
Abstract
Nanocrystalline cobalt ferrite powder has been synthesised by citrate precursor and co-precipitation methods. Structural characterization of the samples has been carried out using powder X-ray diffraction, Fourier transform infrared spectroscopy (FT-IR) and field emission scanning electron microscope (FE-SEM). Distribution of cations among the two interstitial sites (tetrahedral and octahedral sites) has been estimated by analysing the powder X-ray diffraction patterns by employing Rietveld refinement technique, and the results reveal the existence of samples as a mixed type spinel with cubic structure. It is observed that the distribution of cations and structural parameters are strongly dependent on synthesis method and annealing temperature. The vibrational modes of the octahedral and tetrahedral metal complex in the sample have been examined using FT-IR in the wave number range of 390 to 750 cm−1, and it shows an absorption band within this range, which confirms the spinel structure of the sample. The existence of constituents in the sample, i.e., Co, Fe and O has been authenticated using energy dispersive spectrum with the help of a FE-SEM.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Magnetic nanoparticles of spinel ferrites are of great interest in fundamental science especially in addressing the relationship between magnetic properties and their crystal chemistry. Crystal chemistry shows how the chemical composition, internal structure, distribution of cations, and physical properties are linked together, and it gives a clear understanding of magnetic properties of spinel ferrite nanoparticle. Spinel ferrites are isostructural with the mineral MgAl2O4 and have the general molecular formula (M2+) [Fe23+] O42−. Here, M2+ and Fe3+ are the divalent and trivalent cations, respectively. It crystallises in a face-centred cubic structure belonging to the space group composed of large unit cells containing eight formula units [AB2O4]8, i.e. a total of 56 ions per unit cell. The lattice consists of 32 divalent oxygen ions which are in direct contact to one another forming a closed pack face-centred cubic arrangement with 64 tetrahedral interstitial sites (A sites) and 32 octahedral interstitial sites (B sites). Out of these, eight tetrahedral (A sites) and 16 octahedral (B sites) sites are occupied by the divalent and trivalent cations [1]. Thus, the large fraction of empty interstitial sites makes its crystal structure a very open structure conducive to cation migration. Therefore, a whole range of distribution of cations is possible in spinel ferrites which can be represented by the general formula (M2+1−δFe3+ δ )[M2+ δ Fe3+2−δ]O4, where cations inside the round and square brackets are said to occupy the tetrahedral and octahedral site, respectively. Delta (δ) represents the degree of inversion (defined as the fraction of tetrahedral site occupied by Fe3+ cations) which depends on the synthesis method and thermal treatment. If δ = 0, the spinel ferrite is normal; when δ = 1, the spinel ferrite is inverse. However, if 0 < δ < 1, the spinel ferrite is mixed [2]. At room temperature, cobalt ferrite (CoFe2O4) shows some excellent physical properties, such as high coercivity, large magnetic anisotropy, moderate saturation magnetization, high resistivity, and good mechanical and chemical stabilities [3]. It is a hard magnetic material with a magnetic ordering temperature around 520°C [4]. The spinel cobalt ferrite has a cubic symmetry, and it has six crystallographic easy axes (directions) along the cube edges of the crystal, represented as <100>, and four crystallographic hard axes (directions) across the body diagonals denoted as <111> [5–7]. This material is a promising candidate for technological applications at or above room temperature, such as data storage devices, magnetic sensors, actuators, targeted drug delivery, medical diagnosis, etc. [8–10]. However, the nanocrystalline cobalt ferrite is in infant stage to be used in technological applications.
The magnetic properties in this material mainly arise due to strong superexchange interaction. The strength of the superexchange interaction depends on metal cations (Co and Fe) present at the octahedral and tetrahedral sites, as well as its distribution over the two interstitial (tetrahedral and octahedral) sites, as the interaction between is much stronger than the [11, 12]. The distribution of cations over the two interstitial sites is strongly affected by the variation of lattice constants, size of the crystallites, and bond angles and bond lengths between the cations [13]. These parameters are sensitive to synthesis method and annealing temperature. However, to distribute the cations between the two interstitial sites in a controlled way to get a desire magnetic property is still a challenging task.
One requires detailed knowledge of the crystal structure and distribution of cations over the interstitial sites to understand the physical properties. Such detailed structural information including cation distribution over the interstitial sites can be obtained by diffraction technique which gives a volume-averaged picture of the atomic ordering. In contrast, imaging techniques such as transmission electron microscope or atomic force microscopy reveal only structural features projected down on one axis or onto a surface [14]. As magnetic properties of nanocrystalline cobalt ferrite are sensitive to the crystal structure, many researchers have carried out the investigation on nanocrystalline cobalt ferrite (CoFe2O4). Waje et al. [15] have synthesised cobalt ferrite nanoparticles using mechanical alloying method and found that crystallite size depends on the milling time, ball-to-powder ratio, and annealing temperature. Abbas et al. [16] have prepared nanocrystalline CoFe2O4 by co-precipitation and ceramic method. They have found that structural parameters depend upon synthesis method. Thang et al. [17] have prepared fine particle of cobalt ferrite by complexometric method using ethylene diamine tetra acetic acid as a complexing agent and found that CoFe2O4 powder calcined at above 850°C shows single phase crystallographic structure. Zi et al. [18] synthesised cobalt ferrite by a modified chemical co-precipitation route and found that thermal heating changed the cation distribution in tetrahedral-octahedral sites. Very limited efforts have been made to compare the structural properties as well as the distribution of cations over two chemically inequivalent sites in nanocrystalline cobalt ferrite prepared by different synthesis techniques using identical annealing conditions. In order to get better understanding of the effect of synthesis method as well as annealing condition on the structural properties (lattice constants, atomic positions, site occupancies, bond lengths, bond angles, and crystallite sizes) and cation distributions over the tetrahedral and octahedral sites, we have synthesised the nanocrystalline CoFe2O4 by the citrate precursor and co-precipitation methods using identical annealing conditions. Here, we focus on to understand the tunning of cation distribution over the two interstitial sites with increasing annealing temperatures. In the present work, we have shown that increasing the heat treatment, the metastable cation distribution (non-equilibrium) move towards to the stable (equilibrium) cation distribution over the two chemically inequivalent sites. Also, the ionic radii of Co and Fe cation in tetrahedral and octahedral environments are different. So, the distribution of cations over the interstitial sites with the heat treatment may lead to different lattice strains. However, no deviation from cubic symmetry has been observed even in metastable state, which reveals that crystal symmetry is retained with increasing annealing temperature.
Methods
Materials
The reagents, cobalt nitrate (Co(NO3)2·6H2O), iron nitrate (Fe(NO3)3·9H2O), NaOH, and citric acid (C6H8O7.H2O), of analytical grade with very high purity were used to prepare the materials and were from E. Merck Ltd., Mumbai, India.
Synthesis of cobalt ferrite nanocrystals
Citrate precursor method: sample S1
In the citrate precursor method, Co(NO3)2·6H2O and Fe(NO3)3·9H2O were dissolved in deionised water (Milli-Q grade, Millipore Corp., Billerica, USA) to produce a solution with Co/Fe mole ratio of 1:2. An aqueous solution of citric acid was mixed with metal nitrate solutions. The molar ratio of metal nitrate solution-to-citric acid was taken as 1:3. The mixed solution was heated at 80°C with constant stirring using hot plate. The solution became viscous and finally formed brown gel. The gel was dried overnight using an oven at 80°C in order to remove excess water. During the process of drying, the gel swells into a fluffy mass and eventually broke into brittle flakes. The resulting material was heat-treated in air atmosphere at 200°C, 400°C, and 800°C for 2 h per temperature. We name this sample as S1.
Co-precipitation method: sample S2
In the co-precipitation method, an aqueous solution of 0.5 M cobalt nitrate and aqueous solution of 0.5 M iron nitrate was prepared with Fe-to-Co mole ratio of 2:1. An aqueous solution of 2 M NaOH was used as the precipitant agent. The desired amount of aqueous solution of cobalt nitrate and iron nitrate were taken and subjected to stirring using a magnetic stirrer for half an hour. Under stirring, the aqueous solution of NaOH was added dropwise to the mixed solution until a pH = 11.5 was reached. The liquid precipitate was then brought to reaction temperature of 80°C and stirred for 1 h. The product was then cooled to room temperature. The precipitates were filtered and washed several times with deionised water. Finally, the precipitated powders were dried overnight using an oven at 80°C in order to remove excess water. The resulting material was annealed at 200°C, 400°C, and 800°C for 2 h per temperature. We denote this sample as S2.
Characterization
Elemental analysis
The compositions of all the samples were studied by energy dispersive spectroscopy (EDS) with the help of Hitachi S4800 field emission scanning electron microscope (FE-SEM; Hitachi Ltd., Tokyo, Japan).
Powder X-ray diffraction study
The crystalline phase of all the samples was identified at room temperature using Rigaku Miniflex diffractometer (Rigaku Corporation, Tokyo, Japan) with CuK α radiation (λ = 1.5418 Å). All the X-ray diffraction (XRD) patterns were analysed with the help of FullProf programme by employing Rietveld refinement technique. The XRD patterns for all the samples could be refined using the space group [19]. The Rietveld method is a well-established technique for extracting structural details from powder diffraction data. The method employs a least-squares procedure to compare Bragg intensities and those calculated from a possible structural model. In the first step of refinement, the global parameters, such as background and scale factors, were refined. In the next step, the structural parameters such as lattice parameters, profile shape and width parameters, preferred orientation, asymmetry, isothermal parameters, atomic coordinates, and site occupancies were refined in sequence. The fitting quality of the experimental data is assessed by computing the parameters such as the ‘goodness of fit’ χ2 and the R factors (Rp = profile factor, RB = Bragg factor, and RF = crystallographic factor) [20]. When these parameters reached their minimum value, the best fit to the experimental diffraction data is achieved, and the crystal structure is regarded as satisfactory [20]. The site occupancies of the cations in the two interstitial sites (tetrahedral and octahedral sites) are constrained so as to preserve the stoichiometric composition of the materials. The proposed occupancies of the cations in the two interstitial sites (tetrahedral and octahedral) are calculated by considering that the A site has a sum of cationic distribution equal to one and that for B site is two. The initial model and atomic coordinates are taken from literature [19].
The analysis of the crystallite size has been carried out using the broadening of the XRD peaks. Peak broadening comes from several sources, i.e. instrumental effect, finite crystallite size, and strain effect within the crystal lattice [21]. Taking care of all the sources, crystallite size has been calculated using Scherrer's formulae, Williamson-Hall plot, and Rietveld method. We have calculated the crystallite size by the above three methods to compare their sizes different methods. The Scherrer's formula [22] is defined as
where constant k depends upon the shape of the crystallite size (which is equivalent to 0.89, assuming the circular grain), β is the full width at half maximum of the intensity (a.u.) vs. 2θ profile, λ is the wavelength of the CuK α radiation (equal to 0.1542 nm), θ is the Bragg's diffraction angle, and D is the crystallite size. In Scherrer's formula, the average crystallite size has been calculated using Gaussian fit, fitted to the peaks in XRD pattern. D has been taken average for all the peaks. According to the Williamson-Hall method [21], the individual contributions to the broadening of reflections can be expressed as
where 4ε sinθ is the strain effect on the crystallites. A complete expression used in Rietveld method [20] is defined as
where U, V, and W are the usual peak shape parameters; IG is a measure of the isotropic size effect, and DST is the coefficient related to strain.
Spectral measurement
The FT-IR spectroscopy is a very useful technique to deduce the molecular dynamics. The FT-IR absorption bands of the crystalline solid are usually assigned to the vibration of ions in the crystal lattice. The FT-IR spectra of the samples are recorded at room temperature using PerkinElmer (Model 400; PerkinElmer Inc., MA, USA) in the range of 390 to 750 cm−1.
Results and discussion
Elemental analysis
Typical FE-SEM image of the sample S1 annealed at 800°C is shown in Figure 1. The image shows that the particles have an almost homogeneous distribution, and some of them are in agglomerated form. The composition of samples S1 and S2 are determined using the EDS analysis. The EDS patterns reveal the presence of Co, Fe, Au, and O elements in the sample. We have observed no other peaks except the extra gold (Au) peak. The gold peak appears due to the thin coating on the sample surface to make it conducting, which is required to record the SEM picture. It reveals that there is no contamination in the samples. In addition, the EDS results indicate that the atomic ratio of Fe/Co for all samples is maintained at about 2:1.

FE-SEM image of the sample S1 (CoFe 2 O 4 /citrate method) annealed at 800°C.
Structural analysis
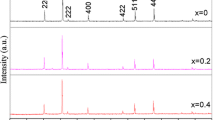
The X-ray diffraction patterns along with Rietveld refined data have been shown in Figures 2a,b,c and 3a,b,c for the samples S1 (prepared by the citrate precursor method) and S2 (prepared by the citrate precursor method), respectively. Each panel (a, b, and c) in the figures represent the samples annealed at 200°C, 400°C, and 800°C, respectively. XRD patterns for the sample S1 annealed at 200°C and 800°C are similar to our earlier publication [23, 24]. Also, the parameters obtained in the current samples are close to our earlier reported values. Here, the experimental data are shown as open circles, and calculated intensities are shown as solid line. The bottom line represents the difference between the measured and the calculated intensities. The allowed Braggs positions for the space group are marked as vertical lines. We have observed that all the experimental peaks are allowed Bragg 2θ positions for space group. In the refinement, the oxygen positions (x = y = z) have been taken as free parameters. However, all other atomic fractional positions have been taken as fixed. Other parameters such as lattice constants, isothermal parameters, occupancies, scale factors, and shape parameters have been taken as free parameters. The background has been corrected by pseudo-Voigt function. Typical fractional positions of the atoms for samples S1 and S2 annealed at 800°C are given in Table 1. The refined XRD patterns show that the samples are in single phase form. The XRD pattern annealed at 200°C shows very broad peaks, which indicate the small crystallite size. Increasing the annealing temperature further, decrease in the broadening of the diffraction peaks suggests the formation of CoFe2O4 crystals with enhanced crystallinity.

Rietveld refined XRD patterns for sample S1 (CoFe 2 O 4 : citrate method) annealed at (a) 200°C, (b) 400°C, and (c) 800°C. The circles represent experimental points, and the solid line represents Rietveld refined data. The bottom line shows the difference between the experimental and refined data. The marked 2θ positions are the allowed Bragg peaks.

Rietveld refined XRD patterns for sample S2 (CoFe 2 O 4 : co-precipitation method) annealed at (a) 200°C, (b) 400°, and (c) 800°C. The circles represent experimental points, and the solid line represents Rietveld refined data. The bottom line shows the difference between the experimental and refined data. The marked 2θ positions are the allowed Bragg peaks.
The various R factors are listed in Table 2. The values of R factor such as Rp are found to be large. Similar high values of R factors for nanocrystalline materials have been observed by the other authors [16, 25]. It could be due to the large noise-to-signal ratio of XRD patterns for nanocrystalline materials. However, we have observed a low value of χ2 (goodness of fit) which justifies the goodness of refinement. In the diffraction pattern of nanocrystalline materials, diffuse scattering is dominant than in those of bulk crystalline materials, and it is due to the large ratio of surface to volume atoms. The diffuse scattering becomes significant at the nanoscale while Bragg scattering gets diminished which leads to decline in crystallinity and large R factors [26].
The refined lattice parameters and unit cell volumes for samples S1 and S2 annealed at 200°C, 400°C, and 800°C have been listed in Table 2 along with the errors in parentheses. The unit cell volume and lattice constants increase with the annealing temperature, irrespective of the method of preparation. It is due to growth of the crystal with increasing temperature. The lattice parameters are comparable to the values reported in the literature [27]. Under identical annealing condition, the lattice constants of samples S1and S2 are different from each other, which reflect the different cation distribution in the samples (discussed in the next section).
Bond lengths and bond angles were calculated using refined lattice parameters and fractional coordinates with the help of Powder Cell programme. The effective bond lengths (R) between two cations were calculated using the formula below [28]:
where, RA-o and RB-o are bond lengths of A and B cations with oxygen anions, respectively. Typical unit cell showing bond length between cations and anions for sample S1 annealed at 800°C has been depicted in Figure 4. The values of bond lengths and bond angles for samples S1 and S2 are listed in Table 3. The bond lengths and bond angles in both the samples S1 and S2 change with the annealing temperature. It is due to the change in the distribution of cations and change in lattice constants with the annealing temperature. In both samples (S1 and S2), effective bond lengths increase with the increase of annealing temperature which occurs due to the increase of lattice constant (expansion of unit cell). We have observed that under identical annealing condition, effective bond lengths are more in sample S2 than those in S1.

The crystal unit cell of nanocrystalline CoFe 2 O 4 (citrate method) annealed at 800°.
Crystallite sizes calculated by Scherrer's formulae, Williamson-Hall plot, and Rietveld method are listed in Table 4 for the samples S1 and S2 annealed at 200°C, 400°C and 800°C. We have observed that for both the samples, crystallite size increases with the increase of annealing temperature. It is due to the growth of the crystal with the increase of annealing temperature. During the thermal annealing, solid-vapour surface of the crystals was replaced by solid-solid interface via diffusion to reduce the overall surface energy which leads to the expansion of volume. The size of the crystallite is less for sample S1 annealed at 200°C and 400°C than S2. At 800°C, sample S1 has a larger crystallite size than sample S2. With the increase in the annealing temperature from 400°C to 800°C, there is a considerable increase in the crystallite size of sample S1; such trend is not observed in sample S2. Although both samples were prepared under identical conditions, the crystallite size was not the same. The crystallite size is related to the relative interdependence between the nucleation and growth steps, and it is strongly affected by the synthesis route which gives different rates of ferrite formation [29–31]. This implies that at 200°C and 400°C, the nucleation rate was higher than the growth rate in sample S1 compared to S2. At the annealing temperature of 800°C, sample S1 has a bigger crystallite size than sample S2, which indicates that nucleation rate is less and growth rate is high which gives high degree of crystallinity. Thus, the citrate precursor method makes the formation of crystallites easier due to suitable growth of the crystals. We see that the crystallite size obtained by Rietveld method is less than that of Williamson-Hall method within the 1% to 2% error bar. It is due to the correction of peak broadening taking into account all instrumental factors in the Rietveld method. Also, crystallite size obtained by Williamson-Hall method is less than that obtained by Scherrer's formula within the same error bar. It is because the strain correction factor has been taken into account in the Williamson-Hall method, whereas it was not taken into account in Scherrers's method.
The cation distribution in 8 tetrahedral and 16 octahedral interstitial sites were estimated using the Rietveld refinement of the occupancy values. The estimated cation distribution and degree of inversion from the Rietveld analysis for samples S1 and S2 are enlisted in Table 5. One can see that Co ions are present on both tetrahedral and octahedral sites, which reveals that the samples are in mixed spinel structure irrespective of the preparation method. The degree of inversion [2] (defined as the fraction of tetrahedral sites (A sites) occupied by Fe3+ ions) of both samples S1 and S2 increases with the annealing temperature.
The site occupancy analysis for sample S1 annealed at 200°C reveals that 25% tetrahedral sites have been occupied by the Co ions and the remaining 75% tetrahedral sites by Fe ions; 38% of the octahedral sites have been occupied by Co ions and the other 62% octahedral sites by Fe ions. Hence, the Rietveld refinement results suggest that the chemical formula for sample S1 annealed at 200°C is (Co0.25Fe0.76)[Co0.75Fe1.24]O4, where cations in round and square bracket occupy the tetrahedral and octahedral sites, respectively. The given chemical formula suggests that there is random distribution of cations (mixed spinel) among the tetrahedral and octahedral in the structure. As the annealing temperature is raised (200°C to 800°C), the Fe ions at the octahedral site continues to move to the tetrahedral site, and Co ions continuously migrate from the tetrahedral to the octahedral sites. At 800°C, Rietveld refinement results suggest that the chemical formula for sample S1 is (Co0.04Fe0.95)[Co0.96Fe1.05]O4 which indicates an almost inverse spinel distribution. Hence, the refinement result shows that for sample S1, the random distribution (metastable or in equilibrium distribution) of cations over the tetrahedral and octahedral sites could be changed into an inverse distribution (stable or equilibrium distribution) by annealing at higher temperature. As the crystallite size of sample S1 annealed at 800°C is about 48 nm and the distribution of cations over the interstitial sites is almost an inverse spinel, it appears that annealing at 800°C, sample S1 might have changed from nanocrystalline form to bulk form. The above observation suggests that at relatively low annealing temperature, the thermal energy is low to overcome the energy barrier to an ordered (stable) cation distribution [11, 32]. As the annealing temperature is increased, there is a redistribution of cation between the tetrahedral and octahedral sites in order to relax the strain at the lattice site. At relatively higher annealing temperature, there is sufficient energy which enables the cations to migrate between the two interstitial sites which lead to an ordered cation distribution. The migration of cations between the interstitial sites continues unless its free energy attains a minimum value. This change of cation distribution is consistent with the change of lattice constant and bond lengths. Similar behaviour has been observed in nanocrystals of Mn-Fe ferrite and MgFe2O4[3, 31, 32]. As there are 8 tetrahedral sites and 16 octahedral sites inside a unit cell and the cation distribution shows the more occupancy of Fe ions in tetrahedral site, it suggests that there would be more and more FeA3+ − FeB3+ interaction with increasing annealing which will enhance the saturation magnetization.
The cation distribution in the tetrahedral and octahedral sites for sample S2 is different from S1. In S2 sample annealed at 200°C, 24% of the tetrahedral sites have been occupied by Co ions and the remaining 76% tetrahedral sites by Fe ions; 38% octahedral sites have been occupied by Co ions and the other 62% octahedral sites by Fe ions. However, increasing the annealing temperature from 200°C to 400°C, the cation distributions in the tetrahedral and octahedral sites remain almost unchanged. Further, increasing the annealing temperature from 400°C to 800°C would result in a significant change in the cation distribution in the tetrahedral and octahedral sites. We observed that annealing at 800°C, S2 is still in mixed (random) spinel phase, whereas S1 approaches towards the inverse spinel phase. Also, under identical annealing conditions, sample S1 has a higher degree of inversion than S2. This could be due to the different nucleation and growth rates in the formation of sample S1 and S2. From the above discussion, it is clear that the distribution of cations in the tetrahedral (A site) and octahedral (B site) sites depends upon the annealing as well as on the synthesis conditions. The details of annealing effect and synthesis condition on the magnetic properties of nanocrystalline spinel cobalt ferrite have been discussed in our recent publication [33].
In bulk cobalt ferrite material, the Co2+ ion shows a strong preference for octahedral site (B site) [34]. Here, we observed that Co2+ ion occupies both the tetrahedral (A site) and octahedral sites (B site). It reveals that in case of nanocrystalline materials, the cation preference over a particular site does not hold well due to surface effects. Thus, the distribution of cations between tetrahedral and octahedral sites in nanocrystalline form is entirely different from the bulk. It suggests that in nanocrystalline form, cation distribution is in metastable state.
FT-IR analysis
The formation of the spinel structure of cobalt ferrite in the nanocrystalline form and its cation distribution is supported by FT-IR analysis. Typical FT-IR spectra for sample S1 annealed at 200°C, 400°C and 800°C are shown in Figure 5a,b,c. The spectra indicate the presence of absorption bands in the range of 390 to 750 cm−1 which is a common feature of the spinel ferrite [35]. The higher frequency absorption band (ν1) lies in the range 500 to 600 cm−1 and is assigned to vibration of the tetrahedral metal complex which consists of a bond between the oxygen ion and the tetrahedral site metal ion (O-MTet), and the lower frequency absorption band (ν2) lies in the range of 400 to 490 cm−1 and is assigned to vibration of the octahedral metal complex which consists of a bond between the oxygen ion and the octahedral site metal ion (O-MOct) [36]. The appearance of higher frequency band is due to the stretching vibration, while the lower frequency band is due to the bending vibration [37]. These band positions are found to be in agreement with the characteristic infrared absorption bands of cobalt ferrite nanocrystals [38]. The ν1 absorption band in the spectra has not appeared properly for the sample annealed at relatively lower temperature (200°C and 400°C) which reflects the presence of local structural disorder due to finite size effect (surface effect) [39]. This fact is in agreement with XRD, which shows the low crystallinity (broadening of the peak) in the sample. However, at relatively higher annealing temperature of 800°C, the appearance of ν1 band becomes sharper due to the minimization of lattice distortion and enhancement of crystallinity (reduction of surface effect). The broadening of ν1 band continuously increases with the annealing temperature which reflects the presence of high degree of crystallinity, as it is evident from large crystallite size at high temperature. It has been observed that as the annealing temperature is increased, the ν1 band continues to shift towards the high wave number. The reasons might be as follows: the tetrahedral site dimension is less compared to the octahedral site, and the absorption band has an inverse relationship with the bond length. The radius of Fe3+ (0.64 Å) is smaller than Co2+ (0.74 Å). It suggests that at the tetrahedral sites, site radius has been decreased due to the increasing occupancy of smaller Fe3+ ions [40]. Also, the increasing annealing temperature shifts the position of the ν1 and ν2 bands, which might be attributed to the changes that occurred in the cation distribution (i.e. migration of cations from tetrahedral to octahedral sites and vice versa) and variations in the cation-oxygen bond length for octahedral and tetrahedral complexes [38]. The FT-IR results are in close agreement to cation distribution obtained by XRD. Similar behaviour has been observed for sample S2. Typical FT-IR spectra for sample S2 annealed at 400°C and 800°C area shown in Figure 6. Thus, the absorption band observed within these specific frequency limits reveals the formation of single phase spinel structure having two interstitial sites.

FT-IR spectra of the sample S1 (CoFe 2 O 4 : citrate method) annealed at various temperatures. (a) 200°C, (b) 400°C, and (c) 800°C.

FT-IR spectra of the sample S2 (CoFe 2 O 4 : co-precipitation method) annealed at different temperatures. (a) 400°C and (b) 800°C.
Conclusions
Different sizes of nanocrystalline cobalt ferrites were successfully synthesised by the citrate precursor and co-precipitation methods. The samples were crystallised to space group with cubic symmetry. It has been observed that the crystallite size increases with the increase in annealing temperature, and the crystallite size can be controlled by controlling the annealing temperature. The distribution of cations between the interstitial sites could be tuned up by a subsequent annealing of the nanocrystalline material. Under identical annealing conditions, the nanocrystalline samples synthesised by the citrate method show high degree of inversion. An ordered cation distribution could be achieved at higher annealing temperature in the citrate precursor-derived sample. Also, we have observed a suitable growth of crystallites for citrate precursor method samples. The lattice parameters, unit cell volume and effective bond lengths increase with the crystallite size. FT-IR absorption bands within the specific frequency limit support the migration of the cations between the octahedral and tetrahedral sites.
References
Goldman A: Modern Ferrite Technology. New York: Springer; 2006.
Gomes JA, Sousa MH, Tourinho FA, Mestnik-Filho J, Itri R, Depeyrot J: Rietveld structure refinement of the cation distribution in ferrite fine particles studied by X-ray powder diffraction. J. Magn. Magn. Mater. 2005, 289: 184–186.
Valenzuela R: Magnetic Ceramics. Cambridge: Cambridge University Press; 1994.
Haneda K, Morrish AH: Noncollinear magnetic structure of CoFe 2 O 4 small particles. J. Appl. Phys. 1988, 63: 4258–4262.
Shenker H: Magnetic anisotropy of cobalt ferrite and nickel cobalt ferrite. Phys Rev 1957, 107: 1246–1248.
Hyeon T: Chemical synthesis of magnetic nanoparticles. Chem. Comm. 2003, 8: 927–932.
Walker M, Mayo PI, Grady KO, Charles SW, Chantrell RW: The magnetic properties of single-domain particles with cubic anisotropy. I. Hysteresis loops. J. Phys.: Condens. Matter. 1993, 5: 2779–2783.
Murdock ES, Simmons RF, Davidson R: Roadmap for 10 Gbit/in2 media: challenges. IEEE Trans. Magn. 1992, 28: 3078–3083.
Yin Y, Alivisatos AP: Colloidal nanocrystal synthesis and the organic–inorganic interface. Nature 2005, 437: 664–668.
Geng BY, Ma JZ, Liu XW, Du QB, Kong MG, Zhang LD: Hydrophilic polymer assisted synthesis of room-temperature ferromagnetic Fe 3 O 4 nanochains. Appl. Phys. Lett. 2007, 90: 043120–043124.
John ZZ, Wang ZL, Chakoumakas BC, Yin JS: Temperature dependence of cation distribution and oxidation state in magnetic Mn-Fe ferrite nanocrystals. J. Am. Chem. Soc. 1998, 120: 1800–1804.
Goodenough JB, Loeb AL: Theory of ionic ordering, crystal distortion and magnetic exchange due to covalent forces in spinels. Phys. Rev. 1955, 98: 391–393.
Jacob J, Khadar MA: Investigation of mixed spinel structure of nanostructured nickel ferrite. J. Appl. Phys. 2010, 107: 114310–114316.
Gateshki M, Petkov V, Pradhan SK, Vogt T: Structure of nanocrystalline MgFe 2 O 4 from X-ray diffraction. Rietveld and atomic pair distribution function analysis. J. Appl. Cryst. 2005, 38: 772–779.
Waje SB, Hashim M, Yusoof WDW, Abbas Z: X-ray diffraction studies on crystallite size evolution of CoFe 2 O 4 nanoparticles prepared using mechanical alloying and sintering. Appl. Surf. Sci. 2010, 256: 3122–3127.
Abbas YM, Mansour SA, Ibrahim MH, Ali SE: Microstructure characterization and cation distribution of nanocrystalline cobalt ferrite. J. Magn. Magn. Mater. 2011, 323: 2748–2756.
Thang PD, Rijnders G, Blank DHA: Spinel cobalt ferrite by complexometric synthesis. J. Magn. Magn. Mater. 2005, 295: 251–256.
Zi Z, Sun Y, Zhu X, Yang Z, Dai J, Song W: Synthesis and magnetic properties of CoFe 2 O 4 ferrite nanoparticles. J. Magn. Magn. Mater. 2009, 321: 1251–1255.
Young RY: The Rietveld Method. Oxford: Oxford University Press; 1996.
Carbonin S, Martignago F, Menegazzo G, Negro A: X-ray single-crystal study of spinels: in situ heating. Phys. Chem. Miner. 2002, 29: 503–514.
Suryanarayana C, Nortan MG: X-ray diffraction: a practical approach. New York: Plenum Publishing Corporation; 1998.
Cullity BD: Elements of X-ray Diffraction. Boston: Addison-Wesley; 1978.
Kumar L, Kar M: Influence of Al3+ ion concentration on the crystal structure and magnetic anisotropy of nanocrystalline spinel cobalt ferrite. J. Magn. Magn. Mater. 2011, 323: 2042–2048.
Kumar L, Kumar P, Kar M: Effect of La3+ substitution on the structural and magnetocrystalline anisotropy of nanocrystalline cobalt ferrite. Ceram. Int. 2012, 38: 4771–4782.
Bhagwat M, Ramaswamy AV, Tyagi AK, Ramaswamy V: Rietveld refinement study of nanocrystalline copper doped zirconia. Mat. Res. Bull. 2003, 38: 1713–1724.
Jovic NG, Masadeh AS, Kremenovic AS, Antic BV, Blanusa JL, Cvjeticanin ND, Goya GF, Antisari MV, Bozin ES: Effect of thermal annealing on structural and magnetic properties of lithium ferrite nanoparticles. J. Phys. Chem. 2009, 113: 20559–20567.
Franco AJ, Silva FC: High temperature magnetic properties of cobalt ferrite nanoparticles. Appl. Phys. Lett. 2010, 96: 172505–172508.
Yazdani A, Jalilian Nosrati MR, Ghasemi R: A new approach to spinel ferrites through mean field approximation. J. Magn. Magn. Mater. 2006, 304: e433-e435.
Berkowitz AE, Schuele W: Magnetic properties of some ferrite micropowders. J. Appl. Phys. 1959, 30: S134-S139.
Chinnasamy CN, Jeyadevan B, Perez OP, Shinoda K, Tohji K, Kasuya A: Growth dominant co-precipitation process to achieve high coercivity at room temperature in CoFe 2 O 4 nanoparticles. IEEE Trans. Magn. 2002, 38: 2640–2644.
Perez OP, Sasaki H, Kasuya A, Jeyadevan B, Tohji K, Hihara T, Sumiyama K: Production of monodispersed magnetic particles by using effective size selection methods at the nanosize level: size-dependent magnetic properties. J. Appl. Phys. 2002, 91: 6958–6963.
Liu C, Zou B, Adam RJ, Zhang JZ: Chemical control of superparamagnetic properties of magnesium and cobalt spinel ferrite nanoparticles through atomic level magnetic couplings. J. Am. Chem. Soc. 2000, 122: 6263–6267.
Kumar L, Kar M: Effect of annealing temperature and preparation condition on magnetic anisotropy in nanocrystalline cobalt ferrite. IEEE Trans. Magn. 2011, 47: 3645–48.
Nlebedim IC, Ranvah N, Williams PI, Melikhov Y, Snyder JE, Moses AJ, Jiles DC: Effect of heat treatment on the magnetic and magnetoelastic properties of cobalt ferrite. J. Magn. Magn. Mater. 2010, 322: 1929–1933.
Waldron RD: Infrared spectra of ferrites. Phys. Rev. 1955, 99: 1727.
Ramankutty CG, Sugunam S: Surface properties and catalytic activity of ferrospinels of nickel, cobalt and copper prepared by soft chemical method. Appl. Catal. A. 2001, 39: 128.
Bhowmik RN, Naresh N: Structure, AC conductivity and complex impedance study of Co 3 O 4 and Fe 3 O 4 mixed spinel ferrites. Int. J. Eng. Sci. Tech. 2010, 2: 40–52.
Kamble RC, Song KM, Koo YS, Hur N: Low temperature synthesis of nanocrystalline Dy3+ doped cobalt ferrite: structural and magnetic properties. J. Appl. Phys. 2011, 110: 053910.
Rana S, Philip J, Raj B: Micelle based synthesis of cobalt ferrite nanoparticles and its characterization using Fourier transform infrared transmission spectrometry and thermogravimetry. Mat. Chem. Phys. 2010, 124: 264–269.
Jacob BP, Thankachan S, Xavier S, Mohammed EM: Effect of Gd3+ doping on the structural and magnetic properties of nanocrystalline Ni-Cd mixed ferrite. Phys. Scr. 2011, 84: 045702.
Acknowledgements
The authors would like to acknowledge the Department of Atomic Energy, Government of India (sanction no.: 2011/20/37P/03/BRNS/076) for the financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
LK carried out the synthesis of all the samples and has done the XRD analysis. PK participated in analyzing the morphology and structure by SEM/EDX and FTIR. AN participated in conducting the XRD. MK guided and edited the present manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Kumar, L., Kumar, P., Narayan, A. et al. Rietveld analysis of XRD patterns of different sizes of nanocrystalline cobalt ferrite. Int Nano Lett 3, 8 (2013). https://doi.org/10.1186/2228-5326-3-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2228-5326-3-8