
Abstract
Maturity-Onset Diabetes of the Young (MODY) is a monogenic form of diabetes, consisting of a heterogeneous group of autosomal dominant inherited disorders. Typical onset is in individuals prior to twenty five years, and presentation can mimic type 1 or 2 diabetes. Molecular genetic testing can allow precise identification of the different MODY sub-types. Making a specific diagnosis of MODY can have important implications for the guidance of appropriate treatment, prognosis and genetic counselling.
We present the cases of three children and their families diagnosed with MODY over the past two years. These families highlight the features of three of the more common MODY subtypes, including two with novel mutations, one of which segregates in a kindred that is strongly affected by both MODY and classic autoimmune mediated diabetes. To date, we have identified a prevalence of MODY in the paediatric diabetes population of the lower South Island, New Zealand, of approximately 2.5%. This prevalence, along with increasing access to molecular genetic testing, highlights the importance of consideration of MODY in atypical diabetes presentations in the paediatric/adolescent population.
Similar content being viewed by others

Background
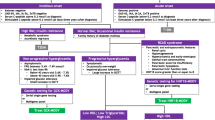
Maturity Onset Diabetes of the Young (MODY) is a monogenic form of diabetes. It consists of a heterogeneous group of autosomal dominant inherited disorders, with typical onset in individuals aged less than twenty five years [1, 2]. There are several sub-types of MODY which can be precisely identified using molecular genetic testing, the four most common of which [3] are outlined in Table 1 (in descending order of frequency). Modes of presentation vary and can mimic either type 1 or 2 diabetes. Making a specific diagnosis of MODY can have important implications for the guidance of appropriate treatment, prognosis and genetic counselling.
The Southern District Health Board paediatric diabetes team provides sole care for approximately 160 children and adolescents spread over the largest geographical region in New Zealand. In this report we present three children and their families who have been diagnosed with MODY over the past two years. We identified two novel mutations and a kindred strongly affected by both MODY and classic autoimmune-mediated diabetes.
Case presentation
Case 1
A 9 year old Caucasian boy with a BMI of 19 kg/m2 (z-score 1.5) presented in 2006 with mild diabetes (two independent fasting blood glucose levels (BGL) >7.0 mmol/L), and was both autoantibody-negative and non-insulin resistant, requiring no immediate treatment. He was initially lost to follow up, but represented in 2010 aged 13 years, along with his sister aged 11 years. Both were asymptomatic but had on-going mild hyperglycaemia (Figure 1). The persistent mild hyperglycaemia, combined with a very significant family history of early onset type two diabetes (T2DM), led to gene screening for a glucokinase (GCK) gene abnormality (MODY 2 subtype). Complicating matters were family members with proven antibody positive (glutamate decarboxylase (GAD) positive) type one diabetes (T1DM), with associated complications. A novel mutation was subsequently identified (Figure 2) in exon 7 of the glucokinase (GCK) gene, in multiple individuals from this kindred (Figure 3). A maternal uncle with latent autoimmune diabetes (LADA) (GAD positive) was identified with this mutation. These children remain well without treatment. Regarding case one, his HbA1c is currently 42 mmol/mol (6%).

Typical BGLs on continuous glucose monitoring - case 1.

One copy of the variant c.698G>A (p.Cys233Tyr) in exon 7 of the GCK gene (Refseq accession number NM_000162).

Family tree - case 1.
Case 2
A 14 yr old Cook Island Maori girl with a BMI of 23 kg/m2 (z-score 1.1) presented with severe non-ketotic hyperglycaemia (BGL 71.7 mmol/L (NR 4–6)) and hypernatraemic dehydration (corrected Na 150 mmol/L (135–145), serum osmolality 364 mosm/kg (275–295)), pH 7.39, and lactate of 5.3 mmol/L (NR 0.5-2). There was evidence of insulin resistance with fasting insulin 336 pmol/L (10–80), C-peptide 1180 pmol/L (350–750), and clinical acanthosis nigricans. Insulin autoantibodies were negative. She had moderate, unexplained intellectual disability with some subtle dysmorphic facial features. There was an extensive family history of T2DM (Figure 4). Initial treatment consisted of insulin up to 3 units/kg/day. A microarray study (Agilent ISCA (v2) 60 K whole genome array) demonstrated a novel 1.3 Mb deletion at chromosome 17q12, this segment includes the HNF1β and multiple other genes. Thus, there is a haplo-insufficiency of HNF1β. Parental studies were normal, showing this to be a de novo deletion. Mutation within the HNF1β gene may cause urogenital abnormalities as well as MODY, but renal and pelvic ultrasonography were normal. The intellectual disability and subtle facial dysmorphism may also be due to the loss of other genes within this deleted segment. Currently her HbA1c is 48 nmmol/mol (6.5%) on insulin 1.25 units/kg/day and 500 mg TDS Metformin.

Case 2 - Family tree.
Case 3
An 11 yr old Caucasian girl with a BMI of 16 kg/m2 (z-score -1.2) presented with a 4 month history of recurrent mucosal candidiasis and mild postprandial hyperglycaemia. Diabetes was confirmed with two random BGLs >11.1 mmol/l. She was well on presentation with a blood pH 7.33, and negative for ketones. Diabetes autoantibody screening was negative, as were clinical and biochemical signs of insulin resistance. Insulin was commenced but her requirement was low at 3 units of isophane daily with aspart as needed with meals (<2 units/day). A significant family history was uncovered (Figure 5) and an HNF1α (MODY 3) gene mutation was suspected and subsequently confirmed on molecular genetic testing (Figure 6). Currently, she is well controlled on Gliclazide 80 mg mane with an HbA1c of 51 mmol/mol (6.8%).

Case 3 – family tree.

Frameshift mutation c.864delGinsCC, or c.864G>C and c.872dupC, (p.Gly292ArgfsX25) in exon 4 of the HNF1α gene (Refseq accession number NM_000545).
Discussion
In the past two years we have identified four children and their families with MODY, covering three subtypes. In each of these cases the diagnosis of MODY has been confirmed by molecular genetic testing. We have identified two novel gene mutations, one in the GCK gene (MODY 2), and one in the HNF1β gene (MODY 5). Clinically, there is diverse range of presentation for MODY subtypes, with our cases highlighting some of this phenotypic variation.
Making a diagnosis of MODY has important implications for treatment, prognosis, and genetic counselling. Glucokinase gene mutations (MODY 2) often require no treatment [1, 2]. In case one, making this diagnosis has led to: reassurance; reduced monitoring; re-diagnosis of many family members; and subsequent acceptance of no treatment for most. HNF1α (MODY 3) and HNF4α (MODY 1) gene mutations usually have a marked sensitivity to oral sulfonylureas [1, 2, 4]. For case three this allowed for cessation of insulin therapy. Unfortunately, as in the father of this patient, an insulin requirement often develops as the disease progresses [5]. In case two, diagnosing an HNF1β (MODY 5) gene deletion led to an explanation of a previously unexplained phenotype, as well as guidance for screening for associated abnormalities.
Case one also highlights the rare occurrence of a kindred that is strongly affected by both MODY and classic autoimmune mediated diabetes. One member of this family now has diabetes consistent with LADA (GAD +ve) combined with the novel GCK gene mutation. This is extremely rare, occurring in <1% of MODY cases [6].
To date (based on these four cases) the Otago/Southland regions in the south of New Zealand have a paediatric MODY prevalence of 2.5% in children with diabetes. While this is based on very small numbers, there is no other data currently published for New Zealand or Australia on MODY prevalence. While similar to the 2.4% paediatric rate reported by Galler et al. [7] for Saxony (Germany), this appears high compared to some worldwide clinic reports [8–10]. Given current estimates of prevalence in adult diabetes of 2-5% [1], it is likely that many MODY cases remain undiagnosed in both paediatric and adult clinics [3, 11]. Vigilance should remain particularly for those with two or more atypical clinical features, such as: no features of insulin resistance; negative β cell autoimmunity; non-ketotic presentations; a strong family history of diabetes (any type); no or unusually low insulin requirement; onset prior to 25 years of age; and unusual or atypical associated phenotypes [1, 2, 4].
Conclusions
In conclusion, our local prevalence, along with increasing access to molecular genetic testing, highlights the importance of considering MODY in atypical diabetes presentations in the paediatric and adolescent diabetes populations.
Consent
Written informed consent was obtained from the patients and the patient’s legal guardians, for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Abbreviations
- MODY:
-
Maturity onset diabetes in young
- BMI:
-
Body mass index
- T2DM:
-
Type 2 Diabetes Mellitus
- GCK:
-
Glucokinase
- HNF1α:
-
hepatocyte nuclear factor 1-alpha gene
- GAD:
-
Glutamate decarboxylase
- T1DM:
-
Type 1 Diabetes Mellitus
- LADA:
-
Latent autoimmune diabetes of adults
- CGMS:
-
Continuous glucose monitoring system
- HNF4α:
-
Hepatocyte nuclear factor 4-alpha gene
- NR:
-
Normal range
- TDS:
-
Three times daily
- HNF1β:
-
Hepatocyte nuclear factor 1-beta gene.
References
Fajans SS, Bell GI, Polonsky KS: Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med 2001,345(13):971–80. Epub 2001/09/29 10.1056/NEJMra002168
Nyunt O, Wu JY, McGown IN, Harris M, Huynh T, Leong GM, et al.: Investigating maturity onset diabetes of the young. The Clinical biochemist Reviews /Australian Association of Clinical Biochemists 2009,30(2):67–74. Epub 2009/07/01
Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S: Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia 2010,53(12):2504–8. Epub 2010/05/26 10.1007/s00125-010-1799-4
Thanabalasingham G, Owen KR: Diagnosis and management of maturity onset diabetes of the young (MODY). BMJ 2011, 343: d6044. Epub 2011/10/21 10.1136/bmj.d6044
Velho G, Vaxillaire M, Boccio V, Charpentier G, Froguel P: Diabetes complications in NIDDM kindreds linked to the MODY3 locus on chromosome 12q. Diabetes Care 1996,19(9):915–9. Epub 1996/09/01 10.2337/diacare.19.9.915
McDonald TJ, Colclough K, Brown R, Shields B, Shepherd M, Bingley P, et al.: Islet autoantibodies can discriminate maturity-onset diabetes of the young (MODY) from Type 1 diabetes. Diabetic medicine : a journal of the British Diabetic Association 2011,28(9):1028–33. Epub 2011/03/15 10.1111/j.1464-5491.2011.03287.x
Galler A, Stange T, Muller G, Nake A, Vogel C, Kapellen T, et al.: Incidence of childhood diabetes in children aged less than 15 years and its clinical and metabolic characteristics at the time of diagnosis: data from the Childhood Diabetes Registry of Saxony, Germany. Hormone research in paediatrics 2010,74(4):285–91. Epub 2010/06/03 10.1159/000303141
Ehtisham S, Hattersley AT, Dunger DB, Barrett TG: British Society for Paediatric E, Diabetes Clinical Trials G. First UK survey of paediatric type 2 diabetes and MODY. Arch Dis Child 2004,89(6):526–9. 10.1136/adc.2003.027821
Awa WL, Schober E, Wiegand S, Herwig J, Meissner T, Schmidt F, et al.: Reclassification of diabetes type in pediatric patients initially classified as type 2 diabetes mellitus: 15 years follow-up using routine data from the German/Austrian DPV database. Diabetes research and clinical practice 2011,94(3):463–7. Epub 2011/10/01 10.1016/j.diabres.2011.09.011
Hotu S, Carter B, Watson PD, Cutfield WS, Cundy T: Increasing prevalence of type 2 diabetes in adolescents. Journal of paediatrics and child health 2004,40(4):201–4. Epub 2004/03/11 10.1111/j.1440-1754.2004.00337.x
Kropff J, Selwood MP, McCarthy MI, Farmer AJ, Owen KR: Prevalence of monogenic diabetes in young adults: a community-based, cross-sectional study in Oxfordshire UK. Diabetologia 2011,54(5):1261–3. Epub 2011/02/26 10.1007/s00125-011-2090-z
Acknowledgements
The authors wish to thank Prof. Robert (Mac) Gardiner for his help with editing the manuscript and also the patients and their families for their forbearance during the diagnostic and treatment process, and for their permission to publish this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BW and PM conceived the manuscript. BW wrote the first draft of the manuscript with assistance from NP. BT and BW medically managed the paediatric cases described from diagnosis through to treatment. PM medically managed the adult family members of the cases described. DP and DL conducted the genetic analyses. All authors contributed to writing and editing the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wheeler, B.J., Patterson, N., Love, D.R. et al. Frequency and genetic spectrum of maturity-onset diabetes of the young (MODY) in southern New Zealand. J Diabetes Metab Disord 12, 46 (2013). https://doi.org/10.1186/2251-6581-12-46
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2251-6581-12-46