
Abstract
Frontotemporal dementia (FTD) is a common dementia syndrome in patients under the age of 65 years with many features overlapping with amyotrophic lateral sclerosis (ALS). The link between FTD and ALS has been strengthened by the discovery that a hexanucleotide repeat expansion in a non-coding region of the C9ORF72 gene causes both familial and sporadic types of these two diseases. As we begin to understand the pathophysiological mechanisms by which this mutation leads to FTD and ALS (c9FTD/ALS), new targets for disease-modifying therapies will likely be unveiled. Putative C9ORF72 expansion pathogenic mechanisms include loss of C9ORF72 protein function, sequestration of nucleic acid binding proteins due to expanded hexanucleotide repeats, or a combination of the two. New animal models and other research tools informed by work in other repeat expansion neurodegenerative diseases such as the spinocerebellar ataxias will help to elucidate the mechanisms of C9ORF72-mediated disease. Similarly, re-examining previous studies of drugs developed to treat ALS in light of this new mutation may identify novel FTD treatments. Ultimately, research consortiums incorporating animal models and well-characterized clinical populations will be necessary to fully understand the natural history of the c9FTD/ALS clinical phenotypes and identify biomarkers and therapeutic agents that can cure the most common form of genetically determined FTD and ALS.
Similar content being viewed by others


Background
Genetic forms of frontotemporal dementia and amyotrophic lateral sclerosis
Frontotemporal dementia (FTD) is a common dementia in people aged under 65 years [1, 2] characterized by impaired social comportment, apathy, lack of empathy, cognitive decline, and appetite changes with neuropathologic and genetic features overlapping with amyotrophic lateral sclerosis (ALS) in a subset of patients. ALS is a neurodegenerative disorder that, in its most common form, causes both upper and lower motor neuron signs with muscle wasting and a rapid progression to death within 3 to 5 years. These two diseases often coexist, with 22% of ALS patients meeting FTD diagnostic criteria and a greater number (48%) manifesting cognitive or behavioral abnormalities of FTD but not the full syndrome [3, 4]. Conversely, 15% of FTD patients display signs of motor neuron disease or ALS [5], suggesting that these diseases lay along the same disease spectrum. A family history of dementia is present in about 40% of FTD cases, with an autosomal dominant pattern of inheritance identifiable in 10% of cases [6]. ALS has an autosomal dominant pattern in up to 10% of cases as well [7]. Previously, most of the known genetic causes of FTD were attributed to mutations on chromosome 17, in genes encoding the microtubule associated-protein tau (MAPT) [8] or progranulin (GRN) [9, 10]. Prior to the discovery of C9ORF72, the most common mutation associated with ALS disease was in superoxide dismutase (SOD1) [11, 12]. Other mutations identified in familial ALS include UBQLN2 [13], TDP43, FUS, OPTN, and VCP.
The discovery of C9ORF72
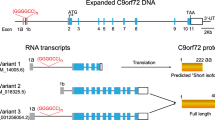
A variety of prior linkage analysis studies of families in which members have developed FTD, ALS or both (FTD-ALS) in an autosomal dominant inheritance pattern suggested linkage to a region on chromosome 9p [14–22]. A collaborative effort between our group at the University of California, San Francisco (UCSF), researchers at the Mayo Clinic, and the University of British Columbia (UBC) led to the discovery in 2011 that a hexanucleotide repeat expansion in a non-coding region, the promoter or the first intron, of the chromosome 9 open reading frame 72 (C9ORF72) gene was the cause of FTD and ALS in the most strongly linked family (Vancouver San Francisco Mayo-20 (VSM-20) family) to chromosome 9p. Analysis of other autosomal dominant FTD kindreds revealed this mutation to be the most common genetic cause of FTD (12% of familial FTD; 3% of sporadic FTD), ALS (23% of familial ALS; 4% of sporadic ALS) or combined FTD-ALS at each of these institutions [12, 16, 23]. At the same time, another group found the same genetic mutation in a Finnish population with higher prevalence (46% of familial ALS; 21% of sporadic ALS) [24]. In the initial studies, the clinical disease phenotypes associated with this mutation most commonly included FTD, ALS, and FTD-ALS [25–27]. Less frequently, other phenotypes, such as the non-fluent variant of primary progressive aphasia (nfvPPA) and semantic variant of primary progressive aphasia (svPPA), both with and without motor neuron disease, have been observed [12, 28, 29] in addition to Alzheimer's disease [30]. At autopsy, examination of these mutation carriers identified frontotemporal lobar degeneration-TAR DNA binding protein-43 (TDP-43; FTLD-TDP) neuropathology in all. The location, morphology, and distribution of TDP-43 immunoreactive inclusions define the TDP subtype of FTD pathology [31] and two subtypes, FTLD-TDP type A and type B, have been reported in association with the C9ORF72 mutation [26–32]. Additionally, immunoreactivity to ubiquilin (UBQLN) and p62 (sequestosome 1), proteins involved in cellular protein degradation pathways, as well as an as yet unidentified protein, have been noted in mutation carriers [12, 16, 26–29, 32–35] and have been hypothesized to be signature pathological features of C9ORF72-related disease [35].
Treatment implications
The discovery of the C9ORF72 mutation has important treatment implications for patients with FTD.
First, this mutation may reveal important mechanistic information about the molecular triggers for FTD and ALS, thus allowing the identification of novel drug targets. In addition, the discovery of C9ORF72 mutations as a cause of FTD may help to resolve some confusing dissociations between two genes that, when mutated, cause ALS but rarely FTD, yet are found at autopsy in the form of insoluble protein deposits in both disorders: TDP-43 and fused in sarcoma (FUS). Since both TDP-43 and FUS are RNA binding proteins, the finding that C9ORF72 expansions have the potential to alter RNA binding protein levels may be particularly important for understanding the biochemical mechanisms underlying FTD-ALS. Specifically, C9ORF72 repeat expansions decrease the levels of TDP-43 or FUS, which could affect RNA transport or processing and may be a key pathophysiological trigger for FTD-ALS. In addition, C9ORF72 mutations could also impair RNA metabolism if the hexanucleotide repeat expansions sequester other nucleic acid binding proteins [23]. Thus, cellular RNA processing and transport mechanisms are likely to be key drug targets for FTD-ALS.
Second, since the C9ORF72 mutation is by far the most prevalent cause of FTD and ALS, accounting for 11.7% of familial FTD, 22.5% of familial ALS, and 4% of sporadic ALS [23], and as much as 46% of familial ALS and 21.1% of sporadic ALS in a Finnish population [24], a treatment developed for C9ORF72 mutation carriers might eventually find a use in both inherited and sporadic forms of these diseases, potentially benefitting a significant pro-portion of patients with both disorders. Both possibilities are discussed in greater detail below.
Drug discovery opportunities afforded by the C9ORF72 mutation
Target identification
Targeting the pathological mechanism responsible for C9ORF72-associated FTD and ALS is a logical first step in leveraging this discovery to develop new treatments for both C9ORF72-associated disease as well as other forms of FTD and ALS. Two non-mutually exclusive mechanisms might explain the pathogenesis of C9ORF72-related FTD-ALS. Expanded repeat disorders in untranslated regions or introns generally can cause disease pathogenesis by loss of function due to decreased protein expression, or by toxic gain of function due to inclusion of multiple repeats within DNA or RNA transcripts [36]. The hexanucleotide expansion can occur in the C9ORF72 gene promoter region that binds to transcription regulatory factors. This can lead to decreased C9ORF72 gene transcription and ultimately protein expression. Consistent with this hypothesis, one of the three RNA splice variant mRNAs from C9ORF72 was decreased in mutation carriers compared to non-carriers in two separate studies [23, 37]. Thus, one target for new FTD drugs might be agents that increase C9ORF72 protein levels, or make up for the loss of C9ORF72 protein function.
Expanded hexanucleotide repeats in RNA transcripts could result in aberrant splicing or generation of RNA fragments that form nuclear inclusions. These foci could sequester RNA-binding proteins in the nucleus and alter regulation and splicing of other genes. As a result, the C9ORF72 hexanucleotide expansion RNA foci could have multi-systemic effects. Such a sequestration mechanism occurs in other non-coding repeat expansion diseases such as myotonic dystrophy (DM1) and fragile X-associated tremor/ataxia syndrome (FXTAS) [38, 39], which have both neuronal and non-neuronal phenotypes. This suggests that a second target for new FTD therapies would be the repeat expansions themselves or the RNA fragment foci that form as a result of the repeat expansions. A final possibility is that RNA-binding protein sequestration by expanded hexanucleotide repeats and haploinsuciency of C9ORF72 protein both contribute to the disease mechanism and could be targets for therapeutic intervention (Figure 1).

Drug development opportunities resulting from the C9ORF72 mutation discovery. The figure shows a general, hypothetical drug development plan with opportunities resulting from the discovery at multiple pre-clinical and clinical development stages. ALS, amyotrophic lateral sclerosis; C9ORF72, chromosome 9 open reading frame 72; FTLD, frontotemporal lobar degeneration; TDP, TAR DNA binding protein.
RNA as a therapeutic target
Clues to identifying which compounds might prove efficacious for C9ORF72-related disease can be found by looking at other neurodegenerative disease models with similar repeat expansion pathophysiology. DM1, FXTAS, and several spinocerebellar ataxias have repeat expansions in non-coding regions that may lead to targeted drug discovery efforts or already have these underway [40]. Examining previously tested drugs (both failed and promising) and drug targets in these disorders might provide starting points for C9ORF72. RNA antisense oligonucleotides have been studied in DM1 [41, 42], were tolerated in a phase I clinical trial for SOD1-related ALS, and could be applied in c9FTD/ALS. These oligonucleotides could act to interrupt sequestration of critical proteins by toxic RNA hexanucleotide repeat expansions or potentially alter the transcription or splicing of C9ORF72. Alternatively, the oligonucleotides could disrupt RNA hairpin structures or other steric conformations that are thought to have toxic effects in other repeat expansion mutation diseases [36, 39, 43].
TDP-43 as a drug target
TDP-43 is another attractive drug target in C9ORF72-related FTD/ALS. Although TDP type A and B have been reported, all autopsy studies of C9ORF72 mutation carriers thus far have been noted to have TDP-43 pathology. Even with the variable FTLD-TDP pathology, a compound that increases clearance or inhibits aggregation of TDP-43 protein could be useful in c9FTD/ALS. One compound that does this is methylene blue, which can decrease TDP-43 aggregation in vitro [44], although, thus far, methylene blue has failed to demonstrate improvements in motor function in TDP-43 mouse models of ALS [45]. Methylene blue may also promote autophagy [46]. Compounds that increase cellular protein turnover via autophagy or the proteasome pathway might also be candidate therapies for C9ORF72-related disease, particularly since there is evidence of accumulation of proteins such as UBQLN and p62 in these cases [33]. Finally, if developed, immunotherapies (vaccines or neutralizing antibodies) targeted towards TDP-43 would be attractive therapies. A variety of such therapies are in development for neurodegenerative diseases with tau, amyloid, and synuclein pathology.
Target validation
In order to determine which mechanism(s) is/are pathogenic, cell-based studies or animal models of C9ORF72-related disease are needed. Transgenic mouse models have been used to study many degenerative diseases, including Alzheimer's disease and ALS, and may ultimately be most useful for developing C9ORF72-targeted therapeutics. In addition, if C9ORF72 homologues exist in Caenorhabditis elegans and Drosophila, these model systems may also be useful for target identification [47]. Induced pluripotent stem cells have also been used to create both patient- and disease-specific cells [48] in order to better study the pathophysiology [49]. High throughput drug screening using cells from C9ORF72 mutation gene carriers, such as those that we have derived from the VSM-20 family, could be used to screen for potential compounds. When therapeutic interventions are identified, patient-specific cell lines can be used to test the toxicology and potential benefit for that individual patient. Given the heterogeneity of C9ORF72 phenotypes, with both slowly and rapidly progressive forms of disease [50], use of patient-specific induced pluripotent stem cells may be particularly useful for C9ORF72-related disease.
Application of current ALS experimental therapeutics to C9ORF72 disease
Other potential agents to consider for treatment of c9FTD/ALS are ones already used or in late stage clinical trials in ALS [51]. Considering the pathological, genetic, and phenotypic similarities now known to be shared with FTD, drugs found to be efficacious for ALS might also be expected to benefit individuals with FTD due to TDP-43, particularly those caused by C9ORF72. Riluzole, a neuroprotective agent thought to block voltage-dependent sodium channels on glutamatergic nerve terminals, is the only US Food and Drug Administration-approved drug to treat ALS and has been shown to reduce mortality, though modestly [52–54], and may be worthwhile testing in preclinical C9ORF72 models. Dexpramipexole, an enantiomer of pramipexole, is thought to have anti-inflammatory properties and was recently found to attenuate the decline in function using the ALS Functional Rating Scale-Revised (ALSFRS) in a dose-dependent manner with good tolerability in ALS [55]. Fingolimod, an anti-inflammatory drug used to treat multiple sclerosis in several countries outside the United States, will soon begin phase II clinical trial in ALS [56] and may also have promise in FTD. Clinical trials of agents that have clearly shown no benefit in ALS, like those with lithium [57], may also be useful in guiding such therapies away from use in FTD due to C9ORF72.
To streamline identification of promising treatments for C9ORF72-related disease, cases from previous ALS clinical trials should be genotyped. Given the high prevalence of the C9ORF72 mutation in ALS, agents that are beneficial for sporadic ALS may also be useful in C9ORF72-associated FTD and FTD-ALS. Such a response might be predicted if post hoc genetic analyses of previous ALS clinical studies showed that C9ORF72 patients clearly benefited from a drug. Even if an overall ALS clinical trial was negative, it remains possible that C9ORF72 carriers could have been a responsive sub-population in whom effects were masked by non-carriers. Similarly, it would be of interest to genotype patients who respond particularly well to a given therapy to assess whether this relates to C9ORF72 gene status.
Identifying disease modifying factors
Studying patients who are carriers of the C9ORF72 mutation with particular attention to the genetic and environmental factors that can slow or alter the disease phenotype is another way to learn about the disease mechanism to identify potential drug targets. An example of a slowly progressive FTD (FTD-SP) phenotype of C9ORF72 disease has been described recently [50]. FTD-SP patients have features of FTD, yet have been noted to have minimal atrophy on structural MRI and little to no progression on sequential neuropsychological measures. Identifying the factors that affect the rate of disease progression like those in FTD-SP patients would provide insight into other targets for potential therapies. An important question that has yet to be answered is whether the number of hexanucleotide repeats affects the C9ORF72 phenotype, similar to other repeat expansion disorders. Preliminary studies have found that normal controls have no more than 23 to 30 repeats of the hexanucleotide (GGGGCC), but carriers of the mutated alleles usually have over 60 [37] and as high as 1,600 [23], although the number of repeats is not easily quantified.
It is also likely that other genes exist that modify the C9ORF72 phenotype. For example, in FTLD-TDP caused by progranulin (GRN) mutations, a number of genes and microRNAs have been identified that alter the disease phenotype [58]. The presence of certain TMEM106B single nucleotide polymorphisms was shown to reduce GRN mutation penetrance possibly by modifying progranulin protein levels [59]. TMEM106B could thus be a target for new therapies for patients with GRN mutations, and similarly, genes that modify C9ORF72 protein levels or function would be good targets for drugs in C9ORF72 mutation carriers.
Studies such as COHORT-HD (Cooperative Huntington's Observational Research Trial) that seek to identify genetic and environmental factors that modify disease progression are being pursued in other repeat expansion diseases such as Huntington's disease [60] and suggest that similar efforts should be pursued in c9FTD/ALS. A large study like this, if employed for C9ORF72, could identify both genetic and epigenetic factors that influence the C9ORF72 hexanucleotide expansion phenotype. Potentially, factors such as the number of hexanucleotide repeats, brain atrophy pattern at baseline, or environmental exposures could be used to identify other targets for C9ORF72 disease modifying agents.
Human clinical trials
In preparing for clinical trials on mutation carriers of C9ORF72, a first step would be to use the C9ORF72 genotype as a biomarker for diagnostic inclusion. If the rate of progression of disease is related to the length of repeats, as seen in other repeat expansion diseases like spinocerebellar ataxias and Huntington's disease, this could also help to select certain populations of C9ORF72 mutation carriers who are expected to progress at the same rate. To determine if a particular agent is modifying the course of C9ORF72 disease or delaying expression of the disease phenotype in a mutation carrier, a biomarker that accurately captures disease progression would be particularly helpful.
A cure for C9ORF72-related disease is more likely if a disease modifying treatment can be initiated early in the course of the disease, ideally before the onset of disease. By following the model of other groups that study autosomal dominant forms of dementia, such as the Dominantly Inherited Alzheimer Network (DIAN), future researchers can emulate methods to study the effect of the C9ORF72 mutation in presymptomatic mutation carriers. DIAN is a clinical research network that studies the presymptomatic events that occur in autosomal dominant Alzheimer's disease gene (mainly presenilin 1 and amyloid precursor protein) carriers to learn about the disease. DIAN has identified changes in neuroimaging and fluid biomarkers that precede the development of AD in these cases, often by 15 years or more. Biomarkers will be crucial to gauge the efficacy of therapeutic agents in clinical trials of disease modifying agents initiated before the patient displays clinically manifest disease. Such a presymptomatic 'prevention' trial is currently planned for DIAN as well as another similar Alzheimer's disease initiative called the Alzheimer's Disease Prevention Initiative. Once biomarkers that capture C9ORF72 disease progression are developed (one possibility might be cerebrospinal fluid TDP-43 measurements), similar C9ORF72 prevention clinical trials might be considered.
Conclusions
The discovery of the hexanucleotide repeat expansion in the C9ORF72 gene is a major step forward in understanding the pathophysiology of the FTD/ALS spectrum of diseases. With this information, the time is ripe for developing treatments that target specific C9ORF72-associated disease mechanisms. Moreover, the link between various inherited neurodegenerative diseases like FXTAS, DM1, spinocerebellar ataxias, and FTD is becoming stronger as more is learned about the pathogenic mechanisms of nucleotide expansion repeat diseases. A possible common mechanism for all FTLD-TDP diseases involving RNA processing abnormalities could also facilitate the identification of novel therapeutic agents. In order to achieve the goals of finding a disease-modifying agent for C9ORF72 FTD/ALS, an appropriate biomarker of disease progression or severity must be identified to be used in human pharmacodynamics and efficacy studies. For example, if TDP-43 is the drug target, then finding an in vivo tool for measuring the burden of pathology, such as a cerebrospinal fluid or imaging biomarker, might be necessary. Likewise, if raising disease-relevant mRNA levels is the goal of a potential compound, it is important to demonstrate that the RNA levels change with disease. Measuring cerebrospinal or plasma RNA levels might be one way to demonstrate such target engagement in human subjects. Future clinical trials could also use measurements of such levels as a surrogate endpoint of efficacy. Further research is required before specific C9ORF72- related compounds can be developed and tested in humans, but the discovery of the C9ORF72 mutation suggests that an important pathophysiological mechanism involves FTLD-TDP RNA processing. This finding may lead to new therapies for FTD, ALS, and possibly other repeat expansion degenerative disorders.
Abbreviations
- ALS:
-
amyotrophic lateral sclerosis
- C9ORF72 chromosome 9 open reading frame :
-
72
- DIAN:
-
Dominantly Inherited Alzheimer Network
- DM1:
-
myotonic dystrophy
- FTD:
-
frontotemporal dementia
- FTD-SP:
-
slowly progressive FTD
- FTLD:
-
frontotemporal lobar degeneration
- FXTAS:
-
fragile X-associated tremor/ataxia syndrome
- GRN:
-
progranulin
- MRI:
-
magnetic resonance imaging
- TDP:
-
TAR DNA binding protein
- UBQLN:
-
ubiquilin
- VSM-20:
-
Vancouver San Francisco Mayo-20 family.
References
Knopman DS, Petersen RC, Edland SD, Cha RH, Rocca WA: The incidence of frontotemporal lobar degeneration in Rochester, Minnesota, 1990 through 1994. Neurology. 2004, 62: 506-508. 10.1212/01.WNL.0000106827.39764.7E.
Ratnavalli E, Brayne C, Dawson K, Hodges JR: The prevalence of frontotemporal dementia. Neurology. 2002, 58: 1615-1621. 10.1212/WNL.58.11.1615.
Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B: Are amyotrophic lateral sclerosis patients cognitively normal?. Neurology. 2003, 60: 1094-1097. 10.1212/01.WNL.0000055861.95202.8D.
Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE: Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005, 65: 586-590. 10.1212/01.wnl.0000172911.39167.b6.
Lomen-Hoerth C, Anderson T, Miller B: The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002, 59: 1077-1079. 10.1212/WNL.59.7.1077.
Goldman JS, Rademakers R, Huey ED, Boxer AL, Mayeux R, Miller BL, Boeve BF: An algorithm for genetic testing of frontotemporal lobar degeneration. Neurology. 2011, 76: 475-483. 10.1212/WNL.0b013e31820a0d13.
Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC: Amyotrophic lateral sclerosis. Lancet. 2011, 377: 942-955. 10.1016/S0140-6736(10)61156-7.
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H: Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998, 393: 702-705. 10.1038/31508.
Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M: Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006, 442: 916-919. 10.1038/nature05016.
Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C: Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006, 442: 920-924. 10.1038/nature05017.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX: Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993, 362: 59-62. 10.1038/362059a0.
Hsiung GY, DeJesus-Hernandez M, Feldman HH, Sengdy P, Bouchard-Kerr P, Dwosh E, Butler R, Leung B, Fok A, Rutherford NJ, Baker M, Rademakers R, Mackenzie IR: Clinical and pathological features of familial frontotemporal dementia caused by C9ORF72 mutation on chromosome 9p. Brain. 2012, 135: 709-722. 10.1093/brain/awr354.
Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, Jiang H, Hirano M, Rampersaud E, Jansen GH, Donkervoort S, Bigio EH, Brooks BR, Ajroud K, Sufit RL, Haines JL, Mugnaini E, Pericak-Vance MA, Siddique T: Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011, 477: 211-215. 10.1038/nature10353.
Pearson JP, Williams NM, Majounie E, Waite A, Stott J, Newsway V, Murray A, Hernandez D, Guerreiro R, Singleton AB, Neal J, Morris HR: Familial frontotemporal dementia with amyotrophic lateral sclerosis and a shared haplotype on chromosome 9p. J Neurol. 2011, 258: 647-655. 10.1007/s00415-010-5815-x.
Gijselinck I, Engelborghs S, Maes G, Cuijt I, Peeters K, Mattheijssens M: Identification of 2 Loci at chromosomes 9 and 14 in a multiplex family with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol. 2010, 67: 606-616. 10.1001/archneurol.2010.82.
Boxer AL, Mackenzie IR, Boeve BF, Baker M, Seeley WW, Crook R, Feldman H, Hsiung GY, Rutherford N, Laluz V, Whitwell J, Foti D, McDade E, Molano J, Karydas A, Wojtas A, Goldman J, Mirsky J, Sengdy P, Dearmond S, Miller BL, Rademakers R: Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry. 2011, 82: 196-203. 10.1136/jnnp.2009.204081.
Vance C, Al-Chalabi A, Ruddy D, Smith BN, Hu X, Sreedharan J, Siddique T, Schelhaas HJ, Kusters B, Troost D, Baas F, de Jong V, Shaw CE: Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain. 2006, 129: 868-876. 10.1093/brain/awl030.
Morita M, Al-Chalabi A, Andersen PM, Hosler B, Sapp P, Englund E, Mitchell JE, Habgood JJ, de Belleroche J, Xi J, Jongjaroenprasert W, Horvitz HR, Gunnarsson LG, Brown RH: A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006, 66: 839-844. 10.1212/01.wnl.0000200048.53766.b4.
Momeni P, Schymick J, Jain S, Cookson MR, Cairns NJ, Greggio E, Greenway MJ, Berger S, Pickering-Brown S, Chiò A, Fung HC, Holtzman DM, Huey ED, Wassermann EM, Adamson J, Hutton ML, Rogaeva E, St George-Hyslop P, Rothstein JD, Hardiman O, Grafman J, Singleton A, Hardy J, Traynor BJ: Analysis of IFT74 as a candidate gene for chromosome 9p-linked ALS-FTD. BMC Neurol. 2006, 6: 44-10.1186/1471-2377-6-44.
Luty AA, Kwok JB, Thompson EM, Blumbergs P, Brooks WS, Loy CT, Dobson-Stone C, Panegyres PK, Hecker J, Nicholson GA, Halliday GM, Schofield PR: Pedigree with frontotemporal lobar degeneration - motor neuron disease and Tar DNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9. BMC Neurol. 2008, 8: 32-10.1186/1471-2377-8-32.
Valdmanis PN, Dupre N, Bouchard JP, Camu W, Salachas F, Meininger V, Strong M, Rouleau G: Three families with amyotrophic lateral sclerosis and frontotemporal dementia with evidence of linkage to chromosome 9p. Arch Neurol. 2007, 64: 240-245. 10.1001/archneur.64.2.240.
Le Ber I, Camuzat A, Berger E, Hannequin D, Laquerrière A, Golfier V, Seilhean D, Viennet G, Couratier P, Verpillat P, Heath S, Camu W, Martinaud O, Lacomblez L, Vercelletto M, Salachas F, Sellal F, Didic M, Thomas-Anterion C, Puel M, Michel BF, Besse C, Duyckaerts C, Meininger V, Campion D, Dubois B, Brice A; French Research Network on FTD/FTD-MND: Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology. 2009, 72: 1669-1676. 10.1212/WNL.0b013e3181a55f1c.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R: Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011, 72: 245-256. 10.1016/j.neuron.2011.09.011.
Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE: A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011, 72: 257-268. 10.1016/j.neuron.2011.09.010.
Sha SJ, Takada LT, Rankin KP, Yokoyama JS, Rutherford NJ, Fong JC, Khan B, Karydas A, Baker MC, DeJesus-Hernandez M, Pribadi M, Coppola G, Geschwind DH, Rademakers R, Lee SE, Seeley W, Miller BL, Boxer AL: Frontotemporal dementia due to C9ORF72 mutations. Neurology. 2012, 79: 1002-1011. 10.1212/WNL.0b013e318268452e.
Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, Vemuri P, Jones D, Lowe V, Murray ME, Dickson DW, Josephs KA, Rush BK, Machulda MM, Fields JA, Ferman TJ, Baker M, Rutherford NJ, Adamson J, Wszolek ZK, Adeli A, Savica R, Boot B, Kuntz KM, Gavrilova R, Reeves A, Whitwell J, Kantarci K, Jack CR, Parisi JE: Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain. 2012, 135: 765-783. 10.1093/brain/aws004.
Mahoney CJ, Beck J, Rohrer JD, Lashley T, Mok K, Shakespeare T, Yeatman T, Warrington EK, Schott JM, Fox NC, Rossor MN, Hardy J, Collinge J, Revesz T, Mead S, Warren JD: Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain. 2012, 135: 736-750. 10.1093/brain/awr361.
Stewart H, Rutherford NJ, Briemberg H, Krieger C, Cashman N, Fabros M, Baker M, Fok A, DeJesus-Hernandez M, Eisen A, Rademakers R, Mackenzie IR: Clinical and pathological features of amyotrophic lateral sclerosis caused by mutation in the C9ORF72 gene on chromosome 9p. Acta Neuropathol. 2012, 123: 409-417. 10.1007/s00401-011-0937-5.
Simón-Sánchez J, Dopper EG, Cohn-Hokke PE, Hukema RK, Nicolaou N, Seelaar H, de Graaf JR, de Koning I, van Schoor NM, Deeg DJ, Smits M, Raaphorst J, van den Berg LH, Schelhaas HJ, De Die-Smulders CE, Majoor-Krakauer D, Rozemuller AJ, Willemsen R, Pijnenburg YA, Heutink P, van Swieten JC: The clinical and pathological phenotype of C9orf72 hexanucleotide repeat expansions. Brain. 2012, 135: 723-735. 10.1093/brain/awr353.
Murray ME, DeJesus-Hernandez M, Rutherford NJ, Baker M, Duara R, Graff-Radford NR, Wszolek ZK, Ferman TJ, Josephs KA, Boylan KB, Rademakers R, Dickson DW: Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathol. 2011, 122: 673-690. 10.1007/s00401-011-0907-y.
Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, Perry RH, Trojanowski JQ, Mann DM, Lee VM: A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011, 122: 111-113. 10.1007/s00401-011-0845-8.
Snowden JS, Rollinson S, Thompson JC, Harris JM, Stopford CL, Richardson AM, Jones M, Gerhard A, Davidson YS, Robinson A, Gibbons L, Hu Q, DuPlessis D, Neary D, Mann DM, Pickering-Brown SM: Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain. 2012, 135: 693-708. 10.1093/brain/awr355.
Brettschneider J, Van Deerlin VM, Robinson JL, Kwong L, Lee EB, Ali YO, Safren N, Monteiro MJ, Toledo JB, Elman L, McCluskey L, Irwin DJ, Grossman M, Molina-Porcel L, Lee VM, Trojanowski JQ: Pattern of ubiquilin pathology in ALS and FTLD indicates presence of C9ORF72 hexanucleotide expansion. Acta Neuropathol. 2012, 123: 825-839. 10.1007/s00401-012-0970-z.
Troakes C, Maekawa S, Wijesekera L, Rogelj B, Siklós L, Bell C, Smith B, Newhouse S, Vance C, Johnson L, Hortobágyi T, Shatunov A, Al-Chalabi A, Leigh N, Shaw CE, King A, Al-Sarraj S: An MND/ALS phenotype associated with C9orf72 repeat expansion: Abundant p62-positive, TDP-43-negative inclusions in cerebral cortex, hippocampus and cerebellum but without associated cognitive decline. Neuropathology. 2011, 32: 505-514.
Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobágyi T, Shaw CE: p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011, 122: 691-702. 10.1007/s00401-011-0911-2.
Krzyzosiak WJ, Sobczak K, Wojciechowska M, Fiszer A, Mykowska A, Kozlowski P: Triplet repeat RNA structure and its role as pathogenic agent and therapeutic target. Nucleic Acids Res. 2012, 40: 11-26. 10.1093/nar/gkr729.
Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S, Engelborghs S, Sieben A, De Jonghe P, Vandenberghe R, Santens P, De Bleecker J, Maes G, Bäumer V, Dillen L, Joris G, Cuijt I, Corsmit E, Elinck E, Van Dongen J, Vermeulen S, Van den Broeck M, Vaerenberg C, Mattheijssens M, Peeters K, Robberecht W: A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2011, 11: 54-65.
Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J: RNAbinding proteins hnRNP A2/B1 and CUGBP1 suppress fragile × CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron. 2007, 55: 565-571. 10.1016/j.neuron.2007.07.021.
Todd PK, Paulson HL: RNA-mediated neurodegeneration in repeat expansion disorders. Ann Neurol. 2010, 67: 291-300. 10.1001/archneurol.2010.3.
Wheeler TM: Myotonic dystrophy: therapeutic strategies for the future. Neurotherapeutics. 2008, 5: 592-600. 10.1016/j.nurt.2008.08.001.
Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT, Thornton CA: Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science. 2009, 325: 336-339. 10.1126/science.1173110.
Mulders SA, van den Broek WJ, Wheeler TM, Croes HJ, van Kuik-Romeijn P, de Kimpe SJ, Furling D, Platenburg GJ, Gourdon G, Thornton CA, Wieringa B, Wansink DG: Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc Natl Acad Sci USA. 2009, 106: 13915-13920. 10.1073/pnas.0905780106.
Kole R, Krainer AR, Altman S: RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov. 2012, 11: 125-140.
Yamashita M, Nonaka T, Arai T, Kametani F, Buchman VL, Ninkina N, Akiyama H, Goedert M, Hasegawa M: Methylene blue and dimebon inhibit aggregation of TDP-43 in cellular models. FEBS Lett. 2009, 583: 2419-2424. 10.1016/j.febslet.2009.06.042.
Audet JN, Soucy G, Julien JP: Methylene blue administration fails to confer neuroprotection in two amyotrophic lateral sclerosis mouse models. Neuroscience. 2012, 209: 136-143.
Congdon EE, Wu JW, Myeku N, Figueroa YH, Herman M, Marinec PS, Gestwicki JE, Dickey CA, Yu WH, Duff K: Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy. 2012, 8: 609-622. 10.4161/auto.19048.
Boxer AL, Gold M, Huey E, Gao FB, Burton EA, Chow T, Kao A, Leavitt BR, Lamb B, Grether M, Knopman D, Cairns NJ, Mackenzie IR, Mitic L, Roberson ED, Van Kammen D, Cantillon M, Zahs K, Salloway S, Morris J, Tong G, Feldman H, Fillit H, Dickinson S, Khachaturian Z, Sutherland M, Farese R, Miller BL, Cummings J: Frontotemporal degeneration, the next therapeutic frontier: Molecules and animal models for frontotemporal degeneration drug development. Alzheimers Dement. 2012,
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S: Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007, 131: 861-872. 10.1016/j.cell.2007.11.019.
Shi Y, Kirwan P, Smith J, MacLean G, Orkin SH, Livesey FJ: A human stem cell model of early Alzheimer's disease pathology in Down syndrome. Sci Transl Med. 2012, 4: 124ra29-10.1126/scitranslmed.3003771.
Khan BK, Yokoyama JS, Takada LT, Sha SJ, Rutherford NJ, Fong JC, Karydas AM, Wu T, Ketelle RS, Baker MC, Hernandez MD, Coppola G, Geschwind DH, Rademakers R, Lee SE, Rosen HJ, Rabinovici GD, Seeley WW, Rankin KP, Boxer AL, Miller BL: Atypical, slowly progressive behavioural variant frontotemporal dementia associated with C9ORF72 hexanucleotide expansion. J Neurol Neurosurg Psychiatry. 2012, 83: 358-364. 10.1136/jnnp-2011-301883.
Zinman L, Cudkowicz M: Emerging targets and treatments in amyotrophic lateral sclerosis. Lancet Neurol. 2011, 10: 481-490. 10.1016/S1474-4422(11)70024-2.
Bensimon G, Lacomblez L, Meininger V: A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994, 330: 585-591. 10.1056/NEJM199403033300901.
Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V: Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. 1996, 347: 1425-1431.
Lacomblez L, Bensimon G, Leigh PN, Guillet P, Powe L, Durrleman S, Delumeau JC, Meininger V: A confirmatory dose-ranging study of riluzole in ALS. ALS/Riluzole Study Group-II. Neurology. 1996, 47 (6 Suppl 4): S242-250.
Cheah BC, Kiernan MC: Dexpramipexole, the R(+) enantiomer of pramipexole, for the potential treatment of amyotrophic lateral sclerosis. IDrugs. 2010, 13: 911-920.
ALSTDI ALS Therapy Development Institute: The ALS Therapy Development Institute to Launch Phase II Clinical Trial of TDI 132 in ALS Patients. [http://www.als.net/Media/5409/News/]
Aggarwal SP, Zinman L, Simpson E, McKinley J, Jackson KE, Pinto H, Kaufman P, Conwit RA, Schoenfeld D, Shefner J, Cudkowicz M, Northeast and Canadian Amyotrophic Lateral Sclerosis consortia: Safety and efficacy of lithium in combination with riluzole for treatment of amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010, 9: 481-488. 10.1016/S1474-4422(10)70068-5.
Ward ME, Miller BL: Potential mechanisms of progranulin-deficient FTLD. J Mol Neurosci. 2011, 45: 574-582. 10.1007/s12031-011-9622-3.
Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus-Hernandez M, Crook R, Hunter T, Ghidoni R, Benussi L, Crook J, Finger E, Hantanpaa KJ, Karydas AM, Sengdy P, Gonzalez J, Seeley WW, Johnson N, Beach TG, Mesulam M, Forloni G, Kertesz A, Knopman DS, Uitti R, White CL, Caselli R, Lippa C, Bigio EH, Wszolek ZK, Binetti G: TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011, 76: 467-474. 10.1212/WNL.0b013e31820a0e3b.
Dorsey ER, Huntington Study Group COHORT Investigators: Characterization of a large group of individuals with Huntington disease and their relatives enrolled in the COHORT study. PLoS One. 2012, 7: e29522-10.1371/journal.pone.0029522.
Acknowledgements
AB is funded by NIH grants R01AG038791, R01AG031278, the John Douglas French Foundation, Alzheimer's Drug Discovery Foundation, the Association for Frontotemporal Degeneration, the Silicon Valley Foundation, the Agouron Institute, the Tau Research Consortium and the Hellman Family Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
SJS declares no competing interests. AB has been a consultant for Bristol Myers Squibb, Genentech, Plexikkon, Phloronol, Envivo and TauRx and receives research support from Allon Therapeutics, Bristol Myers Squibb, Janssen, Forest, Pfizer and Genentech.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
About this article
Cite this article
Sha, S.J., Boxer, A. Treatment implications of C9ORF72. Alz Res Therapy 4, 46 (2012). https://doi.org/10.1186/alzrt149
Published:
DOI: https://doi.org/10.1186/alzrt149