
Abstract
Quality production of the shrimp Penaeus monodon in hatchery operations depends heavily on the evaluation of genetic diversity and population structure of brood stocks. Mitochondrial DNA (mtDNA) sequences have been widely used to study genetic variability and relationships in many crustacean groups, and these same markers may be incorporated into evaluation studies of shrimp broods and populations. For this purpose we looked at variation in mitochondrial D-loop sequences as an indicator of genetic diversity in shrimp populations from a region of India that represents the main sources of new material for brood stocks. In our study of these populations the overall mean genetic diversity was 0.191. The highest level of genetic diversity (0.357) was observed in the Kakinada population, whereas the lowest diversity (0.0171) was observed in the Nellore population. The results also indicate that overall, the populations along the Andhra Pradesh coast are genetically diverse despite the fact that there is considerable gene flow between them. From the results, it is evident that east cost of India shows high genetic diversity among P. monodon broods and no evidence of loss of diversity due to excessive inbreeding. The fact that the genetic variability of these populations has been maintained, despite ten years of dependence on these broods, shows that at the present time there is no indication of over exploitation.
Similar content being viewed by others

Introduction
The genus Penaeus represents an economically important group of shrimps and prawns (Dall et al. 1990; Bailey-Brook and Mass 1992; Rosenberry 2001). Aquaculturing of Penaeus monodon alone accounts for more than 50% of the world’s cultured shrimp (Ronnback 2001). However, because of limited reproductive capacity in captivity, continued culturing is highly dependent on wild caught brood stocks (Spann et al. 1997). Also, in hatchery operations, the identification and evaluation of comparative growth performance of existing stocks is necessary (Benzie 1994). Hence, basic knowledge about genetic markers, levels of genetic diversity and differentiation in broods and populations is imperative for construction of an appropriate genetic based stock enhancement programme and to identify regions that may be over exploited and where artificial recruitment may be required (Kumar et al. 2007).
Mitochondrial DNA (mtDNA) sequences are widely used to study genetic variability in aquaculture species including crustaceans, and these sequences have proved extremely useful in elucidating genetic variability and phylogenetic relationships among many crustacean groups (Cunningham et al. 1992; Chu et al. 2003). These regions may also contain ideal markers for characterizing geographical patterns of genetic variation within and between prawn populations (Simon 1991). The complete mitochondrial genome of P. monodon is around 16 kb (Wilson et al. 2000), of which 991 bp is the long noncoding, ‘AT’ rich control region known as the D-Loop. This region plays a significant role in mitochondrial replication and DNA transcription, and it contains the signals that control many general aspects of RNA and DNA synthesis. Previous reports employing mtDNA D-loop based studies on penaeids have demonstrated the usefulness of this region in genetic variability studies (Chu et al. 2003; Tzeng et al. 2004 and Kumar et al. 2007).
Domestication of P. monodon has been carried out for production of high-quality pond-reared P. monodon brood stocks (Withyachumnarnkul et al. 1998), but the program recently collapsed from a white spot syndrome virus (WSSV) infection. Identification of genetically diverse and geographically differentiated shrimp stocks will be essential for both re-establishing and maintaining effective domestication and breeding programs for P. monodon. In addition, over exploitation of P. monodon may be avoided by continuous monitoring and possible enhancement through the use of natural populations (Klinbunga et al. 2001).
Along the coast line of the area known as Andhra Pradesh (974 Km), the dominant shrimp culture area in India, shrimp hatcheries are clustered in three areas: Vizag (North Andhra), Kakinada (Central Andhra) and Nellore (South Andhra). Out of 280 P. monodon hatcheries in the country, 148 are located in this region (Andhra Pradesh). These produce 7882 million larvae per year, and this represents approximately 63% of total seed production in India. Keeping in mind the importance of shrimp culture to the economy and the degree to which success is primarily dependent on the health of the seed and the brood stock, the present work has been conducted to study the genetic structure and diversity of brood stocks from the Andhra Pradesh (A.P.) area in India to ensure the ability to maintain genetically diverse brood stocks for improved production.
Materials and methods
Sample collection
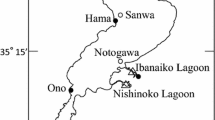
Wild samples of P. monodon brooders were collected from the three regions along the A.P. coast (Figure 1). The study area was demarcated into three regions as Vizag (North Andhra), Kakinada (Central Andhra) & Nellore (South Andhra), where the shrimp hatcheries are clustered. The shrimp broods were collected (30 individuals per population) during January- February, 2011 using mechanised boats having bottom trawl nets specifically designed for shrimp catch. Shrimp caught were washed with clean water and pleopods were dissected and preserved in 95% ethanol at −4°C until they could be further processed.

Sampling locations along the East Coast of India.
Genomic DNA isolation
Genomic DNA was isolated from pleopods following the method described by Sambrook et al. (2005), and the DNA was diluted to obtain a final concentration of 100 ng/μl.
PCR primers
The primers used here for PCR amplification are 12S (F) -5′ AAGAACCAGCTAGGATAAAACTTT 3′ and 1R (R) 5′-GATCAAAGAACATTCTTTAACTAC-3′. These were derived from Chu et al. (2003) and Yang et al. (2006).
PCR amplification reactions
The mtDNA control region was amplified in a 25 μl reaction volume with a final concentration of 1X Taq polymerase buffer and 0.6 U of Taq polymerase, 1.5 mM MgCl2, 2.5 mM dNTPs and 1.5 μM each primer. The thermal profile for hot-start PCR included initial denaturation at 95°C for 5 min, followed by 35 cycles of 20 s at 94°C, 30 s at 48°C, 60 s at 68°C and a final extension of 10 min at 68°C. The PCR product was purified by treatment with exonuclease and shrimp alkaline phosphatase at 37°C for 30 min, and the enzyme inactivation was carried out at 85°C for 15 min. Products were cleaned by ethanol precipitation and sequenced using an ABI Prism DNA analyzer 3730 (Applied Biosystems, USA) and the Big dye cycle sequencing kit.
Data analysis
A total of 81 sequences, each 562 bp in length (on average) from the mtDNA control region, were obtained for analysis. Nine of the original samples with incomplete sequence reads were not included in analysis. The usable sequences were aligned using Bio-edit sequence editor package (Hall 1999), and data analysis was performed using ARLEQUIN version 3.0 (Excoffier et al. 2005) and MEGA 4. The mean nucleotide composition, number of transitions, transversions, indels, number of haplotypes, haplotype diversity (h) and nucleotide diversity (pi) values (Nei 1987) were calculated for all the populations. The haplotype data were analysed phylogenetically by the neighbour-joining (NJ) method using MEGA 5.0 and the genetic distance by the Jukes and Cantor (1969). Support for the tree nodes was assessed by the bootstrap method (1000 replicates). The geographical structuring of population was examined by performing analysis of molecular variance (AMOVA) to partition the total genetic variation into its variance component and to produce FST statistics (Weir and Cockerham 1984).
Results
Genetic diversity and lineages
The partial mitochondrial control region (D-Loop) sequences of 562 bp in length (average) from 81 individuals (NCBI accession nos. JQ863127 to JQ863216) analyzed here show 43 haplotypes with a value of 14.33 for mean haplotype diversity. The overall Jukes-Cantor estimate of nucleotide diversity (pi) for all of the samples analyzed here is 0.452 ± 0.1415, with a mean value for the populations of 0.150 ± 0.141. The average number of differences 18.8443 and the total number of segregating sites is 417 (Table 1).
The Kakinada population contained the largest number of haplotypes (21) and the highest value overall for haplotype diversity (0.931). This was closely followed by the Vizag population where 14 haplotypes produced a diversity value of 0.9195 and finally the Nellore population which had 8 haplotypes and a value for haplotype diversity of 0.864. Most of the haplotypes identified here (42 out of 43) were unique to one of these populations. The one shared haplotype was found in all three of the populations studied here.
Other parameters measuring variation among the three populations are shown in Table 1. A mean expected heterogeneity value of 0.075 was observed among three populations. The individual values ranged from a high of 0.150 in Kakinada to a low of 0.033 in the Vizag population (Table 1). The overall Jukes Cantor (Pi-JC) nucleotide diversity at Vizag population was 0.0350, Kakinada 0.204 and Nellore was 0.036. The average number of pair wise differences (k) is 17.727, with the highest number of differences observed in the Kakinada population which was 37.215 and lowest in Vizag (6.386).
Population structure
For the three populations a total of 417 segregating sites were observed. Also for these populations overall mean numbers of 38 (transition) and 43 (transversion) type substitution mutations were observed. The Kakinada population had the highest numbers of both transitions (74) and transversions (96) whereas lower numbers (22 and 8, respectively) were seen in the Nellore population. An overall average of 81 substitutions were noted with the least number occuring in the Nellore population (30), followed by 43 in Vizag and the highest in the Kakinada region (170). Indels were absent in all three populations (Table 2).
The overall proportions of nucleotides in this dataset are 0.395 (A), 0.409 (T/U), 0.108 (C), and 0.088 (G) based on a total of 286 positions. The transition/transversion rate ratios are k 1 = 24.707 (purines) and k 2 = 31.233 (pyrimidines). The overall transition/transversion bias is R = 5.623, where R = [A*G*k 1 + T*C*k 2 ]/[(A + G)*(T + C)]. All positions containing gaps and missing data were eliminated.
Population level variation
An Analysis of Molecular Variance (AMOVA) was performed in MEGA5.0 for 81 sequences to test for geographic variations/divisions among populations. Results from this analysis showed a percentage of variation attributable to among-population differences of 11.04% whereas most of the variation (88.96%) was attributed to variation within populations (Table 3).
The pairwise Fst comparisons in Table 4 show that the Vizag and Nellore populations are the most differentiated whereas the Vizag and Kakinada populations are the least differentiated. Estimates of Nm to reflect gene flow between populations are given in Table 5. The Nm values between the populations of Vizag and Nellore were relatively lower (3.0486) compared to the highest value of 4.775 seen between the Vizag and Kakinada populations. All of these values, however, are greater than 1.
A Neighbour Joining (NJ) tree was constructed which depicts the overall relationships of the populations studied here (Figure 2). This tree is divided into two lineages. One contains the Nellore population alone while the second lineage contains both the Vizag and Kakinada populations. This overall lineage relationship is also supported by the Neighbor Joining tree run for all individuals (Additional file 1: Figure S1).

Neighbor joining tree for populations.
Discussion
Polymorphisms in mtDNA sequences have been used previously for examining both intraspecific population differentiation and phylogenetic relationships of some penaeid shrimp populations (Benzie 2000; Lavery et al. 2004). This study reports the analysis of genetic variation in the mtDNA D-loop region of P. monodon, a commercially important shrimp species (Bailey-Brook and Mass 1992; Rosenberry 2001), and provides information about the genetic structure and relationships of populations from a region which accounts for the majority (63%) of brood seed production in India (AISHA-All India Shrimp Hatcheries Association 2004; FAO 2004;2006). Because this is the largest shrimp producing and seed supplying region in India, a major goal of this study was to provide baseline data for estimates of genetic diversity and population structure of P. monodon. Analysis of genetic variability and geographic differentiation of such organisms is essential for the development of effective resource management programs (Avise 1994). This type of information is required for maintaining and improving the culture and management efficiency of P. monodon (Carvalho and Hauser 1994; Ward and Grew 1994). In general, relatively low degrees of genetic differentiation have been seen in wild P. monodon, even for those separated over distances of hundreds or thousands of kilometres, except where major biogeographic boundaries act to disrupt gene flow (Benzie et al. 2002).
Among the regions studied here, high levels of mtDNA diversity were observed overall. This is generally consistent with findings from previous studies for decapods and penaeids in general (Silberman et al. 1994; Baldwin et al. 1998; Benzie et al. 2002) although the overall nucleotide and haplotype diversity values obtained in this study are among the highest reported (0.1507 and 0.9049 respectively) for this species. Previously the maximum haplotype diversity reported for P. monodon was 0.682 ± 0.002 (Benzie 2000) and the maximum nucleotide diversity was 0.00334 ± 0.00003 (Klinbunga et al. 1998). We obtained haplotype diversity values of 0.9195, 0.9310 and 0.8634 for the Vizag, Kakinada and Nellore populations, respectively. The genetic diversity for the Kakinada population also appears to be greater compared to that of the other populations. The values we obtained for these P. monodon populations are, however, comparable with those derived from mtDNA d-loop region sequences of the black shrimp Caridina cantonesis, the white shrimp Panaeus setiferus, and the pink shrimp Farfantepenaeus duorarum, (McMillen-Jackson and Bert 20032004; Kumar et al. 2007 and Khamnamtong et al. 2009).
The AMOVA results show that most of the variation (88.96%) detected here is found within populations. Our results also suggest that overall, high levels of gene flow (as reflected by Nm values) are occuring between these populations. Nevertheless, as indicated by the pairwise Fst values, the mixing of lineages in P. monodon in India has clearly not been complete. This could be explained by some ecological or environmental factors such as major physical barriers, pollution or reversals in the monsoon-driven surface water current systems (Dale 1956). Similar findings were reported by Khamnamtong et al. (2009) and Mandal et al. (2012). Also in Australia (Benzie et al. 2002), low levels of population genetic differentiation in wild P. monodon were evident over distances of hundreds or thousands of kilometers, except where major biogeographical boundaries acted to disrupt gene flow.
The NJ trees constructed using control region sequence data also showed a general population structuring according to geographical distribution. However, a number of mixed lineage hapolotypes were found at present in each geographic sample, reflecting some secondary mixing of those haplotypes. This may be explained by the fact that the spawning behavior of P. monodon females can enhance levels of lineage mixing because they migrate offshore when they grow and mature (Motoh 1981). Regardless, the overall relationships shown by the NJ trees that group the Vizag and Kakinada populations together is again consistent with the apparent high levels of gene flow and relatively low levels of genetic differentiation seen between these two populations as compared to the Nellore populaton.
The levels of genetic diversity revealed in the present study using this mtDNA control region might be useful as genetic indicators for aquaculture purposes including planning for selective breeding, maintaining stock diversity and distinguishing hatchery stocks from the wild populations. Some of this diversity may be explained by a high rate of mtDNA mutation as has been suggested for several other penaeid species (Palumbi and Benzie 1991; Baldwin et al. 1998). The basic knowledge of genetic divergence between evolutionary lineages, and the existence of population differentiation between major stocks of Indian P. monodon, suggests that each population should be treated as a separate management unit because it may display unique demographic and dynamic properties (Carvalho and Hauser 1994; Conover et al. 2006).
The assessment of genetic diversity and population structure of P. monodon is critical for appropriate conservation and management purposes. With increased farming and opportunities for future growth in the aquaculture of P. monodon, there is a great concern regarding the loss of wild genetic diversity. For good production, hatchery operators often collect brooders from different parts of the country (AISHA-All India Shrimp Hatcheries Association 2004 and FAO 2006). Similar observations were made by Klinbunga et al. (1998) in Thailand where farmers believe that progeny of the Andaman Sea P. monodon exhibit greater survival and possibly greater growth rates than do progeny from broodstock shrimp caught elsewhere in Thailand. Therefore, genetic monitoring and evaluation of black tiger shrimp can help to identify any negative effects on genetic diversity caused by aquaculture (Naylor et al. 2000; Benzie 2010). Also, maintaining high levels of genetic diversity and population differentiation of P. monodon can help to protect this species from disease epidemics and severe population declines. This would further facilitate the stock improvement programme of this commercially important species through selective breeding. The virtual absence of domesticated specific pathogen free stocks of P. monodon has inhibited breeding programme development and commercial production of this species (Clifford and Preston 2001). Sourcing and spawning of clean founder stocks from wild populations is one means to generate domesticated pathogen free stocks of P. monodon. It is widely accepted that the most economically significant viral pathogens like WSSV, yellow head virus and a host of other pathogens have been introduced into the Asian countries through the careless introduction of live shrimp stocks. Import of disease-free stocks from these regions of India or elsewhere will be beneficial when stocks are used that are free from these and other pathogens and/or viruses.
Conclusions
Information about genetic variability of critical populations and the potential for improvement using biotechnological applications are crucial for the maintenance and future development of shrimp industry. A high level of genetic diversity has been revealed in the present study using the mtDNA control region. The nucleotide and haplotype diversities obtained in this study are among the highest reported for P. monodon populations. The genetic diversity at Kakinada appears to be greater than that of Vizag and Nellore. The relatively high Fst values seen for all of these populations, together with the fact that most of the variation detected here occurs within populations, also indicate that in this region, this species is genetically heterogenous and does not appear to be suffering from extensive inbreeding. The genetic diversity seen here suggests that farmers or hatchery operators can continue to use these populations as sources of natural broodstock from this region of India. Finally, the information obtained here may also be useful for providing genetic markers that can be used for aquaculture purposes such as planning for selective breeding, maintaining stock diversity and distinguishing hatchery stocks from the wild populations.
References
AISHA-All India Shrimp Hatcheries Association: Status of Indian shrimp hatcheries. 7-1-44, Kirlampudi Visakhapatnam - 530 023 Andhra Pradesh, India; 2004:11.
Avise JC: Molecular markers, natural history and evolution. Chapman and Hall, New York; 1994.
Bailey-Brook JH, Mass SM: Penaeid taxonomy, biology and zoogeography. In Marine shrimp culture: principles and practices. Edited by: Fas WA, Lester LJ. Elsevier Science Publishers, Amsterdam; 1992:9-23.
Baldwin JD, Bass AL, Bowen BW, Clark WH: Molecular phylogeny and biogeography of marine shrimp Penaeus. Mol Phylogen Evol 1998, 10: 399-407. 10.1006/mpev.1998.0537
Benzie JAH: Genetic and reproduction research on giant tiger prawn Penaeus monodon : pond reared spawners achieved in Australia. Aust Biotech 1994, 4: 222-224.
Benzie JAH: Population genetic structure in penaeid prawns. Aqua Res 2000, 31: 95-119. 10.1046/j.1365-2109.2000.00412.x
Benzie JAH: Integration of quantitative and molecular genetics in shrimp breeding. Asian Fisheries Sci 2010, 23: 497-523.
Benzie JAH, Ballment E, Forbes AT, Demetriades NT, Sugama K, Haryanti MS: Mitochondrial DNA variation in Indo-Pacific population of the giant tiger prawn Penaeus monodon . Mol Ecol 2002, 11: 2553-2569. 10.1046/j.1365-294X.2002.01638.x
Carvalho GR, Hauser L: Molecular genetics and the stock concept in fisheries. Rev Fish Biol Fish 1994, 4: 326-350. 10.1007/BF00042908
Chu KH, Li CP, Tam YK, Lavery S: Application of mitochondrial control region in population genetic studies of the shrimp Penaeus. Mol Ecol Notes 2003, 3: 120-122. 10.1046/j.1471-8286.2003.00376.x
Clifford HC III, Preston NP: Global Shrimp OP:2001-preliminary report. in: Genetic improvement report. Global Aquat Adv 2001, 4: 20-26.
Conover DO, Clarke LM, Munch SB, Wagner GN: Spatial and temporal scales of adaptive divergence in marine fishes and its implications for conservation. J Fish Biol 2006, 69(C):21-47.
Cunningham CW, Blackstone NW, Buss LW: Evolution of king crabs from hermit crab ancestors. Nature 1992, 355: 539-542. 10.1038/355539a0
Dale WL: Wind and drift currents in the South China Sea. Malays J Trop Geogr 1956, 8: 1-31.
Dall W, Hill BJ, Rothlisberg PC, Staples DJ: The biology of Penaeidae. Adv Mar Biol 1990, 27: 1-484.
Excoffier L, Laval G, Schneider S Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol Bioinform 2005, 1: 47-50.
FAO: Introductions and movement of two penaeid shrimp species in Asia and the Pacific. Edited by: Briggs M, Funge-Smith S, Subasinghe RP, Phillips M. FAO. Fisheries Technical Paper No. 476, Rome; 2004:78.
FAO: Fisheries and aquaculture software. FishStat Plus - Universal software for fishery statistical time series. FAO Fisheries and Aquaculture Department [online], Rome; 2006. http://www.fao.org/fishery/statistics/software/en
Hall TA: BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 1999, 41: 95-98.
Jukes TH, Cantor CR: Evolution of protein molecules. In Mammalian Protein Metabolism. Edited by: Munro HN. Academic Press, New York; 1969:21-132.
Khamnamtong B, Klinbunga S, Menasveta P: Genetic diversity and geographic differentiation of the giant tiger shrimp ( Penaeus monodon ) in Thailand analyzed by mitochondrial COI sequences. Biochem Genet 2009, 47: 42-55. 10.1007/s10528-008-9205-3
Klinbunga S, Penman DJ, McAndrew BJ, Tassanakajon A, Jarayabhand P: Genetic variation, population differentiation, and gene flow of the giant tiger shrimp (P. monodon) inferred from mtDNA RFLP data. In Advances in Shrimp Biotechnology. Edited by: Flegel TW. National Center for Genetic Engineering and Biotechnology, Bangkok, Thailand; 1998:51-59.
Klinbunga S, Siludjai D, Wuthijinda W, Tassanakajon A, Jarayabhand P, Menasveta P: Genetic heterogeneity of the giant tiger shrimp ( Penaeus monodon ) in Thailand revealed by RAPD and mtDNA-RFLP analyses. Mar Biotechnol 2001, 3: 428-438. 10.1007/s10126-001-0055-9
Kumar N, Lakra WS, Majumdar KC, Goswami M, Ravinder K: Genetic diversity in the Indian population of Penaeus monodon (Fabricius 1798) as revealed by mtDNA sequence analysis. Aqua Res 2007, 38: 862-869. 10.1111/j.1365-2109.2007.01740.x
Lavery S, Chan TY, Tam YK, Chu KH: Phylogenetic relationships and evolutionary history of the shrimp genus Penaeus s.l. derived from mitochondrial DNA. Mol Phylogene Evol 2004, 31: 39-49. 10.1016/j.ympev.2003.07.015
Mandal A, Rao D, Karuppaiah D, Gopalakrishnan A, Pozhoth J, Samraj YC, Doyle RW: Population genetic structure of Penaeus monodon, in relation to monsoon current patterns in Southwest, East and Andaman coastal waters of India. Gene 2012, 491(2):149-157. 10.1016/j.gene.2011.10.002
McMillen-Jackson AL, Bert TM: Disparate patterns of population genetic structure and population history in two sympatric penaeid species in the Southeastern United States. Mol Ecol 2003, 12: 2895-2905. 10.1046/j.1365-294X.2003.01955.x
McMillen-Jackson AL, Bert TM: Genetic diversity in the mtDNA control region and population structure in the pink shrimp Farfantepenaeus duorarum . J Crustac Biol 2004, 24: 101-109. 10.1651/C-2372
Motoh H: Studies on the fisheries biology of the giant tiger prawn, Penaeus monodon in the Philippines. Technical paper no. 7, Aquaculture Department, Southeast Asian Department Center, Iloilo; 1981.
Naylor RL, Golfberg RJ, Primavera JH, Kautsky N, Beveridge MCM, Clay J, Folke C, Lubchenco J, Mooney H, Troell M: Effect of aquaculture on world fish supplies. Nature 2000, 405: 1017-1024. 10.1038/35016500
Nei M: Molecular Evolutionary Genetics. Columbia University Press, New York, NY, USA; 1987.
Palumbi SR, Benzie JAH: Large mitochondrial differences between morphologically similar penaeid shrimp. Mol Mar Biol Biotechnol 1991, 1: 27-34.
Ronnback P: Shrimp aquaculture - State of the art. Swedish EIA Centre, Report 1. Swedish University of Agricultural Sciences (SLU), Uppsala; 2001. ISBN 91-576-6113–8
Rosenberry B: World shrimp farming 2000. In Shrimp News International, USA. Edited by: Rosenberry B. Shrimp News International, San Diego, CA, USA; 2001:324.
Sambrook J, Fristisch EF, Maniatis T: Molecular Cloning - A Laboratory Manual. 2nd edition. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA; 2005:6.4-6.12.
Silberman JD, Sarver SK, Walsh PJ: Mitochondrial DNA variation and population structure in the spiny lobster Panulirus argus. Mar Biol 1994, 120: 601-608. 10.1007/BF00350081
Simon C: Molecular systematics at the species boundary: exploiting conserved and variable regions of the mitochondrial genome of animals via direct sequencing from amplified DNA. In Molecular Techniques in Taxonomy. Edited by: Hewitt GM, Johnston AWB, Young JPW. Springer-Verlag, New York; 1991.
Spann KM, Cowley JA, Walker PJ, Lester RJG: A yellow-head-like virus from Penaeus monodon cultured in Australia. Dis Aqua Organ 1997, 31: 169-179.
Tzeng TD, Yeh SY, Hui CF: Population genetic structure of the Kuruma Prawn (P. Japonicus) in East Asia inferred from Mt. DNA sequences. ICES J Mar Sci 2004, 61: 913-920. 10.1016/j.icesjms.2004.06.015
Ward RD, Grew PM: Appraisal of molecular genetic technique in fisheries. Rev F Biol Fish 1994, 4: 300-325. 10.1007/BF00042907
Weir BS, Cockerham CC: Estimating F statistics for the analysis of population structure. Evolution 1984, 38: 1358-1370. 10.2307/2408641
Wilson K, Cahill V, Ballment E, Benzie J: The complete sequence of the mitochondrial genome of the crustacean Penaeus monodon : are malacostracan crustaceans more closely related to insects than to branchiopods? Mol Biol Evol 2000, 17: 863-874. 10.1093/oxfordjournals.molbev.a026366
Withyachumnarnkul B, Boonsaeng W, Flegel TW, Panyim S, Wongteerasupaya C: Domestication and selective breeding of Penaeus monodon in Thailand. Edited by: Felgel T. Proceedings to the special session on advances in shrimp biotechnology, fifth Asian fisheries forum: international conference on fisheries and food security beyond the year 2000, Chiang Mai, Thailand; 1998:73-77.
Yang B, Song XL, Huang J, Shi CY, Liu QH, Liu L: A single-step multiplex PCR for simultaneous detection of white spot syndrome virus and infectious hypodermal and haematopoietic necrosis virus in penaeid shrimp. J Fish Dis 2006, 29: 301-305. 10.1111/j.1365-2761.2006.00713.x
Acknowledgements
Authors are thankful to DBT, Government of India for funding this work and to the anonymous reviewers for suggestions to improve this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GD -experimental design, laboratory work, data analysis and manuscript writing. ACR -Collection of stocks, molecular genetic analysis and related laboratory work. TB- Experimental design, data analysis and manuscript writing. DH experimental design, data analysis, and manuscript writing. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Khedkar, G.D., Reddy, A.C., Ron, T.B. et al. High levels of genetic diversity in Penaeus monodon populations from the east coast of India. SpringerPlus 2, 671 (2013). https://doi.org/10.1186/2193-1801-2-671
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2193-1801-2-671