocytosis-deficiency and significantly reduced Aβ production [13, 31]. Endocytotic pathway abnormalities were evident in the early stage of sporadic AD brain, Down syndrome patients’ brains, and AD animal models; these might contribute to AD pathogenesis by altering trafficking of APP and BACE1. The Down syndrome patients, with an extra copy of chromosome 21, show increased expression of chromosome 21 genes, such as those encoding APP and Regulator of calcineurin 1 (RCAN1), and inevitably develop characteristic AD neuropathology. The Swedish double mutant (KM/NL) APP produced significantly more Aβ (approximately threefold) than wild-type APP [32–35]; however, abolishing the endocytic process of Swedish APP by removing its endocytic motif still resulted in substantially more Aβ than with normal APP. This result indicates that β-cleavage on Swedish APP does not require an intact cytoplasmic domain and Aβ can also be produced in the Golgi during its biosynthetic transport [36]. Furthermore, inhibition of protein transport from the ER to Golgi and redistributing Golgi proteins into the ER by brefeldin A treatment, or retention of APP in the ER with an ER-retrieval signal, significantly reduced but did not abolish intracellular Aβ production over the course of 24 hours [37, 38]. All this evidence indicates that intracellular trafficking of APP and BACE1 clearly plays a central role in APP processing; amyloidogenic cleavage of APP and Aβ production occurred in multiple subcellular organelles, including the ER/ER-Golgi intermediate compartment, the Golgi during its biosynthetic transport, and the endosome/lysome after endocytosis from the PM (Figure 2).

β-Amyloid precursor protein trafficking. β-Amyloid precursor protein (APP) matures through the constitutive secretory pathway from the endoplasmic reticulum to the plasma membrane (PM). The majority of APP is then quickly internalized into early endosomes, where APP is recycled back to the PM or targeted to the lysosomal degradation pathway. Nonamyloidogenic processing (green) mainly occurs at the cell surface, where α-secretase is particularly enriched. Amyloidogenic processing (red) involves APP trafficking through the secretory and recycling pathways where APP interacts with β- and γ-secretases. Amyloid-β (Aβ) is mainly generated in the trans-Golgi network where the γ-secretase complex is enriched.
Co-residence of APP and BACE1 increased amyloid-β protein production
It was reported that APP and BACE1 do not co-localize at the PM [39], a site where APP is generally cleaved by α-secretase [16, 40]. Both APP and BACE1 were sorted into Rab GTPase 5-positive early endosome [39, 41], where intact Aβ can be detected [16, 42]. Before sorting into the endosome/lysome system, however, APP and BACE1 were distinctly regulated along their transport pathways, from the TGN to PM and internalization. For example, Munc18-interacting protein (Mint; also referred to as X11) contributed to the outward transport of APP from the TGN to PM. Phosphorylation of Munc18 facilitated APP-BACE1 interaction and shifted APP to the BACE1-associated microdomains, resulting in increased cleavage of APP by BACE1 and Aβ production [43]. Internalization of APP occurs via recruitment by the adaptor-protein complex AP-2 and Dab2 for clathrin-mediated endocytosis [18, 44, 45]. In contrast to APP, BACE1 is internalized and sorted into Rab GTPase 5-positive early endosome via a route controlled by ADP-ribosylation factor-6 (ARF6) [44]. The short acidic cluster-dileucine motif (DISLL) in the cytosolic tail domain of BACE1 specifically binds to the Golgi-localized γ-ear-containing ARF-binding proteins (GGAs), which mediate the recycling of BACE1 between the TGN and early endosome [46–51]. GGA3 could also modulate BACE1 turnover and stability while sorting it into the lysosome, and decreased GGA3 correlated with increased levels of BACE1 and β-cleavage of APP to produce Aβ [52]. Stimulation of the recycling of cargo from the early endosome back to the PM by overexpression of wild-type rab4, which regulated sorting and cycling of early endosomes [53], decreased Aβ secretion, suggesting that the residence time of APP and/or BACE in early endosomes is essential for β-cleavage [39]. Moreover, BACE1 and APP needed to be in close proximity for cleavage to occur [54, 55]. The lipid raft is known to facilitate the amyloidogenic cleavage of APP. BACE1, but not α-secretase, interacts with proteins in lipid rafts, and all four γ-secretase complex components associate with lipid rafts as well. Addition of a glycophosphatidylinositol anchor targeted BACE1 into lipid rafts, preferentially increasing β-site cleavage on APP and Aβ production [56, 57]. Lipid raft microdomains, with a small size of 10 to 200 nm, form stable and ordered platforms through protein-protein and protein-lipid interactions [58]. They probably also promote the proximity and interaction of APP and BACE1, as well as the γ-secretase complex, thus contributing to Aβ production. Recently, a sterol-linked β-secretase transit-state inhibitor showed more effective inhibition of β-cleavage and Aβ production than free inhibitors [59]. This was not only because this inhibitor was internalized into the endosome, where the β-cleavage occurred, but also because it was enriched in lipid raft microdomains where the interaction between inhibitors and β-secretase was enhanced. Collectively, these data indicate that facilitating the co-residence and/or the interaction between APP and BACE1 leads to increased β-cleavage of APP through a regulated transport mechanism (Figure 3).

Distinctly regulated β-amyloid precursor protein and β-site β-amyloid precursor protein cleaving enzyme 1 trafficking. β-Amyloid precursor protein (APP) and β-site APP cleaving enzyme 1 (BACE1) trafficking is differentially regulated from the site of trans-Golgi network exit, internalization and further sorting into different compartments. Internalization of APP occurs through recruitment of the adaptor-protein complex AP-2 and Dab2 for clathrin-mediated endocytosis. Munc18-interacting protein (Mint; also called X11) is involved in the outward transport of APP from the trans-Golgi network to the plasma membrane. The recycling pathway of BACE1 is mediated by Golgi-localized γ-ear-containing ARF-binding proteins (GGAs). ADP-ribosylation factor-6 (ARF6) mediates sorting of newly internalized BACE1 into Rab5-positive early endosome and GGA3 could modulate BACE1 turnover and stability while sorting it into the lysosome.
BACE1 cleavage of APP at two sites and regulation of β-cleavage site specificity
BACE1 can cleave APP at two different sites to produce secreted forms of APP and a 99- or 89-residue membrane-associated CTF (C99 or C89, respectively). β-Cleavage at the site between Met596 and Asp597 of APP (the Asp1 cleavage site) [21, 60–62] results in the release of C99 and then intact Aβ, Aβ1-40/42. In addition to the Asp1 site, BACE1 can also cleave APP within the Aβ domain between Tyr606 and Glu607 (the Glu11 cleavage site), releasing C89 and then amino-terminally truncated Aβ, Aβ11-40/42. Under normal conditions, the Glu11 cleavage site is the major β-cleavage site [63]. Aβ11-40/42 was the predominant Aβ species in neuronal cultures [21, 64–66]. Both Aβ11-40/42 and p3 can be found in cerebrospinal fluid and media conditioned by cultured cells [66–68].
Preferential cleavage of APP by BACE1 at the Asp1 or Glu11 site is strongly dependent on the APP sequence close to the β-cleavage sites [63] and the BACE1 subcellular localization, and is not simply due to the organelle's pH, oligosaccharide modification, or the amount of APP substrate [69]. BACE1 tagged with KKXXX (where X is any amino acid residue), the ER retrieval motif, on its cytoplasmic tail was retained in the ER, resulting in accumulation of immature BACE1 and leading to β-cleavage of APP predominately at the Asp1 site. Replacing the BACE1 cytoplasmic tail with the intracellular domain of murine furin effectively caused BACE1 to be retained in the TGN, resulting in preferential generation of C89 rather than C99; wild-type BACE1 primarily exists in the TGN and endosomal system, where more C89 than C99 is generated [69]. Targeting BACE1 to lipid rafts via a glycophosphatidylinositol anchor enhanced BACE1 cell surface expression, resulting in the preferential cleavage of APP at the Asp1 site and secretion of more full-length Aβ. Significantly, this shift of the β-cleavage sites was independent of the subcellular localization of APP or the pathogenic KM/NL mutation [56]. C99 can be further cleaved by BACE1 at its Glu11 site to produce C89 [70]. However, when the APP-C99 fragment was targeted to the ER by the KK motif, little C89 was processed compared to the wild-type APP-C99 fragment [71]. Most surprisingly, little Aβ was processed from APP-C99-KK compared to wild-type APP-C99, though APP-C99-KK, rather than wild-type APP-C99, showed colocalization with PS1 in the ER. However, the intracellular Aβ production from C99-KK could be partially restored by addition of brefeldin A [71]. These data demonstrate that C99 and C89 were processed in distinct subcellular organelles; C99 was more likely to be produced in the ER by immature BACE1 whereas C89 was predominantly processed in the downstream apparatus of the ER, TGN and lysosomal system.
Distinct subcellular sites to generate amino-terminal and carboxy-terminal amyloid-β protein fragments
C99 is mainly produced in the ER. The cleavage of C99 to generate Aβ requires co-residence of C99 with activated γ-secretase complex. However, the ER is not the organelle where C99 can be processed by γ-secretase. By retaining C99 in the ER with the KKQN motif or co-expression with a dominant-negative mutant of the Rab1B GTPase to prevent C99 from exiting the ER, Aβ production was almost completely eliminated. In contrast, when C99 was allowed to leave the ER or targeted into the Golgi and other digital compartments with the QLQN motif, it normally gave rise to substantial amounts of Aβ [72]. C99-GFP accumulated in the early endosome after inhibition of γ-secretase activity [73]. Furthermore, inhibiting the exocytosis of constitutively secretory vesicles, rather than inhibiting clathrin-mediated endocytosis, caused the accumulation of C99-GFP in numerous small vesicles beneath the PM. These data suggest that the cleavage of C99 does not occur in the ER, Golgi or constitutively secretory vesicles, but occurs after it is inserted into the cell surface and endocytosis [73, 74].
Assembly of the γ-secretase complex starts at the ER [75, 76]. Aph-1 contributed to the trafficking and maturation of the γ-secretase complex. First, Aph-1 polypeptides formed a stable subcomplex with NCT in the ER, dependent on its GXXXG motif, which is the initial step for the formation of the whole complex [77–79]. This tightly folded subcomplex interacted with nascent PS through the hydrophobic interface formed by the transmembrane domains of Aph-1 and NCT [80]. However, PS preferentially binds to the mature and fully glycosylated NCT [81]; therefore, after NCT passed through the Golgi and fully matured, PS/NCT/Aph-1 formed a trimeric intermediate. Then PS was subjected to endoproteolysis to generate its amino- and CTFs (PS-CTF and PS-NTF, respectively) [82, 83]. Furthermore, Pen-2 interacted with PS-NTF [80]. Finally, a mature and fully glycosylated NCT, a heterodimeric form of PS-NTF/PS-CTF, Pen-2 and Aph-1 exist at the cell surface as a mature and active γ-secretase complex [84] that can interact with C83 and C99 and cleave them to produce the CTF of Aβ. This complex can be isolated and cleave its substrates, C99 and Notch, in in vitro activity assays [27].
PS1 could also play a role in modulating the trafficking of membrane and secretory proteins [85]. Significant amounts of full-length PS1 were found cycling between the ER and Golgi, regulated by COPI-mediated retrograde transport [86]. Recently, the same group reported that the overexpression of PS1 resulted in retention of Aβ-containing CTFs and Aβ in COPI-coated membranes of the vesicular tubular clusters and ER, while increasing the mutant PS1, which was mainly present in post-Golgi, showed the opposite effect on APP trafficking [87]. These data suggest the potential role of PS1 in the trafficking of APP and its derivatives. However, it is still unknown how the C99 is transferred into areas with active γ-secretase complex, and whether C99 can be cleaved into C89 or C83 dependent on encountering β-secretase or α-secretase along its transport pathway.
All these studies indicate that β-secretase and γ-secretase process their substrates in distinct subcellular organelles and, thus, the amino-terminal and CTFs of Aβ are generated in different organelles, and the APP β-cleavage products C99 or C89 need to be transported into the areas with fully mature and active γ-secretase complex.
Conclusion
APP and BACE1 trafficking are essential for APP processing. Enhancing their interaction and/or increasing their chance of co-residence in the same compartment or in close proximity to each other facilitates the β-cleavage of APP. BACE1 cleaves APP at two different sites, the β-site or β’-site, to produce C99 and C89, respectively, and the regulation of β-cleavage site specificity depends on the subcellular localization of BACE1. Targeting BACE1 into the ER or lipid raft microdomains results in the preferential cleavage of APP at the β-site to generate C99 with intact Aβ at its amino terminus. However, C99 has to be further transported into areas, such as the cell surface, with a mature and active γ-secretase complex, where it can be cleaved by γ-secretase to generate the CTF of Aβ.
The trafficking of APP and its processing enzymes, especially BACE1, is essential for APP processing and Aβ production. Understanding how APP and these enzymes are trafficked through their secretory pathways and how the substrates encounter these enzymes with preferential activities in distinct locations will provide important insights that could help in the development of therapeutic drugs in the future. Valid strategies could be to prevent Aβ production by altering the trafficking of APP and BACE1, or to use small molecules to block the accessibilty and interaction between substrates and enzymes.
Abbreviations
- Aβ:
-
Amyloid-β protein
- AD:
-
Alzheimer’s disease
- ADAM:
-
A distintergrin and metalloproteinase
- Aph-1:
-
Anterior pharynx-defective 1
- APP:
-
β-amyloid precursor protein
- BACE1:
-
β-site APP cleaving enzyme 1
- CTF:
-
Carboxy-terminal fragment
- ER:
-
Endoplasmic reticulum
- NCT:
-
Nicastrin
- Pen-2:
-
Presenilin enhance 2
- PM:
-
Plasma membrane
- PS:
-
Presenilin
- sAPPβ:
-
Secreted β-amyloid precursor protein
- TGN:
-
Trans-Golgi network.
References
AA-USA: 2011 Alzheimer’s disease facts and figures. W V Med J. 2011, 107: 82-83.
Divry P, Florkin M: Study histochemique senile plaques. J Belge Neurol Psychiatr. 1927, 27: 643-657.
Alzheimer A, Stelzmann RA, Schnitzlein HN, Murtagh FR: An English translation of Alzheimer's 1907 paper, "Uber eine eigenartige Erkankung der Hirnrinde". Clin Anat. 1995, 8: 429-431. 10.1002/ca.980080612.
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K: Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985, 82: 4245-4249. 10.1073/pnas.82.12.4245.
Glenner GG, Wong CW: Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984, 120: 885-890. 10.1016/S0006-291X(84)80190-4.
Goldgaber D, Lerman MI, McBride OW, Saffiotti U, Gajdusek DC: Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer's disease. Science. 1987, 235: 877-880. 10.1126/science.3810169.
Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B: The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987, 325: 733-736. 10.1038/325733a0.
Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George-Hyslop P, Van Keuren ML, Patterson D, Pagan S, Kurnit DM, Neve RL: Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987, 235: 880-884. 10.1126/science.2949367.
Robakis NK, Ramakrishna N, Wolfe G, Wisniewski HM: Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides. Proc Natl Acad Sci U S A. 1987, 84: 4190-4194. 10.1073/pnas.84.12.4190.
Capell A, Steiner H, Willem M, Kaiser H, Meyer C, Walter J, Lammich S, Multhaup G, Haass C: Maturation and pro-peptide cleavage of beta-secretase. J Biol Chem. 2000, 275: 30849-30854.
Caporaso GL, Takei K, Gandy SE, Matteoli M, Mundigl O, Greengard P, De Camilli P: Morphologic and biochemical analysis of the intracellular trafficking of the Alzheimer beta/A4 amyloid precursor protein. J Neurosci. 1994, 14: 3122-3138.
Koo EH, Squazzo SL, Selkoe DJ, Koo CH: Trafficking of cell-surface amyloid beta-protein precursor. I. Secretion, endocytosis and recycling as detected by labeled monoclonal antibody. J Cell Sci. 1996, 109: 991-998.
Perez RG, Soriano S, Hayes JD, Ostaszewski B, Xia W, Selkoe DJ, Chen X, Stokin GB, Koo EH: Mutagenesis identifies new signals for beta-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Abeta42. J Biol Chem. 1999, 274: 18851-18856. 10.1074/jbc.274.27.18851.
Koo EH, Squazzo SL: Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem. 1994, 269: 17386-17389.
Lai A, Sisodia SS, Trowbridge IS: Characterization of sorting signals in the beta-amyloid precursor protein cytoplasmic domain. J Biol Chem. 1995, 270: 3565-3573. 10.1074/jbc.270.8.3565.
Haass C, Koo EH, Mellon A, Hung AY, Selkoe DJ: Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature. 1992, 357: 500-503. 10.1038/357500a0.
Yamazaki T, Koo EH, Selkoe DJ: Trafficking of cell-surface amyloid beta-protein precursor. II. Endocytosis, recycling and lysosomal targeting detected by immunolocalization. J Cell Sci. 1996, 109: 999-1008.
Nordstedt C, Caporaso GL, Thyberg J, Gandy SE, Greengard P: Identification of the Alzheimer beta/A4 amyloid precursor protein in clathrin-coated vesicles purified from PC12 cells. J Biol Chem. 1993, 268: 608-612.
Parvathy S, Hussain I, Karran EH, Turner AJ, Hooper NM: Cleavage of Alzheimer's amyloid precursor protein by alpha-secretase occurs at the surface of neuronal cells. Biochemistry. 1999, 38: 9728-9734. 10.1021/bi9906827.
Skovronsky DM, Moore DB, Milla ME, Doms RW, Lee VM: Protein kinase C-dependent alpha-secretase competes with beta-secretase for cleavage of amyloid-beta precursor protein in the trans-golgi network. J Biol Chem. 2000, 275: 2568-2575. 10.1074/jbc.275.4.2568.
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M: Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999, 286: 735-741. 10.1126/science.286.5440.735.
Hook VY, Toneff T, Aaron W, Yasothornsrikul S, Bundey R, Reisine T: Beta-amyloid peptide in regulated secretory vesicles of chromaffin cells: evidence for multiple cysteine proteolytic activities in distinct pathways for beta-secretase activity in chromaffin vesicles. J Neurochem. 2002, 81: 237-256. 10.1046/j.1471-4159.2002.00794.x.
Huse JT, Pijak DS, Leslie GJ, Lee VM, Doms RW: Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer's disease beta-secretase. J Biol Chem. 2000, 275: 33729-33737. 10.1074/jbc.M004175200.
Pastorino L, Ikin AF, Nairn AC, Pursnani A, Buxbaum JD: The carboxyl-terminus of BACE contains a sorting signal that regulates BACE trafficking but not the formation of total A(beta). Mol Cell Neurosci. 2002, 19: 175-185. 10.1006/mcne.2001.1065.
Qing H, Zhou W, Christensen MA, Sun X, Tong Y, Song W: Degradation of BACE by the ubiquitin-proteasome pathway. Faseb J. 2004, 18: 1571-1573.
Zhang M, Deng Y, Luo Y, Zhang S, Zou H, Cai F, Wada K, Song W: Control of BACE1 degradation and APP processing by ubiquitin carboxyl-terminal hydrolase L1. J Neurochem. 2012, 120: 1129-1138.
Chyung JH, Raper DM, Selkoe DJ: Gamma-secretase exists on the plasma membrane as an intact complex that accepts substrates and effects intramembrane cleavage. J Biol Chem. 2005, 280: 4383-4392.
Zhang J, Kang DE, Xia W, Okochi M, Mori H, Selkoe DJ, Koo EH: Subcellular distribution and turnover of presenilins in transfected cells. J Biol Chem. 1998, 273: 12436-12442. 10.1074/jbc.273.20.12436.
Lee EB, Zhang B, Liu K, Greenbaum EA, Doms RW, Trojanowski JQ, Lee VM: BACE overexpression alters the subcellular processing of APP and inhibits Abeta deposition in vivo. J Cell Biol. 2005, 168: 291-302. 10.1083/jcb.200407070.
Cataldo AM, Barnett JL, Pieroni C, Nixon RA: Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer's disease: neuropathologic evidence for a mechanism of increased beta-amyloidogenesis. J Neurosci. 1997, 17: 6142-6151.
Selkoe DJ, Yamazaki T, Citron M, Podlisny MB, Koo EH, Teplow DB, Haass C: The role of APP processing and trafficking pathways in the formation of amyloid beta-protein. Ann N Y Acad Sci. 1996, 777: 57-64. 10.1111/j.1749-6632.1996.tb34401.x.
Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ: Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature. 1992, 360: 672-674. 10.1038/360672a0.
Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L: A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992, 1: 345-347. 10.1038/ng0892-345.
Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S: Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996, 2: 864-870. 10.1038/nm0896-864.
Cai XD, Golde TE, Younkin SG: Release of excess amyloid beta protein from a mutant amyloid beta protein precursor. Science. 1993, 259: 514-516. 10.1126/science.8424174.
Haass C, Lemere CA, Capell A, Citron M, Seubert P, Schenk D, Lannfelt L, Selkoe DJ: The Swedish mutation causes early-onset Alzheimer's disease by beta-secretase cleavage within the secretory pathway. Nat Med. 1995, 1: 1291-1296. 10.1038/nm1295-1291.
Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwatsubo T, Lee VM, Doms RW: Alzheimer's A beta(1–42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med. 1997, 3: 1021-1023. 10.1038/nm0997-1021.
Chyung AS, Greenberg BD, Cook DG, Doms RW, Lee VM: Novel beta-secretase cleavage of beta-amyloid precursor protein in the endoplasmic reticulum/intermediate compartment of NT2N cells. J Cell Biol. 1997, 138: 671-680. 10.1083/jcb.138.3.671.
Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K: Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A. 2006, 103: 11172-11177. 10.1073/pnas.0603838103.
Sisodia SS: Beta-amyloid precursor protein cleavage by a membrane-bound protease. Proc Natl Acad Sci U S A. 1992, 89: 6075-6079. 10.1073/pnas.89.13.6075.
Sannerud R, Declerck I, Peric A, Raemaekers T, Menendez G, Zhou L, Veerle B, Coen K, Munck S, De Strooper B, Schiavo G, Annaert W: ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc Natl Acad Sci U S A. 2011, 108: E559-E568. 10.1073/pnas.1100745108.
Golde TE, Estus S, Younkin LH, Selkoe DJ, Younkin SG: Processing of the amyloid protein precursor to potentially amyloidogenic derivatives. Science. 1992, 255: 728-730. 10.1126/science.1738847.
Sakurai T, Kaneko K, Okuno M, Wada K, Kashiyama T, Shimizu H, Akagi T, Hashikawa T, Nukina N: Membrane microdomain switching: a regulatory mechanism of amyloid precursor protein processing. J Cell Biol. 2008, 183: 339-352. 10.1083/jcb.200804075.
Lee J, Retamal C, Cuitino L, Caruano-Yzermans A, Shin JE, van Kerkhof P, Marzolo MP, Bu G: Adaptor protein sorting nexin 17 regulates amyloid precursor protein trafficking and processing in the early endosomes. J Biol Chem. 2008, 283: 11501-11508. 10.1074/jbc.M800642200.
Burgos PV, Mardones GA, Rojas AL, DaSilva LL, Prabhu Y, Hurley JH, Bonifacino JS: Sorting of the Alzheimer's disease amyloid precursor protein mediated by the AP-4 complex. Dev Cell. 2010, 18: 425-436. 10.1016/j.devcel.2010.01.015.
He X, Li F, Chang WP, Tang J: GGA proteins mediate the recycling pathway of memapsin 2 (BACE). J Biol Chem. 2005, 280: 11696-11703. 10.1074/jbc.M411296200.
He X, Chang WP, Koelsch G, Tang J: Memapsin 2 (beta-secretase) cytosolic domain binds to the VHS domains of GGA1 and GGA2: implications on the endocytosis mechanism of memapsin 2. FEBS Lett. 2002, 524: 183-187. 10.1016/S0014-5793(02)03052-1.
He X, Zhu G, Koelsch G, Rodgers KK, Zhang XC, Tang J: Biochemical and structural characterization of the interaction of memapsin 2 (beta-secretase) cytosolic domain with the VHS domain of GGA proteins. Biochemistry. 2003, 42: 12174-12180. 10.1021/bi035199h.
Shiba T, Kametaka S, Kawasaki M, Shibata M, Waguri S, Uchiyama Y, Wakatsuki S: Insights into the phosphoregulation of beta-secretase sorting signal by the VHS domain of GGA1. Traffic. 2004, 5: 437-448. 10.1111/j.1600-0854.2004.00188.x.
von Arnim CA, Tangredi MM, Peltan ID, Lee BM, Irizarry MC, Kinoshita A, Hyman BT: Demonstration of BACE (beta-secretase) phosphorylation and its interaction with GGA1 in cells by fluorescence-lifetime imaging microscopy. J Cell Sci. 2004, 117: 5437-5445. 10.1242/jcs.01422.
Wahle T, Prager K, Raffler N, Haass C, Famulok M, Walter J: GGA proteins regulate retrograde transport of BACE1 from endosomes to the trans-Golgi network. Mol Cell Neurosci. 2005, 29: 453-461. 10.1016/j.mcn.2005.03.014.
Tesco G, Koh YH, Kang EL, Cameron AN, Das S, Sena-Esteves M, Hiltunen M, Yang SH, Zhong Z, Shen Y, Simpkins JW, Tanzi RE: Depletion of GGA3 stabilizes BACE and enhances beta-secretase activity. Neuron. 2007, 54: 721-737. 10.1016/j.neuron.2007.05.012.
van der Sluijs P, Hull M, Webster P, Male P, Goud B, Mellman I: The small GTP-binding protein rab4 controls an early sorting event on the endocytic pathway. Cell. 1992, 70: 729-740. 10.1016/0092-8674(92)90307-X.
Kinoshita A, Fukumoto H, Shah T, Whelan CM, Irizarry MC, Hyman BT: Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J Cell Sci. 2003, 116: 3339-3346. 10.1242/jcs.00643.
Yan R, Han P, Miao H, Greengard P, Xu H: The transmembrane domain of the Alzheimer's beta-secretase (BACE1) determines its late Golgi localization and access to beta-amyloid precursor protein (APP) substrate. J Biol Chem. 2001, 276: 36788-36796. 10.1074/jbc.M104350200.
Vetrivel KS, Barman A, Chen Y, Nguyen PD, Wagner SL, Prabhakar R, Thinakaran G: Loss of cleavage at beta'-site contributes to apparent increase in beta-amyloid peptide (Abeta) secretion by beta-secretase (BACE1)-glycosylphosphatidylinositol (GPI) processing of amyloid precursor protein. J Biol Chem. 2011, 286: 26166-26177. 10.1074/jbc.M111.260471.
Cordy JM, Hussain I, Dingwall C, Hooper NM, Turner AJ: Exclusively targeting beta-secretase to lipid rafts by GPI-anchor addition up-regulates beta-site processing of the amyloid precursor protein. Proc Natl Acad Sci U S A. 2003, 100: 11735-11740. 10.1073/pnas.1635130100.
Pike LJ: Rafts defined: a report on the Keystone Symposium on Lipid Rafts and Cell Function. J Lipid Res. 2006, 47: 1597-1598. 10.1194/jlr.E600002-JLR200.
Rajendran L, Schneider A, Schlechtingen G, Weidlich S, Ries J, Braxmeier T, Schwille P, Schulz JB, Schroeder C, Simons M, Jennings G, Knolker HJ, Simons K: Efficient inhibition of the Alzheimer's disease beta-secretase by membrane targeting. Science. 2008, 320: 520-523. 10.1126/science.1156609.
Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J: Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc Natl Acad Sci U S A. 2000, 97: 1456-1460. 10.1073/pnas.97.4.1456.
Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, Seubert P, Suomensaari SM, Wang S, Walker D, Zhao J, McConlogue L, John V: Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999, 402: 537-540. 10.1038/990114.
Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME: Membrane-anchored aspartyl protease with Alzheimer's disease beta-secretase activity. Nature. 1999, 402: 533-537. 10.1038/990107.
Deng Y, Wang Z, Wang R, Zhang X, Zhang S, Wu Y, Staufenbiel M, Cai F, Song W: Amyloid-beta protein (Abeta) Glu11 is the major beta-secretase site of beta-site amyloid-beta precursor protein-cleaving enzyme 1(BACE1), and shifting the cleavage site to Abeta Asp1 contributes to Alzheimer pathogenesis. Eur J Neurosci. 2013, 37: 1962-1969. 10.1111/ejn.12235.
Creemers JW, Ines Dominguez D, Plets E, Serneels L, Taylor NA, Multhaup G, Craessaerts K, Annaert W, De Strooper B: Processing of beta-secretase by furin and other members of the proprotein convertase family. J Biol Chem. 2001, 276: 4211-4217. 10.1074/jbc.M006947200.
Li Y, Zhou W, Tong Y, He G, Song W: Control of APP processing and Abeta generation level by BACE1 enzymatic activity and transcription. FASEB J. 2006, 20: 285-292. 10.1096/fj.05-4986com.
Gouras GK, Xu H, Jovanovic JN, Buxbaum JD, Wang R, Greengard P, Relkin NR, Gandy S: Generation and regulation of beta-amyloid peptide variants by neurons. J Neurochem. 1998, 71: 1920-1925.
Selkoe DJ, Teplow DB, Schenk D, Koo EH, Lieberburg I, Ostaszewski BL, Mellon A, Vigo-Pelfrey C, Hung AY, Schlossmacher MG, Haass C: Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992, 359: 322-325. 10.1038/359322a0.
Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C, Mccormack R, Wolfert R, Olfert R, Selkoe D, Lieberburg I, Schenk D: Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature. 1992, 359: 325-327. 10.1038/359325a0.
Huse JT, Liu K, Pijak DS, Carlin D, Lee VM, Doms RW: Beta-secretase processing in the trans-Golgi network preferentially generates truncated amyloid species that accumulate in Alzheimer's disease brain. J Biol Chem. 2002, 277: 16278-16284. 10.1074/jbc.M111141200.
Liu K, Doms RW, Lee VM: Glu11 site cleavage and amino-terminally truncated A beta production upon BACE overexpression. Biochemistry. 2002, 41: 3128-3136. 10.1021/bi015800g.
Cupers P, Bentahir M, Craessaerts K, Orlans I, Vanderstichele H, Saftig P, De Strooper B, Annaert W: The discrepancy between presenilin subcellular localization and gamma-secretase processing of amyloid precursor protein. J Cell Biol. 2001, 154: 731-740. 10.1083/jcb.200104045.
Maltese WA, Wilson S, Tan Y, Suomensaari S, Sinha S, Barbour R, McConlogue L: Retention of the Alzheimer's amyloid precursor fragment C99 in the endoplasmic reticulum prevents formation of amyloid beta-peptide. J Biol Chem. 2001, 276: 20267-20279. 10.1074/jbc.M007238200.
Kaether C, Schmitt S, Willem M, Haass C: Amyloid precursor protein and Notch intracellular domains are generated after transport of their precursors to the cell surface. Traffic. 2006, 7: 408-415. 10.1111/j.1600-0854.2006.00396.x.
Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM: Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008, 58: 42-51. 10.1016/j.neuron.2008.02.003.
Kim SH, Yin YI, Li YM, Sisodia SS: Evidence that assembly of an active gamma-secretase complex occurs in the early compartments of the secretory pathway. J Biol Chem. 2004, 279: 48615-48619. 10.1074/jbc.C400396200.
Capell A, Beher D, Prokop S, Steiner H, Kaether C, Shearman MS, Haass C: Gamma-secretase complex assembly within the early secretory pathway. J Biol Chem. 2005, 280: 6471-6478. 10.1074/jbc.M409106200.
Niimura M, Isoo N, Takasugi N, Tsuruoka M, Ui-Tei K, Saigo K, Morohashi Y, Tomita T, Iwatsubo T: Aph-1 contributes to the stabilization and trafficking of the gamma-secretase complex through mechanisms involving intermolecular and intramolecular interactions. J Biol Chem. 2005, 280: 12967-12975.
LaVoie MJ, Fraering PC, Ostaszewski BL, Ye W, Kimberly WT, Wolfe MS, Selkoe DJ: Assembly of the gamma-secretase complex involves early formation of an intermediate subcomplex of Aph-1 and nicastrin. J Biol Chem. 2003, 278: 37213-37222. 10.1074/jbc.M303941200.
Hu Y, Fortini ME: Different cofactor activities in gamma-secretase assembly: evidence for a nicastrin-Aph-1 subcomplex. J Cell Biol. 2003, 161: 685-690. 10.1083/jcb.200304014.
Fraering PC, LaVoie MJ, Ye W, Ostaszewski BL, Kimberly WT, Selkoe DJ, Wolfe MS: Detergent-dependent dissociation of active gamma-secretase reveals an interaction between Pen-2 and PS1-NTF and offers a model for subunit organization within the complex. Biochemistry. 2004, 43: 323-333. 10.1021/bi035748j.
Edbauer D, Winkler E, Haass C, Steiner H: Presenilin and nicastrin regulate each other and determine amyloid beta-peptide production via complex formation. Proc Natl Acad Sci U S A. 2002, 99: 8666-8671.
Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS: Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996, 17: 181-190. 10.1016/S0896-6273(00)80291-3.
Capell A, Grunberg J, Pesold B, Diehlmann A, Citron M, Nixon R, Beyreuther K, Selkoe DJ, Haass C: The proteolytic fragments of the Alzheimer's disease-associated presenilin-1 form heterodimers and occur as a 100-150-kDa molecular mass complex. J Biol Chem. 1998, 273: 3205-3211. 10.1074/jbc.273.6.3205.
Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, Selkoe DJ: Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc Natl Acad Sci U S A. 2003, 100: 6382-6387. 10.1073/pnas.1037392100.
Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, Qian X, Ginty DD, Price DL, Borchelt DR, Wong PC, Sisodia SS: Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 1998, 21: 1213-1221. 10.1016/S0896-6273(00)80637-6.
Rechards M, Xia W, Oorschot VM, Selkoe DJ, Klumperman J: Presenilin-1 exists in both pre- and post-Golgi compartments and recycles via COPI-coated membranes. Traffic. 2003, 4: 553-565. 10.1034/j.1600-0854.2003.t01-1-00114.x.
Rechards M, Xia W, Oorschot V, van Dijk S, Annaert W, Selkoe DJ, Klumperman J: Presenilin-1-mediated retention of APP derivatives in early biosynthetic compartments. Traffic. 2006, 7: 354-364. 10.1111/j.1600-0854.2006.00388.x.
Acknowledgements
We thank Drs Yili Wu and Philip Ly for their critical comments and help in drawing the schematic figures. This work was supported by Canadian Institutes of Health Research (CIHR) Operating Grant CCI-117952. WS is the holder of the Tier 1 Canada Research Chair in Alzheimer's Disease. XZ is the recipient of the Chinese Scholarship Council award.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Zhang, X., Song, W. The role of APP and BACE1 trafficking in APP processing and amyloid-β generation. Alz Res Therapy 5, 46 (2013). https://doi.org/10.1186/alzrt211
Published:
DOI: https://doi.org/10.1186/alzrt211