
Abstract
Early diagnosis of lung cancers and distinction between the tumor types (Small Cell Lung Cancer (SCLC) and Non-Small Cell Lung Cancer (NSCLC) are very important to increase the survival rate of patients. Herein, we propose a diagnostic system based on sequence-derived structural and physicochemical attributes of proteins that involved in both types of tumors via feature extraction, feature selection and prediction models. 1497 proteins attributes computed and important features selected by 12 attribute weighting models and finally machine learning models consist of seven SVM models, three ANN models and two NB models applied on original database and newly created ones from attribute weighting models; models accuracies calculated through 10-fold cross and wrapper validation (just for SVM algorithms). In line with our previous findings, dipeptide composition, autocorrelation and distribution descriptor were the most important protein features selected by bioinformatics tools. The algorithms performances in lung cancer tumor type prediction increased when they applied on datasets created by attribute weighting models rather than original dataset. Wrapper-Validation performed better than X-Validation; the best cancer type prediction resulted from SVM and SVM Linear models (82%). The best accuracy of ANN gained when Neural Net model applied on SVM dataset (88%). This is the first report suggesting that the combination of protein features and attribute weighting models with machine learning algorithms can be effectively used to predict the type of lung cancer tumors (SCLC and NSCLC).
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Introduction
Lung cancer, as a leading cause of death worldwide, starts from the lungs and may spreads to other organs of the body and has a low survival rate of just 15% (Ganesan et al. 2010a 2010b Nomori 2011). It is estimated that at least 1.2 million people were infected with the disease, equivalent to the 12.3% of total number of cancer diagnosed annually, with a mortality rate of 1.1 million people per year (Parkin 2001 Webb-Robertson et al. 2010). As the treatments for each type of lung cancers are different (Motohiro et al. 2002), so if a patient is correctly diagnosed in early stage, the chance for one to be cured will increase (Zhou et al. 2002). The cancer’s types have already been divided into two groups based on pathological and morphological observations: non-small cell lung cancer (NSCLC) (80.4%) and small cell lung cancer (16.8%) (Travis et al. 1995). The diagnosis of tumor type is performed by histology, immunohistochemistry or pathology based on either cells morphology or protein expression. In addition, the underlying genetic aberrations or biological processes may contribute to the cancer malignancy process which cannot be revealed by histological appearance of a tumor (Khan et al. 2001). In order to improve the survival rate, need for an early type detection method of lung tumors have been raised (Delarue and Starr 1967), and this research carried to address this need based on data mining tools.
So far many different techniques such as Chest Radiograph (x-ray), Computed Tomography (CT), Magnetic Resonance Imaging (MRI) and Sputum Cytology have been used for lung cancer classification (Grondin and Liptay 2002 Schaefer-Prokop and Prokop 2002). However, most of these techniques are either expensive and time consuming or applicable only in the advanced stages, when the survival rate of patients is very limited (Fatma et al. 2012). During the last decades, computer-aided cancer classification systems along with the rapid developments of image processing and pattern recognition techniques have been proposed as suitable tools. On the other hand, many researches have looked into the bioinformatics models and data mining algorithms as alternative choices. To improve the accuracy and the speed of lung cancer diagnosis based on radiology, an artificial neural network via hybrid lung cancer detection system named HLND designed (Chiou YSP and Ligomenides 1993). In the other study, a system based on a parameterized two-level convolution artificial neural network have been developed to do this important task (Lin et al. 1996), an automatic method based on the subtraction between two serial mass chest radiographs, which was used in the detection of new lung nodules designed by Hayashibe et al. (Hayashibe et al. 1996). Abe et al. evaluated another computer-aided diagnosis (CAD) system with automatic detection of pulmonary nodules for lung cancer screening with computed tomography (CT) (Abe et al. 2005). Penedo and et al. employed two artificial neural network, one for detecting suspicious regions in a low-resolution image and the other for dealing with the curvature peaks of the suspicious regions, which was used in the detection of lung nodules (Penedo et al. 1998). In the diagnostic systems of lung cancer with computer-aided, the rate of false negative identification should be kept as low as possible to improve the rate of overall identification on the highest possible rate (Zhou et al. 2002).
Machine learning is an automatic and intelligent learning technique that employs variety of statistical tools to “learn” from past data and then use the prior training to classify new data, identify new patterns or predict novel trends (Mitchell 1997). These techniques have been widely used to solve many real world and complex problems (Kerhet et al. 2010). Since their introduction to the bioinformatics community, machine learning approaches helped to accelerate several major researches such as bimolecular structure prediction, gene finding, genomics and proteomics (Zycinski et al. 2011). As these techniques are efficient and inexpensive in solving bioinformatics problems, the applications of them in bioinformatics are becoming popular and continuing to develop (Liu 2004). In recent years machine learning methods have been widely used in prediction especially in medical diagnosis and interestingly. Almost all of these algorithms used in cancer prediction and prognosis employed supervised learning. Furthermore, most of these supervised learning algorithms belonged to a specific category of classifiers that classify on the basis of conditional probabilities or conditional decisions (Cruz and Wishart 2006 Ganesan et al. 2010a 2010b).
One of the most instrumental type of machine learning techniques are Support Vector Machines (SVM ) which were introduced by Vladimir Vapnik and his colleagues (Bazzani et al. 2001 Baumes et al. 2006). SVM s are used for binary classification to find a hyper plane which separates the d-dimensional data perfectly into its two classes (Parsaei and Stashuk 2012 Boswell 2002). In contrast to other classifiers, SVM searches for the hyper plane that maximizes the distance from the hyper plane to the nearest examples in each class. An attractive feature of SVM is that it can map linearly inseparable data into higher dimensional space where they can be linearly separated. This work is executable with introduction of “kernel induced feature space” notion. Recently, SVM has gained much attention as a useful tool for image recognition (Guan et al. 2009 Avci 2012). The use of SVM, like any other machine learning technique, involves two basic steps namely training and testing. The first step involves feeding known data to the SVM along with previously known decision. It is from the training set that an SVM gets its intelligence to classify unknown data (Van Belle et al. 2011). Several studies have already used the performance of Bayesian classifier; artificial neural net and SVM for differentiating obstructive lung diseases, and SVM gained the best performance for classification (Lee et al. 2009). It has been shown that SVM provide better overall quantification for interstitial lung disease differentiation in high-resolution computerized tomography images (Lim et al. 2011).
The Naive Bayes (NB) classifier technique is based on the so-called Bayesian theorem and is particularly suited when the dimensionality of the inputs is high. A Naive Bayes classifier considers that all attributes (features) independently contribute to the probability of a certain class. This classifier can be trained so efficiently in a supervised learning method and works much better in many complex real-world situations, especially in the computer-aided diagnosis (Gorunescu 2006 Belciug 2008 Dumitru 2009). Despite its simplicity, Naive Bayes can often outperform more sophisticated classification methods. The Bayesian approach allows scientists to combine new data with their existing knowledge or expertise. Using a training dataset, the Bayesian classifiers determine the probability of associating certain classes at certain instances given the values of the predictor variables. Naive Bayes classifier provides performances equivalent to other machine learning techniques with low computational effort and high speed (Dumitru 2009).
Herein, regarding the importance of early classification and prediction of lung tumor types in successful treatment of this disease, several machine learning algorithms employed to predict the type of lung cancers based on structural and physicochemical attributes of proteins. Some studies have used sequence-derived structural and physicochemical descriptors in machine learning prediction of structural and functional classes (Dubchak et al. 1999 Karchin et al. 2002 Cai et al. 2003 Cai et al. 2004 Han et al. 2004a 2004b), protein-protein interactions (Bock and Gough 2001 Bock and Gough 2003 Lo et al. 2005 Chou and Cai 2006), subcellular locations (Chou 2000 Chou and Cai 2004 Chou and Shen 2006 Guo and Lin 2006), peptides containing specific properties (Schneider and Wrede 1994 Cui et al. 2007), microarray data (Brown et al. 2000) and protein secondary structure prediction (Ward et al. 2003). Ong and et al. showed that currently used descriptors are generally useful for classifying proteins and the prediction performance may be enhanced by combinations of descriptors (Ong et al. 2007); in this experiment, the same datasets as previously reported was used (Hosseinzadeh et al. 2012). Previously feature selection, tree induction and clustering models had been used to classify lung tumors based on important protein features. Follow up of previse work, the application of three machine learning models practiced here to introduce accurate prediction tools for lung cancer types based on important attributes of related proteins.
Results
Data preparation and feature selection
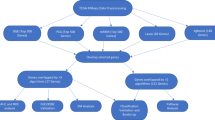
Proteins that involved in two types of lung tumors obtained from conversion of gene symbols defined by microarray analysis in the GSEA db, using DAVID server. The list of genes associated with two types of lung tumors and those that were common between them showed in Table 1.
Data cleaning
In original dataset, 59 records classified as SCLC, 30 records belonged to NSCLC class and 25 other records to COMMON tumor classes. For each record 1497 features computed and after removing duplicate, useless and correlated attributes, the number of protein features for each record decreased to 1089 features (less than 28% removed) and this cleaned dataset named as Final Cleaned database (FCdb).
Feature selection
Twelve attributes weighting models applied on FCdb which gave each feature a weight between 0 to 1. Features that gained weight values higher than 0.50 with at least 50% of weighting algorithms regarded as important protein features. Figure 1 showed the most important protein attributes selected by more than 50 percent of attribute weighting algorithms (Information gain, Information gain ratio, Rule, Deviation, Chi Squared, Gini index, Uncertainty, Relief, SVM and PCA). Dispersions of features’ weight values by two other weighting models (SAM and Maximum Relevance) have illustrated in the Figure 2 and Figure 3.

The most important protein attributes selected by more than fifty percent of attribute weighting algorithms. As is evident, the features of distribution descriptor (F5.3), dipeptide composition (F1.2) and autocorrelation (F3.1) were defined important by 80% of attribute weighting models.

Dispersion of protein attributes that gained weight value between 0 to 1 by attribute weighting model of SAM (the index of protein attributes exactly defined in Additional file 1 : Table S1).

Dispersion of protein attributes that gained weight value between 0 to 1 by attribute weighting model of Maximum Relevance (the index of protein attributes exactly defined in Additional file 1 : Table S1).
Classification and prediction
Support vector machine approach
Gained accuracies and Kappa values for each SVM model (while Gamma and C set as 0.0065 and 10, respectively and ran with X-validation approach) on 13 datasets (FCdb and 12 datasets that obtained from attribute weighting application: Information gain, Information gain ratio, Rule, Deviation, Chi Squared, Gini index, Uncertainty, Relief, SVM, PCA, SAM and MR) illustrated in the Table 2. Furthermore, Table 3 shows the results of running seven SVM and wrapper validation methods on datasets that derived from attribute weighting (this model cannot be applied on main dataset, FCdb, as required attribute weighted datasets). When X-validation (ten-fold cross validation) applied, the average accuracy ranged from 32.27% (SVM Hyper) to 67.36% (for SVM and SVM Linear), while the lowest and highest accuracies accounted for the same algorithms (30.0% and 81.67%, respectively). The Kappa index was followed the same pattern, the lowest came from SVM Hyper (-6.10%) and the highest from SVM and SVM Liner (69.09%). With Wrapper validation application, the average accuracies ranged from 33.21% (for SVM Hyper) to 69.53% (SVM) and the minimum and maximum accuracies (23.86% and 71.97%) were again for the same models, respectively (Figure 5).
Artificial neural network
The results of running three models of ANN (Auto MLp, Neural Net and Perceptron) on 13 datasets given in the Table 4. The most accurate model was Neural Net when applied on SVM dataset with accuracy of 87.73%. The ranges of accuracies in three models of artificial neural network were respectively 52–86, 53–83 and 31 – 59 percents. The percentage of Kappa in the Auto MLP model except for PCA and Deviation datasets started from 46% to went up to 77%. The maximum and minimum percent of Kappa in the Neural Net model (except for PCA and Deviation datasets) were respectively 80% and 43%. Kappa index in Perceptron model was lower than two other models and its maximum got to to 26%.
Naïve bayes
As shown in the Table 5, the performance of NB models was lower when compared to SVM and ANN algorithms. The best accuracy gained with NB models was 77% and belonged to Naïve Bayes model ran on SVM and Maximum Relevance datasets. The Bayes Kernel model performance on 13 datasets differ from 54% to 70% and the same ranged from 44% to 77% when Naïve Bayes model applied (except for FCdb). The Kappa index again was lower than accuracy and its maximum and minimum values were 63% and 11%.
Discussion
Lung cancer is considered as the main cause of cancer death worldwide, and detection of this disease in its early stages is difficult because symptoms appear only at advanced stages causing the mortality rate to be high(Fatma et al. 2012). The 5-year survival rate of localized stage, when the cancer does not spread to additional sites like lymph nodes or other parts of body, is about 50%. Various factors influencing 5-year survival rate such as the stage of cancer, the type of cancer, general health, etc. Early detection of lung cancer is the leading factor decreasing mortality rate and increasing in survival rate (Fontana et al. 1986). Histologically, about 80% of lung cancer are from NSCLC class and just 20% are identified as SCLC cancers (Hu et al. 2002). The pathological distinction between NSCLC and SCLC tumors is so important because patients with NSCLC tumor are treated differently from those with SCLC tumors (Garber et al. 2001). Detection of lung cancer in its early stage is the key in curing patient and automated diagnosis would play crucial roles in this matter (Ganesan et al. 2010a 2010b).
So far many scientists tried to propose new methods to classify the types of lung cancer in early stages (Edwards et al. 2000 Petersen and Petersen 2001 Beadsmoore and Screaton 2003 Boffa 2011 Anagnostou et al. 2012 Gilad et al. 2012 West et al. 2012). In some studies, bioinformatics or data mining models have been used. For example, a training–testing approach has been used to test the reliability of cDNA microarray-based classifications of resected human NSCLCs analyzed (Yamagata et al. 2003). Artificial neural networks have already been widely exploited in computer-aided lung cancer diagnosis, classifing of individual lung cancer cell lines (SCLC and NSCLC) based on DNA methylation markers by using linear discriminant analysis and artificial neural networks (Marchevsky et al. 2004). Neural network have also been used for lung cancer diagnosis to help oncologists to plan for a better medication and early diagnosis (Ganesan et al. 2010a 2010b). The color and texture features from images have also been used as tools for the classification of lung cancer using artificial neural network (Almas and Bariu, 2012). Furthermore, lung cancer gene expression database analysis incorporated prior knowledge with support vector machine-based classification method into cancer classification (Guan et al. 2009). The use of machine learning in cancer detection and prediction is investigated in another study (Lipson et al. 1961). Machine learning techniques like artificial neural network and decision tress are used for cancer detection for nearly 20 years (Galeotti et al. 1986 Campanella 1992 Liu et al. 2006). The potential of using machine learning methods for detecting lung cancer cells or tumors via X-rays, Computed Tomography (CT) has been elaborated in other studies (van Ruth et al. 2003 Kancherla and Mukkamala 2011 2012). They have also been used for tumor classification or cancer detection using microarray data or gene expression are Fisher Linear Discriminant analysis (Brown and Botstein 1999), K Nearest Neighbor (KNN) (Dudoit et al. 2002), (SVM ) (Peterson and Ringner 2002), boosting, and self-organizing maps (SOM) (Eisen et al. 1998), hierarchical clustering (Tamayo et al. 1999), and graph theoretic approaches (Sakas et al. 2007).
A significant number of researchers have worked on the ensemble of the multiple classifiers to improve the performance of classification of cancer (Abbass 2002 Zhou et al. 2002 Futschik et al. 2003 Santos-Garcia et al. 2004 Hong-HeeWon 2007). The ensemble classifier increases not only the performance of the classification, but also the confidence of the results. Zhou and et al. propose an automatic pathological diagnosis procedure named NED, which utilizes artificial neural network ensemble to identify lung cancer cells in the images of the specimens of needle biopsies (Zhou et al. 2002).
Regarding the importance of distinction between lung cancer tumors and need for finding new simple and effective methods for lung cancer types’ detection, the classification and prediction of lung cancers based on structural and physicochemical descriptors of proteins have been proposed by using machine learning models, as sequence-derived structural and physicochemical descriptors may be highly useful for representing and distinguishing proteins or peptides of different structural irrespective of sequence similarity (Cai et al. 2003 Han et al. 2004a 2004b Lo et al. 2005 Li et al. 2006) too long sentence.
The first step for fulfilling to this idea is identification and selection of most important and appropriate features. PROFEAT is a very trusty server for computing sequence-derived structural and physicochemical descriptors (Rao et al. 2011), so 1497 attributes of SCLC and NSCLC proteins computed. Twelve different attribute weighting models applied to final cleaned dataset; as each algorithm uses a specific pattern to define the most important features, thus, the results may be different (Baumgartner et al. 2010 Ebrahimi et al. 2011 Ebrahimie et al. 2011 Hosseinzadeh et al. 2012). As is shown in Figure 1, inline with our previous published study (Hosseinzadeh et al. 2012), the most important feature groups that selected by 80% of models were from F5.3 (distribution descriptors), F1.2 (dipeptide composition) and F3.1 (autocorrelation) groups. Furthermore, Figure 2 and Figure 3 show that by running two additional weighting models, F1.2 (dipeptide composition), F2.1 (autocorrelation) and F3.1 (autocorrelation) were also selected as important features. These features can be effectively used to distinguish between different types of lung tumors. As is proven in the other study, feature selection reduces the number of features and improves the accuracy (Kancherla et al. 2009), here also the potential use of feature selection to improve the accuracy and efficiency of lung cancer detection is confirmed.
In next step, classification and prediction of lung tumors based on structural and physicochemical properties of associated proteins performed and several prediction models such as SVM, ANN and NB used. Seven prediction models of support vector machines (SVM, LibSVM, SVM Linear, SVM Evolutionary, SVM PSO, SVM Fast Large Margin and SVM Hyper Hyper) applied on 13 datasets (main dataset, FCdb, and 12 other datasets that generated from attribute weighting algorithms: Information gain, Information gain ratio, Rule, Deviation, Chi Squared, Gini index, Uncertainty, Relief, SVM, PCA, SAM and Maximum Relevance). Two validation algorithms, X-validation and X-wrapper, applied on datasets to calculate the models performance and accuracies (Tables 2 and 3). The findings showed SVM Hyper performance was the worst and this model even was less accurate than chance models (average 33.21%). Two other models (SVM and SVM Linear) showed the best performance and their accuracies reached up to 82%. When the results of two validation methods (X-Validation and Wrapper Validation) compared, generally the performance of X-Validation was better than X-Wrapper Validation, although the Wrapper performed better when applied on SVM , SVM Linear and SVM Fast models. The best accuracies gained when X-Validation applied on dataset created from SVM attribute weighting but for Wrapper-Validation the datasets were Deviation, Relief and Rule. The results suggested that either SVM or SVM Linear would be the best candidate algorithms to predict lung cancer if they apply on SVM datasets.
The results of three ANN application (Auto MLp, Neural Net and Perceptron) showed Neural Net was the best and the most accuracte model when it agained applied on SVM dataset, while the worst performance belonged to Perceptron model; the accuracies of Auto MLp and Perceptron models were high and nearly at the same levels (86% and 58%) when they applied on Information Gain and SVM datasets. Generally the Kappa indexes were less accurate, the best index obtained from three models Auto MLp, Neural Net and Perceptron were respectively 77%, 80% and 26%; therefore the best index gained from Neural Net model, too.
As shown in Table 5, the best accuracy and Kappa index of Naïve base and Naïve base kernel models gained when they ran on Maximum Relevance dataset (77%), and again the indices were lower. The results confirmed that Naïve base model was is better than Naïve base kernel.
Conclusions
Comparing the performances of three types of machine learning models (SVM , ANN and NB) to predict and detect the type of lung tumors based on structural and physicochemical attributes of proteins showed that the Neural Net model ran on SVM dataset gained the best accuracy (88%). Our results showed the potential use of feature selection and prediction models can be effectively used as a simple application. The results also showed that attribute weighting can be beneficiary both to processing time and getting more accurate results.
Dipeptide composition, Moran autocorrelation and distribution descriptor were the most important protein features selected by weighting tools. To our best knowledge, the findings of this study for the first time showed that protein features in combination with machine learning algorithms can be effectively used to determine any types of lung cancer tumors.
Materials and methods
Data preparation and feature selection
As shown in our previous study (Hosseinzadeh et al. 2012), the over represented genes in any type of lung tumors obtained from microarray GSEA db (Gene Set Enrichment Analysis database); a well-known database used for storing the results of experimental microarray analysis and determines whether contains a section of Molecular Signatures Database (MSigDB) that is a collection of annotated gene sets for use with GSEA software. It made possible to search for gene sets, examine gene sets and their annotations and download them (Subramanian et al. 2005). A list of appropriate gene lists defined and downloaded (for more details in (Hosseinzadeh et al. 2012). The gene sets converted to protein accession numbers by using DAVID server (http://david.abcc.ncifcrf.gov) and protein sequences extracted from UniProt Knowledgebase (Swiss-Prot and TrEmble) afterwards.
Structural and physicochemical attributes
Seven types of of proteins features that were involved in three classes of lung tumors (SCLC, NSCLC and COMMON) were calculated by using PROFEAT web server facilities. These features included of (1) amino acid composition, dipeptide composition. (2) Normalized Moreau–Broto autocorrelation; (3) Moran autocorrelation; (4) Geary autocorrelation; Autocorrelation descriptors are defined from the distribution of amino acid properties along the sequence. The amino acid indices used in these autocorrelation descriptors included hydrophobicity scales, average flexibility indices, polarizability parameter, and free energy of solution accessible surface area in trepeptide, residue volume, steric parameter, and relative mutability. (5) the composition (C), transition (T) and distribution (D) of various structural and physicochemical properties; These descriptors are derived for each of the following physicochemical properties: hydrophibicity, polarity, polarizibility, charge, secondary structures, and normalized Van der Waals volume. (6) sequence- order-coupling number, quasi sequence-order attributes; The quasi-sequence-order descriptors are derived from both the Schneider-Wrede physicochemical distance matrix and the Grantham chemical distance matrix between the 20 amino acids. (7) pseudo amino acid composition; Instead of using the conventional amino acid composition to represent the sample of a protein, Chou proposed the pseudo amino acid (PseAA) composition in order to include the sequence-order information. Therefore one thousands and ninety seven protein features or attributes computed by PROFEAT web server (http://jing.cz3.nus.edu.sg/cgi-501bin/prof/prof.cgi). An index of Fi.j.k.l is used to represent the lth descriptor value of the kth descriptor of the jth feature in the ith feature group, which serves as an easy reference to the PROFEAT manual provided in the server homepage (Li et al. 2006). A complete list of these feature indices and their complete definition is shown in the Addtional file 1: Table S1 (Hosseinzadeh et al. 2012).
Running data mining models
A dataset of 1497 features of three groups of protein was imported into Rapid Miner software (Rapid Miner 5.0.001, Rapid-I GmbH, Stochumer Str. 475, 44227 Dortmund, Germany), and the type of tumor was set as target or label attribute.
Data cleaning
Duplicate and useless features removed and the new database was labeled as the Final Cleaned database (FCdb).
Attribute weighting
To identify the most important features, 12 attribute weightings algorithms applied to the FCdb (they were: weight by Information gain, weight by Information Gain ratio, weight by Rule, weight by Deviation, weight by Chi squared statistic, weight by Gini index, weight by Uncertainty, weight by Relief, weight by SVM (Support Vector Machine) and weight by PCA (Principle Component Analysis) (for more details see (Hosseinzadeh et al. 2012)). Two more attribute weighting models of SAM and MR are also applied in this study with the following definition:
Weight by Significance analysis of microarrays:SAM is a statistical technique; introduced in 2001; which used to determine whether changes in gene expression are statistically significant or not. With the advent of DNA microarrays it is now possible to measure the expression of thousands of genes in a single hybridization experiment. Generated data is huge and introducing such a model is essential.
Weight by Maximum Relevance: The Max-Dependency feature selection can be efficiently implemented as the Minimum Redundancy and Maximum Relevance (mRMR) algorithm. Significantly outperforms the widely used max-relevance selection method: mRMR features cover a broader feature space with fewer features. mRMR is very efficient and useful for gene selection and many other applications. Both relevance and redundancy estimation are low dimensional problems (i.e. involving only 2 variables). This is much easier than directly estimating multivariate density or mutual information in the high dimensional space, this algorithm is faster speed and more reliable estimation.
Attribute selection
After running attribute weighting models on FCdb, each protein attribute gained a weight (between 0 and 1) showing its importance with regards to the target attribute (type of tumors). All variables that obtained weight values higher than 0.50 were selected and 12 new datasets created. These newly formed datasets were named according to their attribute weighting models.
Classification and prediction
In our previous study, after running feature selection, several decision tree induction models and unsupervised clustering algorithms employed to identify the most important protein attributes and obtaining the best classification of lung tumors based of them, but here in this study we used machine learning methods to predict the type of lung tumor based on machine learning and training capabilities.
Support vector machine approach
SVM s are popular and powerful supervised data classification and prediction techniques with associated learning algorithms which analyze data and recognize patterns. Basic SVM takes a set of input data and predicts, for each given input, which of two possible classes forms the output, making it a non-probabilistic binary linear classifier. Given a set of training examples, each marked as belonging to one of two categories, a SVM training algorithm builds a model that assigns new examples into one category or the other. Herein we used seven models of SVM algorithms (SVM, LibSVM, SVM Linear, SVM Evolutionary, SVM PSO, SVM Fast Large Margin and SVM Hyper Hyper) on 13 datasets to predict the type of lung tumors based on sequence-derived structural and physicochemical descriptors of proteins that involved in different types of lung tumors. LIBSVM is an integrated software for support vector classification, (C-SVC, nu-SVC), regression (epsilon-SVR, nu-SVR) and distribution estimation (one-class SVM). It supports multi-class classification. Linear SVM is the newest extremely fast machine learning (data mining) algorithm for solving multiclass classification problems from ultra large data sets that implements an original proprietary version of a cutting plane algorithm for designing a linear support vector machine. Linear SVM is a linearly scalable routine meaning that it creates an SVM model in a CPU time which scales linearly with the size of the training data set. Evolutionary support vector machines (ESVMs) are novel techniques, these methods incorporate the learning engine of the up to date SVMs but develop the coefficients of the decision function by means of evolutionary algorithms (EAs) (Stoean, Stoean et al. 2011). PSO (Particle Swarm Optimization) algorithms make particles with fitness values which are evaluated by the fitness function to be optimized. PSO is initialized with a group of random particles (solutions) and then searches for most efficient particles by updating each generation (Ardjani and Sadouni 2010). Applies a fast margin learner based on the linear support vector learning scheme proposed by R.-E. Fan, K.-W. Chang, C.-J. Hsieh, X.-R. Wang, and C.-J. Lin. Although the result is similar to those delivered by classical SVM or logistic regression implementations, this linear classifier is able to work on data set with millions of examples and attributes. It is well-known that SVM can be properly used for two-way classification. Hyper SVM (Shyu and Liao model solve this problem that how can we decide which parameter order can be changed to reproduce a new classification. This model is included a Huffman-Tree like mechanism, called hyper SVM 2011). Briefly, main database (FCdb) transformed to SVM format and scaled by grid search (to avoid attributes in greater numeric ranges dominating those in smaller numeric ranges) and to find the optimal values for operator parameters. Dataset divided into 10 parts and 9 parts used as training set and the last part as testing set.
Validation methods
To prevent over-fitting problems, X-validation and X-wrapper validation methods applied and the procedure repeated for 12 different testing sets (Information gain, Information gain ratio, Rule, Deviation, Chi Squared, Gini index, Uncertainty, Relief, SVM, PCA, SAM and Maximum Relevance) and then the average of accuraies and Kappa indices computed. The performance evaluator operator used for classification tasks (in cases where the label attribute has a binominal value type) and for polynominal classification tasks. Other polynominal classification tasks such as Polynominal Classification Performance Evaluator (PCPE) operator employed and accuracy and Kappa statistics calculated.
Kernel trick models
In addition to performing linear classification, SVM s can efficiently perform non-linear classification using what is called the kernel trick, implicitly mapping their inputs into high-dimensional feature spaces. The original optimal hyperplane algorithm proposed was a linear classifier. However, later on it was suggested a way to create nonlinear classifiers by applying the kernel trick to maximum-margin hyperplanes. The resulting algorithm is formally similar, except that every dot product is replaced by a nonlinear kernel function. This allows the algorithm to fit the maximum-margin hyperplane in a transformed feature space. For machine learning algorithms, the kernel trick is a way of mapping observations from a general set S into an inner product space V (equipped with its natural norm), without ever having to compute the mapping explicitly, in the hope that the observations will gain meaningful linear structure in V. Linear classifications in V are equivalent to generic classifications in S. The trick to avoid the explicit mapping is to use learning algorithms that only require dot products between the vectors in V, and choose the mapping such that these high-dimensional dot products can be computed within the original space, by means of a kernel function (Figure 4). Therefore, we applied the kernel types of C-SCV, radial and dot on the datasets to find the best accuracy.

The mechanism of Kernel trick models. These machines are used to compute a non-linearly separable function into a higher dimension linearly separable function.

Average performances of two validation methods (X-Validation and Wrapper-Validation) applied on seven different SVM algorithms ( SVM , SVM Linear, SVM Lib, SVM Evolutionary, SVM POS, SVM Hyper and SVM Fast).
Artificial neural network (ANN)
Three models of artificial neural networks algorithms ran on 13 datasets (FCdb and 12 datasets that obtained from attribute weighting models). The models were Auto MLp ( multilayer perceptron), Neural Net and Perceptron (Single-layer Neural Networks). The term of "Perceptrons" was coined by Frank Rosen Blatt in 1962 and is used to describe the connection of simple neurons into networks. In computational geometry, the Single-layer Neural Networks (Perceptrons) is an algorithm for supervised classification of an input into one of two possible outputs. It is a type of linear classifier, i.e. a classification algorithm that makes its predictions based on a linear predictor function combining a set of weights with the feature vector describing a given input. For the moment we will concentrate on Single Layer Perceptrons. A multilayer perceptron (MLP) is a feed forward artificial neural network model that maps sets of input data onto a set of appropriate output. An MLP consists of multiple layers of nodes in a directed graph, with each layer fully connected to the next one. Except for the input nodes, each node is a neuron (or processing element) with a nonlinear activation function. MLP utilizes a supervised learning technique called back propagation for training the network (Rosenblatt 1961). MLP is a modification of the standard linear perceptron and can distinguish data that is not linearly separable (Cybenko 1989). The accuracy and Kappa values from running these three ANN models on 13 datasets illustrated in Table 4.
Naïve Bayes
A Naïve Bayes classifier is a simple probabilistic classifier based on applying Bayes' theorem with strong (naïve) independence assumptions. A more descriptive term for the underlying probability model would be "independent feature model". In simple terms, a Naïve Bayes classifier assumes that the presence (or absence) of a particular feature of a class is unrelated to the presence (or absence) of any other feature, given the class variable. This classifier has been widely used before (for more details see (West 2003 Baseri et al. 2011). Two models of Naïve Bayse (returns classification model using estimated normal distributions) and Naïve bayse kernel (returns classification model using estimated kernel densities) (Beiki et al. 2012) used and the model accuracy in predicting the type of lung tumor calculted.
References
Abbass HA: An evolutionary artificial neural networks approach for breast cancer diagnosis. Artif Intell Med 2002, 25(3):265-281. 10.1016/S0933-3657(02)00028-3
Abe Y, Hanai K, Nakano M, Ohkubo Y, Hasizume T, Kakizaki T, Nakamura M, Niki N, Eguchi K, Fujino T, Moriyama N: A computer-aided diagnosis (CAD) system in lung cancer screening with computed tomography. Anticancer Res 2005, 25(1B):483-488.
Almas P, Bariu KS: Detection and classification of lung cancer using artificial neural network. IJACECT 2012, 1(1):62-67.
Anagnostou VK, Dimou AT, Botsis T, Killiam EJ, Gustavson MD, Homer RJ, Boffa D, Zolota V, Dougenis D, Tanoue L, Gettinger SN, Detterbeck FC, Syrigos KN, Bepler G, Rimm DL: Molecular classification of nonsmall cell lung cancer using a 4-protein quantitative assay. Cancer 2012, 118(6):1607-1618. 10.1002/cncr.26450
Ardjani F, Sadouni K: Optimization of SVM multiclass by particle swarm (PSO-SVM). IJMECS 2010, 2(2):32. 10.5815/ijmecs.2010.02.05
Avci E: A new expert system for diagnosis of lung cancer: GDA-LS_SVM. J Med Syst 2012, 36(3):2005-2009. 10.1007/s10916-011-9660-y
Baseri S, Towhidi M, Ebrahimie E: A modified efficient empirical bayes regression model for predicting phenomena with a large number of independent variables and fewer observations; examples of its application in human disease, protein bioinformatics, and microarray gene expression profiling. Advanced Studies in Biology 2011, 3: 181-204.
Baumes LA, Serra JM, Serna P, Corma A: Support vector machines for predictive modeling in heterogeneous catalysis: a comprehensive introduction and overfitting investigation based on two real applications. J Comb Chem 2006, 8(4):583-596. 10.1021/cc050093m
Baumgartner C, Lewis GD, Netzer M, Pfeifer B, Gerszten RE: A new data mining approach for profiling and categorizing kinetic patterns of metabolic biomarkers after myocardial injury. Bioinformatics 2010, 26(14):1745-1751. 10.1093/bioinformatics/btq254
Bazzani A, Bevilacqua A, Bollini D, Brancaccio R, Campanini R, Lanconelli N, Riccardi A, Romani D: An SVM classifier to separate false signals from microcalcifications in digital mammograms. Phys Med Biol 2001, 46(6):1651-1663. 10.1088/0031-9155/46/6/305
Beadsmoore CJ, Screaton NJ: Classification, staging and prognosis of lung cancer. Eur J Radiol 2003, 45(1):8-17. 10.1016/S0720-048X(02)00287-5
Beiki AH, Saboor S, Ebrahimi M: A new avenue for classification and prediction of olive cultivars using supervised and unsupervised algorithms. PLoS One 2012, 7(9):e44164. 10.1371/journal.pone.0044164
Belciug S: Bayesian classifcation vs. k-nearest neighbor classifcation for the non-invasive hepatic cancer detection. Research notes in artifcial intelligence and digital communications 2008, 31-35.
Bock JR, Gough DA: Predicting protein–protein interactions from primary structure. Bioinformatics 2001, 17(5):455-460. 10.1093/bioinformatics/17.5.455
Bock JR, Gough DA: Whole-proteome interaction mining. Bioinformatics 2003, 19(1):125-134. 10.1093/bioinformatics/19.1.125
Boffa DJ: The revised stage classification system for primary lung cancer. Clin Chest Med 2011, 32(4):741-748. 10.1016/j.ccm.2011.08.013
Boswell D: Introduction to Support Vector Machines. 2002.
Brown MP, Grundy WN, Lin D, Cristianini N, Sugnet CW, Furey TS, Ares M Jr, Haussler D: Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc Natl Acad Sci USA 2000, 97(1):262-267. 10.1073/pnas.97.1.262
Brown PO, Botstein D: Exploring the new world of the genome with DNA microarrays. Nat Genet 1999, 21(1 Suppl):33-37.
Cai CZ, Han LY, Ji ZL, Chen X, Chen YZ: SVM-Prot: Web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res 2003, 31(13):3692-3697. 10.1093/nar/gkg600
Cai CZ, Han LY, Ji ZL, Chen YZ: Enzyme family classification by support vector machines. Proteins 2004, 55(1):66-76. 10.1002/prot.20045
Campanella R: Membrane lipids modifications in human gliomas of different degree of malignancy. J Neurosurg Sci 1992, 36(1):11-25.
Chiou YSP LY, Ligomenides PA: Neural network image analysis and classification in hybrid lung nodule detection (HLND) system. Proceedings of the IEEE-SP Workshop on Neural Networks for Signal Processing 1993.
Chou KC: Prediction of protein subcellular locations by incorporating quasi-sequence-order effect. Biochem Biophys Res Commun 2000, 278(2):477-483. 10.1006/bbrc.2000.3815
Chou KC, Cai YD: Prediction of protein subcellular locations by GO-FunD-PseAA predictor. Biochem Biophys Res Commun 2004, 320(4):1236-1239. 10.1016/j.bbrc.2004.06.073
Chou KC, Cai YD: Predicting protein-protein interactions from sequences in a hybridization space. J Proteome Res 2006, 5(2):316-322. 10.1021/pr050331g
Chou KC, Shen HB: Hum-PLoc: a novel ensemble classifier for predicting human protein subcellular localization. Biochem Biophys Res Commun 2006, 347(1):150-157. 10.1016/j.bbrc.2006.06.059
Cruz JA, Wishart DS: Applications of machine learning in cancer prediction and prognosis. Cancer Inform 2006, 2: 59-77.
Cui J, Han LY, Lin HH, Zhang HL, Tang ZQ, Zheng CJ, Cao ZW, Chen YZ: Prediction of MHC-binding peptides of flexible lengths from sequence-derived structural and physicochemical properties. Mol Immunol 2007, 44(5):866-877. 10.1016/j.molimm.2006.04.001
Cybenko G: Approximation by superpositions of a sigmoidal function. MCSS 1989, 2(4):303-314.
Delarue NC, Starr J: A review of some important problems concerning lung cancer. The importance of complete preoperative assessment in bronchogenic carcinoma. Can Med Assoc J 1967, 96(1):8-20.
Dubchak I, Muchnik I, Mayor C, Dralyuk I, Kim SH: Recognition of a protein fold in the context of the structural classification of proteins (SCOP) classification. Proteins 1999, 35(4):401-407. 10.1002/(SICI)1097-0134(19990601)35:4<401::AID-PROT3>3.0.CO;2-K
Dudoit S, Fridlyand J, Speed T: Comparison of discrimination methods for the classification of tumors using gene expression data. J Am Statistical Assoc 2002, 97(457):77-87. 10.1198/016214502753479248
Dumitru D: Prediction of recurrent events in breast cancer using the Naive Bayesian classifcation. Annals of University of Craiova, Math Comp Sci Ser 2009, 36(2):92-96.
Ebrahimi M, Lakizadeh A, Agha-Golzadeh P, Ebrahimie E: Prediction of thermostability from amino acid attributes by combination of clustering with attribute weighting: a new vista in engineering enzymes. PLoS One 2011, 6(8):e23146. 10.1371/journal.pone.0023146
Ebrahimie E, Ebrahimi M, Sarvestani NR: Protein attributes contribute to halo-stability, bioinformatics approach. Saline Systems 2011, 7(1):1. 10.1186/1746-1448-7-1
Edwards SL, Roberts C, McKean ME, Cockburn JS, Jeffrey RR, Kerr KM: Preoperative histological classification of primary lung cancer: accuracy of diagnosis and use of the non-small cell category. J Clin Pathol 2000, 53(7):537-540. 10.1136/jcp.53.7.537
Eisen MB, Spellman PT, Brown PO, Botstein D: Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 1998, 95(25):14863-14868. 10.1073/pnas.95.25.14863
Fatma T, Naoufel W, Hussain A-A, Rachid S: Lung cancer detection by using artificial neural network and fuzzy clustering methods. Am J Biomed Eng 2012, 295-298.
Fontana RS, Sanderson DR, Woolner LB, Taylor WF, Miller WE, Muhm JR: Lung cancer screening: the Mayo program. J Occup Med 1986, 28(8):746-750. 10.1097/00043764-198608000-00038
Futschik ME, Reeve A, Kasabov N: Evolving connectionist systems for knowledge discovery from gene expression data of cancer tissue. Artif Intell Med 2003, 28(2):165-189. 10.1016/S0933-3657(03)00063-0
Galeotti T, Borrello S, Minotti G, Masotti L: Membrane alterations in cancer cells: the role of oxy radicals. Ann N Y Acad Sci 1986, 488: 468-480. 10.1111/j.1749-6632.1986.tb46579.x
Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen GD, Perou CM, Whyte RI, Altman RB, Brown PO, Botstein D, Petersen I: Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A 2001, 98(24):13784-13789. 10.1073/pnas.241500798
Gilad S, Lithwick-Yanai G, Barshack I, Benjamin S, Krivitsky I, Bocker Edmonston T, Bibbo M, Thurm C, Horowitz L, Huang Y, Feinmesser M, Steve Hou J, St Cyr B, Burnstein I, Gibori H, Dromi N, Sanden M, Kushnir M, Aharonov R: Classification of the four main types of lung cancer using a MicroRNA-based diagnostic assay. J Mol Diagn 2012, 14(5):510-517. 10.1016/j.jmoldx.2012.03.004
Gorunescu F: Data Mining: Concepts, models and techniques. Cluj- Napoca: Blue Publishing House; 2006.
Grondin SC, Liptay MJ: Current concepts in the staging of non-small cell lung cancer. Surg Oncol 2002, 11(4):181-190. 10.1016/S0960-7404(02)00050-6
Guan P, Huang D, He M, Zhou B: Lung cancer gene expression database analysis incorporating prior knowledge with support vector machine-based classification method. J Exp Clin Cancer Res 2009, 28: 103. 10.1186/1756-9966-28-103
Guo J, Lin Y: TSSub: eukaryotic protein subcellular localization by extracting features from profiles. Bioinformatics 2006, 22(14):1784-1785. 10.1093/bioinformatics/btl180
Han LY, Cai CZ, Ji ZL, Cao ZW, Cui J, Chen YZ: Predicting functional family of novel enzymes irrespective of sequence similarity: a statistical learning approach. Nucleic Acids Res 2004, 32(21):6437-6444. 10.1093/nar/gkh984
Han LY, Cai CZ, Lo SL, Chung MC, Chen YZ: Prediction of RNA-binding proteins from primary sequence by a support vector machine approach. RNA 2004, 10(3):355-368. 10.1261/rna.5890304
Hayashibe R, Asano N, Hirohata H, Okumura K, Kondo S, Handa S, Takizawa M, Sone S, Oshita S: An automatic lung cancer detection from X-ray images obtained through yearly serial mass survey. ICIP 1996, 1: 343-346.
Hong-HeeWon S-BC: Cancer classification using ensemble of neural networks with multiple significant gene subsets. Springer Science?+?Business Media; 2007:243-250.
Hosseinzadeh F, Ebrahimi M, Goliaei B, Shamabadi N: Classification of lung cancer tumors based on structural and physicochemical properties of proteins by bioinformatics models. PLoS One 2012, 7(7):e40017. 10.1371/journal.pone.0040017
Hu YC, Sidransky D, Ahrendt SA: Molecular detection approaches for smoking associated tumors. Oncogene 2002, 21(48):7289-7297. 10.1038/sj.onc.1205805
Kancherla K, Mukkamala S: Lung cancer detection using labeled sputum sample: multi spectrum approach. Modern Approaches in Applied Intelligence 2011, 446-458.
Kancherla K, Mukkamala S: Feature selection for lung cancer detection using SVM based recursive feature elimination method. Evolutionary Computation, Machine Learning and Data Mining in Bioinformatics 2012, 168-176.
Kancherla MK, Aveeshaveeshti S, Cousins J: Labeling of cancer cells in sputum for the early detection of lung cancer using tetrakis carboxy phenyl porphine (TCPP). IICAI 2009, 1503-1518.
Karchin R, Karplus K, Haussler D: Classifying G-protein coupled receptors with support vector machines. Bioinformatics 2002, 18(1):147-159. 10.1093/bioinformatics/18.1.147
Kerhet A, Small C, Quon H, Riauka T, Schrader L, Greiner R, Yee D, McEwan A, Roa W: Application of machine learning methodology for PET-based definition of lung cancer. Curr Oncol 2010, 17(1):41-47.
Khan J, Wei JS, Ringner M, Saal LH, Ladanyi M, Westermann F, Berthold F, Schwab M, Antonescu CR, Peterson C, Meltzer PS: Classification and diagnostic prediction of cancers using gene expression profiling and artificial neural networks. Nat Med 2001, 7(6):673-679. 10.1038/89044
Lee Y, Seo JB, Lee JG, Kim SS, Kim N, Kang SH: Performance testing of several classifiers for differentiating obstructive lung diseases based on texture analysis at high-resolution computerized tomography (HRCT). Comput Methods Programs Biomed 2009, 93(2):206-215. 10.1016/j.cmpb.2008.10.008
Li ZR, Lin HH, Han LY, Jiang L, Chen X, Chen YZ: PROFEAT: a web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res 2006, 34(Web Server issue):W32-W37.
Lim J, Kim N, Seo JB, Lee YK, Lee Y, Kang SH: Regional context-sensitive support vector machine classifier to improve automated identification of regional patterns of diffuse interstitial lung disease. J Digit Imaging 2011, 24(6):1133-1140. 10.1007/s10278-011-9367-0
Lin JS, Lo SB, Hasegawa A, Freedman MT, Mun SK: Reduction of false positives in lung nodule detection using a two-level neural classification. IEEE Trans Med Imaging 1996, 15(2):206-217. 10.1109/42.491422
Lipson RL, Baldes EJ, Olsen AM: Hematoporphyrin derivative: a new aid for endoscopic detection of malignant disease. J Thorac Cardiovasc Surg 1961, 42: 623-629.
Liu H, Kho AT, Kohane IS, Sun Y: Predicting survival within the lung cancer histopathological hierarchy using a multi-scale genomic model of development. PLoS Med 2006, 3(7):e232. 10.1371/journal.pmed.0030232
Liu Y: Active learning with support vector machine applied to gene expression data for cancer classification. J Chem Inf Comput Sci 2004, 44(6):1936-1941. 10.1021/ci049810a
Lo SL, Cai CZ, Chen YZ, Chung MC: Effect of training datasets on support vector machine prediction of protein-protein interactions. Proteomics 2005, 5(4):876-884. 10.1002/pmic.200401118
Marchevsky AM, Tsou JA, Laird-Offringa IA: Classification of individual lung cancer cell lines based on DNA methylation markers: use of linear discriminant analysis and artificial neural networks. J Mol Diagn 2004, 6(1):28-36. 10.1016/S1525-1578(10)60488-6
Mitchell T: Machine Learning. New York: McGraw Hill; 1997.
Motohiro A, Ueda H, Komatsu H, Yanai N, Mori T: Prognosis of non-surgically treated, clinical stage I lung cancer patients in Japan. Lung Cancer 2002, 36(1):65-69. 10.1016/S0169-5002(01)00459-7
Ganesan N, Venkatesh K, Rama MA, Malathi Palani A: Application of neural networks in diagnosing cancer disease using demographic data. IJCA 2010, 1:?.
Ganesan N, Venkatesh K, Rama MA, Malathi Palani A: Application of neural networks in diagnosing cancer disease using demographic data. IJCA 2010, Volume 1 – No. 26: 0975-8887.
Nomori H: Primary lung cancer. Kyobu Geka 2011, 64(8 Suppl):702-709.
Ong SA, Lin HH, Chen YZ, Li ZR, Cao Z: Efficacy of different protein descriptors in predicting protein functional families. BMC Bioinformatics 2007, 8: 300. 10.1186/1471-2105-8-300
Parkin DM: Global cancer statistics in the year 2000. Lancet Oncol 2001, 2(9):533-543. 10.1016/S1470-2045(01)00486-7
Parsaei H, Stashuk DW: SVM-based validation of motor unit potential trains extracted by EMG signal decomposition. IEEE Trans Biomed Eng 2012, 59(1):183-191.
Penedo MG, Carreira MJ, Mosquera A, Cabello D: Computer-aided diagnosis: a neural-network-based approach to lung nodule detection. IEEE Trans Med Imaging 1998, 17(6):872-880. 10.1109/42.746620
Petersen I, Petersen S: Towards a genetic-based classification of human lung cancer. Anal Cell Pathol 2001, 22(3):111-121.
Peterson C, Ringner M: Analysis tumor gene expression profiles. Artif Intell Med 2002, 28(1):59-74.
Rao HB, Zhu F, Yang GB, Li ZR, Chen YZ: Update of PROFEAT: a web server for computing structural and physicochemical features of proteins and peptides from amino acid sequence. Nucleic Acids Res 2011, 39(Web Server issue):W385-W390.
Rosenblatt FX: Principles of Neurodynamics: Perceptrons and the Theory of Brain Mechanisms. Washington DC: Spartan Books; 1961.
Sakas DE, Panourias IG, Simpson BA: An introduction to neural networks surgery, a field of neuromodulation which is based on advances in neural networks science and digitised brain imaging. Acta Neurochir Suppl 2007, 97(Pt 2):3-13.
Santos-Garcia G, Varela G, Novoa N, Jimenez MF: Prediction of postoperative morbidity after lung resection using an artificial neural network ensemble. Artif Intell Med 2004, 30(1):61-69. 10.1016/S0933-3657(03)00059-9
Schaefer-Prokop C, Prokop M: New imaging techniques in the treatment guidelines for lung cancer. Eur Respir J Suppl 2002, 35: 71s-83s.
Schneider G, Wrede P: The rational design of amino acid sequences by artificial neural networks and simulated molecular evolution: de novo design of an idealized leader peptidase cleavage site. Biophys J 1994, 66(2 Pt 1):335-344.
Shyu F-M, Liao H-Y: A hyper SVM model for multiple classifications. Natural Computation (ICNC). 2011 Seventh International Conference on, IEEE 2011.
Stoean R, Stoean C, Lupsor M, Stefanescu H, Badea R: Evolutionary-driven support vector machines for determining the degree of liver fibrosis in chronic hepatitis C. Artif Intell Med 2011, 51(1):53-65. 10.1016/j.artmed.2010.06.002
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP: Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005, 102(43):15545-15550. 10.1073/pnas.0506580102
Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander ES, Golub TR: Interpreting patterns of gene expression with self-organizing maps: methods and application to hematopoietic differentiation. Proc Natl Acad Sci U S A 1999, 96(6):2907-2912. 10.1073/pnas.96.6.2907
Travis WD, Travis LB, Devesa SS: Lung cancer. Cancer 1995, 75(1 Suppl):191-202.
Van Belle V, Pelckmans K, Van Huffel S, Suykens JA: Support vector methods for survival analysis: a comparison between ranking and regression approaches. Artif Intell Med 2011, 53(2):107-118. 10.1016/j.artmed.2011.06.006
van Ruth S, Baas P, Zoetmulder FA: Surgical treatment of malignant pleural mesothelioma: a review. Chest 2003, 123(2):551-561. 10.1378/chest.123.2.551
Ward JJ, McGuffin LJ, Buxton BF, Jones DT: Secondary structure prediction with support vector machines. Bioinformatics 2003, 19(13):1650-1655. 10.1093/bioinformatics/btg223
Webb-Robertson BJ, Cannon WR, Oehmen CS, Shah AR, Gurumoorthi V, Lipton MS, Waters KM: A support vector machine model for the prediction of proteotypic peptides for accurate mass and time proteomics. Bioinformatics 2010, 26(13):1677-1683. 10.1093/bioinformatics/btq251
West L, Vidwans SJ, Campbell NP, Shrager J, Simon GR, Bueno R, Dennis PA, Otterson GA, Salgia R: A novel classification of lung cancer into molecular subtypes. PLoS One 2012, 7(2):e31906. 10.1371/journal.pone.0031906
West M: Bayesian factor regression models in the “large p, small n” paradigm. Bayesian statistics 2003, 7(2003):723-732.
Yamagata N, Shyr Y, Yanagisawa K, Edgerton M, Dang TP, Gonzalez A, Nadaf S, Larsen P, Roberts JR, Nesbitt JC, Jensen R, Levy S, Moore JH, Minna JD, Carbone DP: A training–testing approach to the molecular classification of resected non-small cell lung cancer. Clin Cancer Res 2003, 9(13):4695-4704.
Zhou ZH, Jiang Y, Yang YB, Chen SF: Lung cancer cell identification based on artificial neural network ensembles. Artif Intell Med 2002, 24(1):25-36. 10.1016/S0933-3657(01)00094-X
Zycinski G, Barla A, Verri A: SVS: data and knowledge integration in computational biology. Conf Proc IEEE Eng Med Biol Soc 2011, 2011: 6474-6478.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no conflict of interest.
Authors’ contributions
Designed the experiments and proposed the ideas: ME, FH. Data prepared: FH Performed the experiments: ME, FH. Analyzed the data: ME, FH. Contributed reagents/materials/analysis tools: BG. Wrote the paper: ME, FH AHK. Designed the experiments and managed the research, designed the data mining models, supervised FH and AHK to run the models and edited the manuscript: ME. Carried out the machine learning models: AHK, drafted the first manuscript: FH. Helped FH in running the models, preparing the figures and tables: AHK. Co-supervised FH in running the models, extraction of protein attributes: BG. All authors read and approved the final manuscript.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hosseinzadeh, F., KayvanJoo, A.H., Ebrahimi, M. et al. Prediction of lung tumor types based on protein attributes by machine learning algorithms. SpringerPlus 2, 238 (2013). https://doi.org/10.1186/2193-1801-2-238
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2193-1801-2-238