
Abstract
In this work, we have investigated one-sided and two-sided fully saturated hydrocarbons with C:H formula derived from a single sheet of graphene, which are called table-like and chair-like graphanes. The effects of full hydrogenation of graphene sheet, graphane, were studied using generalized gradient approximation of the density functional theory and the pseudopotential method. Total energy calculations show that chair-like conformer of graphane is more favorable than table-like one. A change of hybridization from sp2 to sp3 for chair-like conformer of graphane has been achieved. After full geometric relaxation of graphene and two phases of graphane, we obtain the adsorption geometry, adsorption energies, charge transfer, energy gap, density of states, quantum conductivity, and current for graphene and graphane. We show that the band gap induced by hydrogenation of graphene sheet in table-like conformer can greatly improve the electrical characteristics of graphene-based field effect transistors and its on/off ratio. We identify that graphene is acting as an acceptor after saturation with hydrogen.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Graphene, a one-atom-thick carbon sheet, is a transparent single layer of sp2 hybridized carbon atoms arranged in a hexagonal honeycomb structure with a strong bond in plane σ and a weaker one in π. It has chemical stability and physical properties like a conductor with high charge carrier mobility. Undoped graphene is a zero-gap semiconductor. Electronic states near the Fermi energy of a graphene sheet at energies close to K and K′ points in momentum space form Dirac cones. So, at low energy, linear energy dispersion relation results zero mass quasiparticles that provide ultrahigh mobility for the graphene carriers and high conductivity. Thus, graphene flakes are suitable to construct field effect transistors (FETs) [1–3]. The biggest hurdle for graphene to be used as an electronic material is the lack of an energy gap in its electronic spectra and its minimal conductivity. Since the Dirac fermions are massless, electrical current does not block by tunning the gate voltage. The lack of a band gap limits the usage of two-dimensional graphene for digital switching [3, 4], where high on/off ratios are necessary.
Hydrogen-saturated graphene makes the two carbon sublattices inequivalent, which opens an energy gap at Dirac points [5, 6]. The origin of this gap is the breaking of sublattice symmetry, owing to the C-H interaction that generates mass for the Dirac fermions that open an energy gap at Dirac points. By this way, one can solve the complication of using graphene in electronic devices. Graphane, fully hydrogenated graphene, is a semiconductor and an extended two-dimensional covalently bonded hydrocarbon. Two configurations of graphane with a C:H ratio of 1:1 are called chair-like conformer and table-like conformer. When the graphene monolayer is patterned on substrates, only one side of the graphene is accessible for hydrogenation. The structure is called table-like conformer. On the other hand, if the graphene sheet is suspended, the hydrogen atoms can be hosted on either side of the graphene sheets. The structure is called chair-like conformer.
In this paper, we have investigated the structures and electronic properties of graphene and the two configurations of graphane using first-principle plane wave method within the density functional theory (DFT). Also, electronic transport calculations of pristine graphene and graphane have been investigated. We compare the electrical transport properties of these two systems. So, the organization of this paper is as follows: In ‘Electronic transport and maximally localized Wannier functions’ section, an approach to calculate the transport properties and Wannier functions from first principle calculations is presented. In the next section, the computational details are discussed. In ‘Results and discussion’ section, we investigate the structural and electronic properties of the graphene and the two configurations of graphane, using density functional theory within the generalized gradient approximation (GGA). Finally, in ‘Calculation of electrical conductivity’ section, the electrical conductance of pristine graphene and graphane are studied.
Electronic transport and maximally localized Wannier functions
The zero-temperature conductance through a region of non-interacting electrons in the theory of electronic transport is related to the scattering properties of the region itself by Landauer formula [7]:
where g is the quantum conductance and Γ(Ef) is the transmission function which is proportional to the probability that an electron injected from one end of the conductor with the Fermi energy, Ef, will propagate to the other end.
The transmission function can be expressed in terms of the retarded and advanced Green's functions, GCr and GCa, of the conductor and the couplings of the conductor to left and right leads, ΓL and ΓR, using the Fisher and Lee formulas:
Calculation of the quantum conductance via Green's functions relies on a localized orbital representation, so Bloch orbitals that are delocalized have to be transformed into localized function orbital, the basis to construct matrix elements of the Hamiltonian. Maximally localized Wannier functions (WFs) are proposed for this methodology. A finite set of N WFs in cell R associated with band n given in terms of Bloch functions can be constructed by following the Marzari and Vanderbilt method [8–12]:
where , a Wannier function labeled by the Bravais lattice vector , is usually defined via a unitary transformation of the Bloch functions of the n th band; V is the volume of the unit cell, and the integration is performed over the entire Brillouin zone.
Spread operator, Ω, that shows the spatial delocalization of WFs is defined as:
The value of spread obviously depends on the choice of unitary matrices, . Minimizing Ω in terms of , we obtain the matrices that transform the first principles into the maximally localized WFs.
The construction of matrix elements of the Hamiltonian on maximally localized WFs, basis to calculate quantum conductance, can be obtained from the unitary rotations with the matrices .
The electronic current is evaluated using the Landauer formula:
where nF is the Fermi distribution function. At equilibrium, the current is proportional to the conductance g(E),
where (μL and μR) are the chemical potential energies of the left and right leads respectively.
Computational details
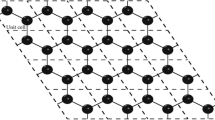
Our calculations are based on the use of DFT and the ab initio pseudopotential plane-wave method using the PWSCF code [13] of the Quantum ESPRESSO distribution. We performed the calculations with generalized gradient approximation Perdew-Burke-Ernzerhof (non-NLCC) pseudopotentials [14]. Pseudopotentials with 2s22p2 and 1s1 valence electron configurations for C and H atoms are used respectively. For the pseudopotential generation, Brillouin zone integrations were performed using a 16 × 16 × 1 Monkhorst and Pack special point grids [15] using Gaussian smearing technique with a smearing width of 0.01 Ry to smoothen the Fermi distribution. The Kohn-Sham orbitals are expanded in a plane wave basis set. The energy cut-offs for the wave function and for the charge density are 45 and 400 Ry, respectively. These values are used because of the ultrasoft pseudopotentials for H and C, and the imposed periodic boundary conditions. A graphene sheet is simulated by a relatively small hexagonal cell of two atoms, with lattice parameters a = 2.46 Å, b = 2.46 Å in the xy planes, and c = 12 Å. We represent the vacuum above graphene with an empty space of 12 Å that is large enough to have no interaction between the graphene's layers (Figure 1a). The sampling of the Brillouin zone was done using a 16 × 16 × 1 grid and was tested to give convergent results for the total energy. To calculate the density of states (DOS), we have used a 50 × 50 × 1 grid. The convergence in energy and force are set to 10−6 eV and 0.001 eV/Å, respectively.

Structure of graphene and graphane. (a) Garphene sheet, (b) chair-like graphane, and (c) table-like graphane.
The amount of charge transfer between graphene sheet and H atoms can be estimated by projecting the charge density onto the atomic orbitals. We define the charge transfer as the difference between the Löwdin charges [16] for isolated graphene sheet and graphene sheet with H atoms. From this result, one can determine whether H atom acts as an acceptor or as a donor. The donor behavior of H atoms causes the Fermi level of graphene sheet with H atoms to shift upward, and its acceptor behavior causes the Fermi level of graphene sheet with H atoms to shift downward with respect to the Fermi level of graphene sheet.
Results and discussion
The configurations of the H-graphene systems were optimized through fully relaxing the atomic structures until the remaining forces were smaller than 0.001 eV/Å. The lattice parameter in the xy plane, a, is obtained 2.54 Å after cell relaxation of the chair-like graphane, in which the hydrogen atoms bound at two inequivalent carbon atoms in the graphene lattice are at opposite surface of the graphene sheet. The orientations of H atoms are adsorbed with each carbon atom as shown in Figure 1b. The bond lengths of the adsorbed H-C and C-C are 1.112 and 1.537 Å, and the H-C-C and C-C-C angles are calculated to be 107.4° and 111.5°, respectively, which are in good agreement with other theoretical studies [5, 6, 17, 18]. These angles resemble the tetrahedral angle of 109° in diamond, which show sp3-like features.
A unit cell of graphane containing two carbon atoms and two hydrogen atoms is used to represent crystalline graphene with the in-plane lattice constant a that is obtained 2.752 Å after cell relaxation for table-like graphane. The orientations of H atoms are adsorbed above each carbon atom as shown in Figure 1c. The bond lengths of the H-C and C-C are 1.075 and 1.604 Å, and the H-C-C and C-C-C angles are calculated to be 90° and 120°, respectively.
To verify the effects of the adsorption of H atoms on the graphene sheet's electronic properties, the total electronic DOS of the systems are calculated; Figures 2 and 3 show the DOS for representative systems.

DOS of graphene. The Fermi level is set to zero.

DOS of chair-like and table-like conformer of graphane. The Fermi level is set to zero.
The binding of hydrogen induces a charge transfer between hydrogen and carbon atoms. There is a total charge transfer of 0.153 e and 0.177 between graphene surface and H atoms per cell for chair-like and table-like conformer within GGA calculations, respectively, which is in good agreement with previous work [5]. Comparison of the Fermi energy of graphene (Ef = −1.0922 eV) with the Fermi energies of the two configurations of graphane (Eftable-like = −0.2550 eV and Efchair-like = −0.1862 eV) shows that graphene sheet with H atoms acts as an acceptor. The acceptor behavior of chair-like conformer is stronger than that of the table-like one. The graphene sheet in both cases acts as acceptor since carbon is slightly more electronegative than hydrogen.
The projected DOS (PDOS) plot near the Fermi level shows the characteristics of sp3 for the chair-like feature. The characteristics of sp2 and p z (l = 1, m = 0) orbital which is distinct and contributes mostly near the Fermi level are obtained via PDOS plot for the table-like feature (Figures 4 and 5) (2p x and 2p y orbitals have similar contributions in the PDOS plots).

PDOS of graphene. The Fermi level is set to zero.

PDOS of chair-like and table-like conformer of graphane. The Fermi level is set to zero.
Symmetry between two representative carbon atoms in the unit cell is broken during hydrogenation of the chair-like configuration, and a transition from sp2 to sp3 hybridization happens which induces the out-of-plane corrugation of 0.46 Å that is in good agreement with theoretical and experimental results [18, 19]. The total DOS and PDOS of these systems show that they modulate the electronic property in different manners. The hydrogenation through p z orbital opens an energy gap of 3.08 eV which is smaller than the previous pseudopotential result of 3.5 eV [5, 6] and 0.59 eV in comparison with the zero-gap nature of graphene for chair-like and table-like conformers within GGA calculations, respectively. Charge density distributions for graphene and the two features of graphane in [0 0 1] plane are showed in Figure 6. Figure 6a shows graphene's charge density, in which blue points refer to the position of the carbon atoms. Figure 6b shows the chair-like graphane's charge density, in which an out-of-plane corrugation is induced after hydrogenation. Here, three blue points in each hexagon have a lighter blue color than other ones; therefore, carbon atom is hybridized with sp3 characteristic. Pink color around the blue points is related to the charge density of H atoms. Figure 6c shows the table-like graphane's charge density, showing no corrugation in carbon atom layer, so in this case, the hybridization of carbon atom has an sp2 characteristic like that of graphene.

Charge density distributions. (a) Graphene; (b) and (c) two features of graphane in [0 0 1] plane.
The adsorption energy (Ead) is defined as the difference between the energy of the fully relaxed graphene sheet with the adsorbed H atoms on it (Etotal) and the sum of the graphene sheet (Esheet) and the isolated H atom (EH) energies:
The comparison result of the adsorption energies of the two phases of graphane indicates that the chair-like phase is energetically more stable and favorable than the table-like one, which is in good agreement with the previous theoretical studies [5, 6] (the negative adsorption energy corresponds to the exothermic reaction). The adsorption energies are −2.565 and −0.389 eV per H-C pair.
Contour plots of the total charge density calculations in [0 1 0] - [0 0 1] plane and along the lines joining both the C-C and C-H atoms are performed. Our results in Figure 7 show a degree of covalency for the C-C and C-H bonds.

Contour plots of total charge density distributions. (a) Graphene; (b) and (c) two features of graphane in [0 1 0] - [0 0 1] plane.
Figure 7a shows the C-C π orbital in graphene. Figure 7b shows that upon hydrogenation, the π orbital in the graphene is broken, and a sp z orbital is formed by H s orbital and C p z orbital. The charge density along the neighboring C atoms is typical of the diamond structure. Figure 7c shows a nearly π orbital around C-C bond. It is also shown that a large amount of charge is condensed around the carbon atoms.
Calculation of electrical conductivity
Calculations of the quantum conductance have been obtained using the WanT package [20], which are performed using the Fisher-Lee formula. The bulk transmittance for the system is considered with homogeneous Monkhorst-Pack grids for Brillouin zone summation. We have investigated the electrical transmission of the two-dimensional pristine graphene and table-like graphane. The WFs' centers for occupied σ bands are chosen at every bond center, and the centers of the WFs for π bands are considered on every carbon atom for graphene (Figure 8) and similar for graphane C atoms with additional WFs on H atoms. At first, the WFs and the spreads of the WFs are obtained from the calculations.

Isosurfaces. Isosurface of π WF in graphene (left). Isosurface of σ WF in graphene (right).
After calculation in pristine graphene sheet, the WFs are symmetric and are centered both at the mid-point of C-C bonds and on carbon atoms, while in the presence of H atoms, the interactions between hydrogen and carbon atoms induce a polarization which modifies the WFs, such that their distributions are unsymmetrical near the mid-point of the C-C bonds and on the carbon atoms (Figure 9).

Isosurface of π WF in graphane (left); isosurface of σ WF in graphane (right).
Calculation of quantum conductance and current at low biases are done. Although the chair-like graphane is more stable than the table-like one, we only consider the table-like graphane to calculate the conductance properties since it has a small energy gap. As can be seen in Figure 10, quantum conductance of graphane is weaker than that of graphene. At the Fermi level, the conductivity of graphane is less than that of pristine graphene. This is due to the donor behavior of the H atoms, which causes the Fermi level of graphane shift upward with respect to the Fermi level of the pristine graphene. In Figure 10, the I/V curves of the two cases have been depicted. They are almost linear and ohmic; this theoretical model is in good agreement with the measurements for graphene [1–3, 19]. Calculating the slopes of these curves, we obtain the conductance of pristine graphene and graphane to be 0.617 and 0.086 μA/V, respectively.

Calculated quantum conductance (the Fermi level is set to zero; left) and current for pristine graphene and graphane in terms of bias voltage (right).
Bordering of the bandstructure near the Fermi level at K point in momentum space after forming graphane induces non-zero mass for quasiparticles that provide lower mobility for the graphene carriers and lower conductivity. The minimum conductivities are calculated as 1.25 and 0.14 e2/h for graphene and graphane, respectively. Minimum current at zero bias voltage is 4.04 × 10−10 μA for graphene that limits the usage of two-dimensional graphene for digital switching, where high on/off ratios are necessary [1–4]. The minimum current is decreased by placing H atoms on graphene sheet, 0.48 × 10−10 μA. As a result, the table-like graphene has higher on/off ratio with respect to the pristine graphene, which is in good agreement with experimental results [21].
Conclusions
Using the plane-wave pseudopotential method and the density functional theory within the generalized gradient approximation, we have investigated the electronic structure and quantum conductance of graphene and graphane, fully hydrogen passivated graphene. In contrast to graphene that is a gapless semimetal, graphane is a semiconductor with a wide energy gap. We optimized the position of H atoms on graphene sheet for two conformers of graphane. One phase of graphane induces an out-of-plane displacement for carbon atoms of 0.46 Å and changes hybridization of carbon atoms to sp3, and opens a band gap of 3.08 eV. In this case, the adsorption energy of H on graphene sheet is larger than 1 eV, corresponding to a strong chemisorption. Another phase of graphane keeps the sp2 hybridization of carbon atoms, although it reduces its characteristic with respect to graphene, and opens a band gap of 0.59 eV. The adsorption energy of H on graphene sheet for this case indicates that the adsorption is a weak chemisorption. After full relaxation, the adsorption energy and the charge transfer properties between the H atoms and graphene sheet are determined for both conformers. Such strong and weak interactions are also evident in their DOS structures, which show changes after the adsorption. The charge transfers from the H atom to graphene sheets were observed. The donor behavior of H atoms causes the Fermi level of graphene sheet with adsorbed H atoms to shift upward. So, H atoms act as donor by the results. The results show that the graphene sheet is more stable in former phase. The resulting inequivalence of the two carbon sites leads to the opening of a non-zero gap at the Dirac points of graphene and to the generating of a non-zero mass for the Dirac fermions, which causes the decrease of the minimum conductance (nine orders of magnitude). The induced band gap can greatly improve the characteristics of graphene-based FETs and its on/off ratio.
In summary, the total charge transfers, out-of-plane corrugation of graphene after hybridization, the adsorption energies, I/V curves, the conductances, the minimum conductivities, and the minimum currents of pristine graphene and graphane have been investigated in this work and compared with experimental results.
References
Kristó f T, Tian F, Pei Z, Aniruddha K, Chuanxin L, Huili X, Michelle K, Debdeep J: Graphene Transistors, Physics and Applications of Graphene – Experiments. InTech, New York;
Standley B, Bao W, Zhang H, Bruck J, Lau CN, Bockrath M: Graphene-based atomic-scale switches. Nano Lett. 2008,8(10):3345. 10.1021/nl801774a
Yujie R, Shanshan C, Weiwei C, Yanwu Z, Chaofu Z, Ruoff RS: Controlling the electrical transport properties of graphene by in situ metal deposition. Appl. Phys. Lett. 2010, 97: 053107. 10.1063/1.3471396
Yoo K, Seo EK, Kim SJ, Kim W, Park MG, Yu H, Chanyong H: Electrical transport properties of graphene-covered-Cu wires grown by chemical vapor deposition. Curr. Appl. Phys. 2012, 12: 115–118. 10.1016/j.cap.2011.05.013
Alzahrani AZ, Srivastava GP: Structural and electronic properties of H-passivated graphene. Appl. Surf. Sci. 2010, 256: 5783–5788. 10.1016/j.apsusc.2010.03.088
Sofo JO, Chaudhari AS, Barber GD: A two-dimensional hydrocarbon. Phys. Rev. B 2007, 75: 153401.
Landauer R: Electrical resistance of disordered one-dimensional lattices. Philos. Mag. 1970, 21: 863–867. 10.1080/14786437008238472
Calzolari A, Marzari N, Souza I, Nardelli MB: Ab initio transport properties of nanostructures from maximally localized Wannier functions. Phys. Rev. B 2004, 69: 035108.
Marzari N, Vanderbilt D: Maximally localized generalized Wannier functions for composite energy bands. Phys. Rev. B 1997, 56: 12847. 10.1103/PhysRevB.56.12847
Lee YS, Marzari N: Cycloaddition functionalizations to preserve or control the conductance of carbon nanotubes. Phys. Rev. Lett. 2006, 97: 116801.
Lee YS, Marzari N: Cycloadditions to control bond breaking in naphthalenes, fullerenes, and carbon nanotubes: a first-principles study. J. Phys. Chem. C 2008, 112: 4480. 10.1021/jp073067i
Cantele G, Lee YS, Ninno D, Marzari N: Spin channels in functionalized graphene nanoribbons. Nano Lett. 2009, 9: 3425. 10.1021/nl901557x
Giannozzi P, Baroni S, Bonini N, Calandra M, Car R, Cavazzoni C, Ceresoli D, Chiarotti GL, Cococcioni M, Dabo I, Dal Corso A, de Gironcoli S, Fabris S, Fratesi G, Gebauer R, Gerstmann U, Gougoussis C, Kokalj A, Lazzeri M, Martin-Samos L, Marzari N, Mauri F, Mazzarello R, Paolini S, Pasquarello A, Paulatto L, Sbraccia C, Scandolo S, Sclauzero G, Seitsonen AP, et al.: QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. Cond. Matt. 2009, 21: 395502. 10.1088/0953-8984/21/39/395502
Perdew JP, Burke K, Ernzerhof M: Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77: 3865. 10.1103/PhysRevLett.77.3865
Monkhorst HJ, Pack JD: Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13: 5188. 10.1103/PhysRevB.13.5188
Löwdin PO: On the non‐orthogonality problem connected with the use of atomic wave functions in the theory of molecules and crystals. J. Chem. Phys. 1950, 18: 365. 10.1063/1.1747632
Boukhvalov DW, Katsnelson MI, Lichtenstein AI: Hydrogen on graphene: Electronic structure, total energy, structural distortions and magnetism from first-principles calculations. Phys. Rev. B 2008, 77: 035427.
Xue K, Zhiping X: Strain effects on basal-plane hydrogenation of graphene: A first-principles study. Appl. Phys. Lett. 2010, 96: 063103. 10.1063/1.3298552
Elias DC, Nair RR, Mohiuddin TMG, Morozov SV, Blake P, Halsall MP, Ferrari AC, Boukhvalov DW, Katsnelson MI, Geim AK, Novoselov KS: Control of graphene's properties by reversible hydrogenation: evidence for graphane. Science 2009, 323: 610. 10.1126/science.1167130
Ferretti A, Bonferroni B, Calzolari A, Buongiorno Nardelli M: WanT. (2012). Accessed 15 April 2011 http://www.wannier-transport.org
Balog R, Jørgensen B, Nilsson L, Andersen M, Rienks E, Bianchi M, Fanetti M, Lægsgaard E, Baraldi A, Lizzit S, Sljivancanin Z, Besenbacher F, Hammer B, Pedersen TG, Hofmann2 P, Hornekær L: Bandgap opening in graphene induced by patterned hydrogen adsorption. Nat. Mater 2010, 9: 315–319. 10.1038/nmat2710
Acknowledgment
The authors gratefully acknowledge the financial and other support of this research provided by the Islamic Azad University, Islamshahr Branch, Tehran, Iran.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
MP carried out DFT calculations, provided guidance at transport stage of the study and drafted the manuscript. VG participated in the transport calculations and reviewed the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Pashangpour, M., Ghaffari, V. Investigation of structural and electronic transport properties of graphene and graphane using maximally localized Wannier functions. J Theor Appl Phys 7, 9 (2013). https://doi.org/10.1186/2251-7235-7-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2251-7235-7-9