
Abstract
In the pediatric population, B-acute lymphoblastic leukemia (B-ALL) is the most prevalent childhood hematological malignancy, as well as the leading cause of childhood cancer-related mortality. Advances in cytogenetics utilizing array-based technologies and next-generation sequencing (NGS) techniques have revealed exciting insights into the genetic basis of this disease, with the hopes of developing individualized treatment plans for affected children. In this comprehensive review, we discuss our current understanding of childhood (pediatric) B-ALL and highlight the most recent genetic advances and their therapeutic implications.
Similar content being viewed by others


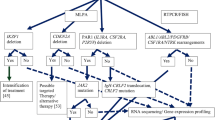
Introduction
ALL is a malignant clonal proliferation of lymphoid progenitor cells, most commonly of the B-cell lineage (B-ALL). In the pediatric population, ALL accounts for 81% of childhood leukemias; leukemia overall accounts for one third of cancers diagnosed in children between ages 0–14 years [1]. In the United States, the majority of ALL cases occur in ages 1–4, with an incidence rate in this group of 8 per 100,000, and preponderance for males over females [1]. The long-term rates of event-free survival (EFS) for childhood B-ALL approach 90%, although infants are associated with poorer prognosis and lower EFS rates [2–4].
This review discusses our current understanding of childhood B-ALL and highlights recent genetic advances and their therapeutic implications. Genetic classification of B-ALL is paramount for risk stratification and in treatment evaluation, especially within the context of clinical trial enrollment. At the forefront of pediatric oncologic research is the Children’s Oncology Group (COG) in North America, as well as the International Berlin-Frankfurt-Münster (BFM) Study Group in Europe, whose work has played a significant role in disease-specific research and therapeutic developments. Treatment protocols, including clinical trials, are the mainstay treatment for children with hematological and solid tumor malignancies. Cytogenetics advances of B-ALL have led to the discovery of numerous additional genetic changes, including mutations involving key cellular pathways in lymphoid development, tumor suppression, and cell cycle regulation. Uncovering the prognostic significance of these genetic aberrations is fundamental for risk stratification and ultimately individualized treatment.
Clinical presentation
ALL is a disease of the bone marrow. Clinical suspicion for ALL arises with signs and symptoms reflective of bone marrow failure (pancytopenia) and/or extramedullary disease. The most common symptoms include fatigue, pallor, bone pain, arthritis, limping, easy bruising/bleeding, and/or petechiae. Physical examination may show lymphadenopathy and hepatosplenomegaly [5]. Extramedullary involvement may be seen in the central nervous system (CNS) with signs of increased intracranial pressure including headache [6]. In boys, testicular involvement may present as a testicular mass. Adverse prognostic factors include high leukocyte count on presentation, age <1 year or >10 years, and adverse cytogenetics [7, 8].
Histology
Bone marrow aspirate studies show a preponderance of lymphoblasts with high nuclear to cytoplasmic (N:C) ratio, finely dispersed nuclear chromatin, and prominent nucleoli. On occasion, vacuolated cytoplasm and cytoplasmic pseudopods can be seen. Bone marrow core biopsies demonstrate diffuse replacement of normal marrow elements by uniform sheets of round to oval lymphoblasts with indented to convoluted nuclei [9] (Figure 1). No lower limit of blast percentage is required to establish the diagnosis, but most treatment protocols define ALL by the presence of greater than or equal to 25% blasts. By immunophenotype, B-lymphoblasts demonstrate universal positivity for B-cell markers including CD19, cytoplasmic CD79a, and cytoplasmic CD22; as well as positivity for surface CD22, CD24, PAX5 and TdT; with variable expression of CD10, CD20 and CD34 [9]. CD10 is often negative in infant ALL (age <1 year) with MLL (11q23) gene rearrangements [10].

Evaluation of a boy with abdominal pain, night sweats, increased fatigue, petechiae, and a white blood cell count of 113 × 103 /μL. A. Bone marrow core biopsy (100×) showed diffuse replacement of normal marrow elements by uniform sheets of round to oval lymphoblasts with indented to convoluted nuclei. B. Touch preparation of core biopsy material showed lymphoblasts with high nuclear to cytoplasmic (N:C) ratio, finely dispersed nuclear chromatin, and prominent nucleoli. C-D. Peripheral blood smear (100×) showed lymphoblasts with high N:C ratio and cytoplasmic pseudopods.
Recurrent genetic abnormalities
Approximately 75% of childhood ALL cases harbor recurrent genetic abnormalities, including aneuploidy or structural chromosomal arrangements, detected by conventional karyotyping and fluorescence in situ hybridization (FISH) [11]. Translocations t(9;22)(q34;q11) [BCR-ABL1], t(12;21)(p13;q22) [ETV6-RUNX1 (TEL-AML1)], hyperdiploidy, and translocation t(4;11)(q21;q23) [MLL-AFF1(AF4)] in infants, are found at the highest frequency in childhood B-ALL [11]. Other recurrent cytogenetic abnormalities include hypodiploidy and translocation t(1;19)(q23;p13) [TCF3-PBX1 (E2A-PBX1)] [11]. Advances in cytogenetics utilizing array-based technologies and NGS have uncovered additional submicroscopic DNA alterations affecting genes involved in normal hematopoiesis, tumor suppression, apoptosis, and cell cycle regulation, including IKZF1, CRLF2, PAX5, and FLT3 (Table 1). Advanced techniques have revealed new insights into well-known recurrent abnormalities, and have more importantly elucidated new gene targets involved in aberrant hematopoiesis and relapse. Overall, the utilization of these newly identified genetic alterations has clinical utility for diagnosis, risk stratification, and targeted therapy.
Aneuploidy
High hyperdiploidy
High hyperdiploidy (51–65 chromosomes) is one of the most common cytogenetic abnormalities observed in childhood B-ALL (Figure 2). It is seen in 25-30% of total childhood B-ALL cases, with the highest frequency in the 1 to 4 year age range [12, 13]. High hyperdiploidy is characterized by a nonrandom gain of chromosomes, including + X, +4, +6, +10, +14, +17, +18, and +21 [13, 14]. The diagnosis confers a good prognosis in childhood B-ALL, with EFS rates of approximately 80% and overall survival (OS) rates of 90% (reviewed in [13]). Despite favorable outcomes, 20% of children relapse and 10% eventually succumb to the disease [15]. SNP array analysis of high hyperdiploid B-ALL has shown that almost 80% of cases display further genetic abnormalities in addition to characteristic chromosomal gains; although these additional aberrations have no definitive clinical ramification [14].

Evaluation of a 3 year-old boy with hyperdiploid B-ALL. A. Abnormal male hyperdiploid karyotype with extra copies of chromosomes X, 2, 9, 14. B. FISH analysis detected +9q, +14q and +21q in 96% , 93.7% and 96% of the nuclei examined, respectively. In addition, 12p deletion was observed in 89% of the nuclei examined, suggestive of an underlying complex aneuploid (most likely hyperdiploid) karyotype.
Recently, rare alleles of PRDM9 (which encodes a meiosis-specific histone H3 methyltransferase that controls activation of recombination hotspots) have been reported to be associated with the development of high hyperdiploid and infant B-ALL [16, 17]. Furthermore, it was even postulated that PRDM9 activity during the early stages of meiosis in the parental germline could lead to genomic instability and development of childhood B-ALL [16].
Hypodiploidy
Hypodiploidy is characterized by fewer than 46 chromosomes and is seen in 5-8% of total B-ALL cases [18, 19]. The current high risk COG protocol AALL1131 denotes hypodiploidy as less than 44 chromosomes. The majority of hypodiploid B-ALL contain 45 chromosomes. The remainder of hypodiploidly cases are much rarer and include high-hypodiploid (40–44 chromosomes), low-hypodiploid (33–39 chromosomes), and near-haploid (24–29 chromosomes) groups [19, 20]. In general, hypodiploidy with less than 40 chromosomes confers a poor prognosis. The 3-year EFS for near-haploid and low-hypodiploid B-ALL is 30% [19, 20]. Hypodiploid cases have also been shown to undergo reduplication, resulting in a hyperdiploid karyotype (so called “masked hypodiploid ALL”), which may preclude the correct genetic classification and therefore render an inappropriate treatment regimen [20, 21].
A recent genomic profiling study of hypodiploid ALL cases identified multiple recurrent genetic alterations, distinguishing near haploid from low-hypodiploid ALL [22]. Near-haploid ALL cases showed alterations targeting genes in receptor tyrosine kinase (RTK) signaling and Ras signaling (NF1) pathways, as well as high frequency alterations in the IKAROS gene family, particularly IKZF3 (Aiolos) which encodes for the zinc finger transcription factor AIOLOS [22]. Low-hypodiploid cases showed genetic alterations of TP53, RB1, and IKZF2 (Helios) [22]. In the TP53 mutated cases, non-tumor cells also harbored TP53 mutations, suggesting an inherited basis of disease and a possible manifestation of Li-Fraumeni syndrome (LFS) [23]. Both low-hypodiploid and near-haploid ALL showed activation of Ras-signaling and PI3K (phosphoinositide 3-kinase)-signaling pathways that were sensitive to PI3K inhibitors such as rapamycin in vitro, suggesting that PI3K inhibitors could be explored as a therapeutic treatment option [22].
Recurrent translocations
ETV6-RUNX1 (TEL-AML1)
The most common chromosomal rearrangement in B-ALL is t(12;21)(p13;q22), encoding for ETV6-RUNX1 (TEL-AML1) [11] (Figures 3, 4 and 5). It occurs in 25% of children with B-ALL and confers an excellent prognosis [24, 25]. Both ETV6 and RUNX1 transcription factors are required for normal hematopoiesis [26, 27]. The ETV6-RUNX1 fusion protein is thought to disrupt the normal expression of RUNX1-regulated genes by converting RUNX1 to a transcriptional repressor [28].

Evaluation of a 2 year-old girl presenting with fevers. A. Bone marrow core biopsy (100×) showing sheets of round to oval lymphoblasts. B. Bone marrow aspirate (100×) showing lymphoblasts with cytoplasmic vacuoles. C. Representative flow cytometry histogram. The CD45(dim) gated population contained excess B-lymphoblasts (81% of total), positive for CD10, CD19, CD34, CD38, CD79a, HLA-DR, and TdT. D. Abnormal female karyotype with unbalanced rearrangements of 1p, a derivative chromosome 12 (due to an unbalanced translocation between chromosomes 1p and 12p), and a derivative chromosome 21 (due to an unbalanced translocation between chromosomes 12 and 21), resulting in ETV6-RUNX1 fusion. E-F. Abnormal FISH signal pattern consistent with ETV6-RUNX1 (TEL-AML1) fusion, indicative of t(12;21) translocation.

Evaluation of a 3 year-old girl with pancytopenia. A. Bone marrow core biopsy (40×) showing sheets of lymphoblasts. B-C. Representative flow cytometry histograms. The CD45(dim) gated population contained excess and abnormal B-lymphoblasts (85% of the total), positive for CD10, CD13, CD19, CD22, CD34, CD38, HLA-DR, plus intracellular CD79a, intracellular CD22, and TdT. D. Abnormal composite female karyotype with monosomy 16, trisomy 21, and deletions of 6q and 9q. E. FISH analysis detected the ETV6-RUNX1 (TEL-AML1) fusion, indicative of t(12;21) translocation. In addition, 4.4% of these abnormal cells showed an extra copy of the RUNX1 locus, suggestive of an underlying +21q.

Evaluation of a 7 year-old boy with B-ALL. A. Abnormal male karyotype with a deletion of 11q and trisomy 21. B. FISH analysis demonstrated an abnormal signal pattern consistent with ETV6-RUNX1 (TEL-AML1) fusion, indicative of t(12;21) translocation, as well as +21q and 11q- (MLL deletion).
TCF3-PBX1 (E2A-PBX1)
The t(1;19)(q23;p13) rearrangement and its unbalanced variant der(19)t(1;19)(q23;p13) are commonly seen in B-ALL [11] (Figure 6). The resultant TCF3-PBX1 (E2A-PBX1) fusion protein is comprised of the transactivation domains of TCF3 and a DNA binding domain of the homeobox protein PBX1, converting PBX1 into a transactivating factor and reducing expression of the TCF3 encoded transcription factors E12 and E47, required for early lymphoid development [29, 30]. The translocation occurs in 6% of childhood B-ALL and is historically associated with poorer outcomes [31]. However, advances in treatment have improved clinical outcomes of children with this abnormality and the translocation is now thought to confer an intermediate prognosis [32].

Evaluation of an 18 year-old female with B-ALL. A. Variably cellular marrow (100×) with clusters of B-lymphoblasts and reduced multilineage hematopoiesis. B. Representative flow cytometry histogram. The CD45(dim) gated population comprised approximately 4% of total cells and contained no excess blasts. C. Abnormal female karyotype demonstrating t(17;19) translocation. D. FISH analysis detected an abnormal signal pattern compatible with TCF3 (19p13) rearrangement.
BCR-ABL1 (Philadelphia chromosome)
B-ALL harboring the t(9;22)(q34;q11) translocation or ‘Philadelphia chromosome’ (Ph + B-ALL), encodes the fusion gene BCR-ABL1. This translocation is present in 3-5% of childhood B-ALL cases [33] and is associated with older age, higher leukocyte count, and more frequent CNS involvement at time of diagnosis [34]. The translocation fuses the 5’ sequence of the breakpoint cluster region (BCR) on chromosome 22 to the 3’ sequence of the ABL1 gene on chromosome 9. The resultant oncoprotein is a constitutively active nonreceptor tyrosine kinase, responsible for leukemogenesis. The BCR region contains two breakpoint areas, including a major BCR (M-bcr) area commonly seen in chronic myelogenous leukemia (CML), and a minor BCR (m-bcr) area seen in pediatric B-ALL. In 90% of childhood B-ALL cases, fusion genes created by breaks in m-bcr encode for a 190 kDa fusion protein (p190) [35].
The use of ABL1 tyrosine kinase inhibitors (TKIs), such as imatinib, has been revolutionary in the treatment of Ph + B-ALL. Once associated with dismal outcomes, use of TKIs combined with intensive chemotherapy has improved 3-year EFS in children and adolescents with Ph + B-ALL with no appreciable increase in toxicity [36]. The COG AALL0622 clinical trial, substituting the second generation TKI, dasatinib, for imatinib, is under investigation [37].
MLL gene rearrangements
MLL (mixed-lineage-leukemia) gene rearrangements at 11q23 are present in 80% of all infant B-ALL cases and 10% of all childhood B-ALL [38, 39]. The MLL gene encodes for a protein with histone methyltransferase activity, which is essential for hematopoietic regulation of HOXA and MEIS1 gene expression [40]. The most common gene rearrangements include t(4;11)(q21;q23) encoding MLL-AFF1(AF4), t(9;11)(p22;q23) encoding MLL-MLLT3(AF9), t(11;19)(q23;p13.3) encoding MLL-ENL, and t(10;11)(p13-14;q14-21) encoding MLL-MLLT10(AF10) [41, 42]. About 50% of MLL rearrangements show the t(4;11)(q21;q23) translocation. In general, MLL rearrangements are associated with adverse outcomes, with an EFS of approximately 35% [43], largely due to cellular drug resistance [44, 45].
IGH@ translocations
Rearrangements of the immunoglobulin heavy chain locus (IGH@) on chromosome 14q32 are rare in B-ALL, occurring in <5% of cases [46]. IGH@ rearrangements occur more frequently in adolescents and appear to have poor clinical outcomes. The most common IGH@ partners include CRLF2 (cytokine receptor-like factor 2) at the pseudoautosomal region 1 (PAR1) of Xp22.3/Yp11.3 (resulting in overexpression of CRLF2) [47], ID4 (inhibitor of DNA binding 4) at 6p22 [48], and members of the CEBP (CCAAT/enhancer binding protein) family [49, 50]. Translocations between IGH@ and EPOR (erythropoietin receptor) at 19p13 have also been reported [51–53], with other remaining translocations appearing sporadic [46].
Intrachromosomal amplification of chromosome 21 (iAMP21)
iAMP21 is defined as the presence of three or more copies of the RUNX1 gene within a morphologically abnormal chromosome 21 [54, 55]. Amplified regions on chromosome 21 are found within a 5.1 Mb region containing RUNX1, miR-802, and genes mapping to the Down syndrome critical region. iAMP21 occurs in approximately 2% of older children with B-ALL, and is associated with poorer outcomes when treated with standard therapy, and also increased risk for early and late relapse [56]. The five-year EFS is approximately 29%, with an OS of 71% [57]. Recent studies have shown that treatment of iAMP21 patients as high-risk provides a significant improvement in outcome [58]. One recurrent abnormality in iAMP21, seen in 35% of children with iAMP21, includes the P2RY8-CRLF2 fusion (created by a focal deletion at the PAR1 region at Xp22.3/Yp11.3) which results in the overexpression of CRLF2 [47]. Gain of the X chromosome, and abnormalities affecting the genes IKZF1 (Ikaros), CDKN2A, PAX5, ETV6, and RB1 have also been associated with iAMP21 [56]. Lastly, individuals born with the constitutional Robertsonian translocation rob(15;21)(q10;q10)c have a 2700-fold increased risk of developing B-ALL with iAMP21 [59].
Submicroscopic genetic alterations
The role of IKZF1 in BCR-ABL1 and BCR-ABL1-like ALL
“BCR-ABL1-like” B-ALL, seen in 15% of childhood B-ALL, has recently been defined by gene expression profiling (GEP). BCR-ABL1-like B-ALL shows a similar GEP to Ph + B-ALL, but in the absence of the BCR-ABL1 rearrangement [60, 61]. These cases are associated with poor outcomes and increased relapse risk [62–64]. Deletions and sequence mutations in IKZF1 (Ikaros) at 7p13, which encodes for the lymphoid transcription factor IKAROS, are associated with approximately 70% and 40% of BCR-ABL-positive and BCR-ABL1-like B-ALL, respectively [60, 63]. Despite improvements in outcomes for children with Ph + B-ALL that has resulted from combination TKI and chemotherapy, recent studies have demonstrated that Ph + ALL harboring IKZF1 deletions are associated with unfavorable outcomes; irrespective of imatinib treatment [65]. In addition to frequent IKZF1 (Ikaros) abnormalities, up to 50% of BCR-ABL1-like ALL harbor rearrangements in CRLF2, with concurrent Janus kinase family (JAK) mutations [66, 67]. BCR-ABL1-like ALL has also shown resistance to L-asparaginase, and to a lesser extent, daunorubicin; although intensified therapy could give more suitable treatment options [60].
Additional BCR-ABL1-like ALL studies have shed light on the heterogenous pathogenesis of ALL. In 2013, a genome-wide association study (GWAS) of BCR-ABL1-like ALL identified a susceptibility locus for BCR-ABL1-like ALL (GATA3, rs3824662) [68]. This locus was shown to be associated with CRLF2 rearrangements, JAK mutations, and deletions in IKZF1 (Ikaros) [68].
Transcriptome and whole-genome sequencing of BCR-ABL1–like ALL has also identified other genetic alterations involved in the activation of kinase signaling, including EBF1-PDGFRB, comprised of the transcription factor EBF1 (early B-cell factor 1) and the receptor tyrosine kinase PDGFRB (platelet-derived growth factor receptor β), resulting from 5q33q33 microdeletion [53, 69]. Several reports suggest that the use of TKIs to treat B-ALL harboring the EBF1-PDGFRB rearrangement may be of clinical benefit [70, 71].
JAK mutations and CRLF2 rearrangements
The role of cytokine receptors and JAK family members are playing increasingly larger roles in B-ALL studies. The JAK family encodes four nonreceptor tyrosine kinases (JAK1, JAK2, JAK3, TYK2) involved in cytokine-mediated signaling (JAK-STAT pathway) [72]. Mutations occur in about 10% of high-risk childhood B-ALL cases [73]. In the setting of BCR-ABL1-like B-ALL, JAK mutations are also associated with concomitant IKZF1 (Ikaros) and CDKN2A/B alterations, and correlate with worse outcomes [74, 75]. JAK2 mutations are also associated with CRLF2 rearrangements (as described above), and are described in 60% of Down syndrome (Trisomy 21)-associated ALL [76, 77]. Approximately 40% of CRLF2- rearranged cases can harbor JAK2 mutations [78].
Abnormalities involving JAK2 (9p24) most often arise via point mutations involving the pseudokinase domain R683 [79, 80]; however, rare cases of JAK2 rearrangements have also been identified [79, 81, 82]. In JAK2 rearrangements, dimerization or oligomerization of JAK2 is induced without ligand binding, resulting in constitutive activation of downstream pathways in leukemic cells. Alterations in CRLF2, (Xp22.3/Yp11.3), occur in up to 8% of unscreened childhood B-ALL cases and up to 15% of high-risk B-ALL patients [83–86]. CRLF2 rearrangements result in constitutive activation of the STAT5 pathway, resulting in leukemogenesis. Additionally, abnormal PI3K/mTOR pathway signaling has also been implicated [87].
The JAK2 inhibitor, ruxolitinib, has been shown to reduce tumor burden in xenograft mouse models harboring BCR-JAK2 [t(9;22)(p24;q11.2)] [88], and has demonstrated promising results in the treatment of CRLF2-rearranged, JAK2-mutated leukemic cells in vitro [87]. Additionally, the PI3K inhibitor, rapamycin, has been shown to control leukemic burden [88]. Clinical trial NCT01251965, utilizing ruxolitinib in refractory or relapsed ALL or AML (acute myelogenous leukemia), is currently ongoing.
More recently, another potential molecular target in JAK2-mutated B-ALL was revealed. In a mouse model, overexpression of mutant JAK2 led to downstream upregulation of prosurvival Bcl-2 gene family members, and combined use of the Bcl-2/Bcl-xL inhibitor ABT-737 with JAK2 inhibitors prolonged disease regression time [89].
PAX5 deletions and rearrangements
PAX5 (9p13), a member of the paired box gene family, is a transcription factor necessary for normal hematopoietic development [90]. In childhood B-ALL, mutations in PAX 5 have been detected in 32% of cases by genome-wide analysis [75]. PAX5 rearrangements occur with incidence of about 2.5%, with numerous reported rearrangements including ETV6 (12p13) and JAK2 (9p24) [91]. Recently, a heterozygous germline PAX5 variant, c.547G > A, encoding p.Gly183Ser, was identified in two unrelated families with autosomal dominant B-ALL, suggesting that PAX5 mutations may play a role in the inherited susceptibility of B-ALL [92].
FLT3 mutations
FLT3 (fms-tyrosine kinase 3) on chromosome 13q12 is frequently mutated in MLL-rearranged and high hyperdiploid B-ALL [93]. Infants with MLL- rearrangements have been shown to be sensitive to the FLT3 inhibitor, PKC412 (midostaurin), suggesting that multitarget kinase inhibition may present as novel therapeutic modalities [94]. COG study AALL0631, utilizing the FLT3 inhibitor lestaurtinib (CEP701) along with standard chemotherapy, is currently ongoing in infants with MLL rearrangements.
The role of epigenetics
The role of epigenetic regulation in B-ALL has gained considerable attention. In 2013, the first integrated genome-wide analysis in childhood ALL, incorporating cytosine methylation profiling, DNA copy number alterations (CNA) and GEP, was reported [95]. Recurrent epigenetic alterations were identified across all B-ALL subtypes studied, suggesting that certain epigenetic events are required for leukemic transformation [95]. Moreover, genes frequently affected by structural abnormalities were shown to be targets for aberrant DNA methylation [95]. Additionally, global histone modification profiling revealed a distinct molecular chromatin signature in several ALL cell lines, subsequently noted to harbor abnormalities in NSD2, encoding for a methyltransferase [96]. Targeted investigation of patient samples revealed approximately 7.5% of childhood B-ALL harbored mutations in WHSC1/NSD2 (particularly p.E1099K) but were enriched the B-ALL subtypes ETV-RUNX1 (20%) and TCF3-PBX1 (15%) [96]. More recently, NGS targeted exome profiling identified a number of epigenetic regulators, including CREBBP and SETD2, mutated in 25% of B-ALL samples at time of diagnosis [97]. Further analysis revealed that mutations in SETD2 were enriched in MLL-rearranged (22%) and the ETV-RUNX1 (13%) subtypes of B-ALL, and that over 50% of matched relapsed cases, regardless of subtype, demonstrated enrichment of mutations in epigenetic regulators (discussed more below) [97].
Epigenetic studies have also shown insights into our understanding of MLL- rearranged B-ALL. MicroRNAs (miRNAs), short noncoding RNAs involved in the regulation of signaling pathways of cell differentiation, proliferation, and apoptosis, have been shown to promote leukemogenesis through aberrant epigenetic activity [98]. The presence of these epigenetic aberrancies suggests that histone deacetylase (HDAC), DNA methyltransferase (DNMT), and/or histone methyltransferase (HMT) inhibitors may play a therapeutic role in MLL-rearranged B-ALL [99, 100]. Notably, selective DOT1L HMT inhibitors, such as EPZ-004777, have been shown to selectively destroy MLL-rearranged cells in mouse models [101, 102]. Clinical trial NCT01684150 is currently evaluating the use of the DOT1L HMT inhibitor, EPZ-5676, in adults with MLL-rearrangements.
Relapsed ALL
Despite overall progress in treatment, relapsed B-ALL has a dismal prognosis with an overall survival of 30% [103]. Several genetic subtypes and aberrations are associated with high treatment failure risk, including CDKN2A/B, ETV6, and IKZF1 (Ikaros) mutations. Relapse occurs across the spectrum of all B-ALL subtypes, with some cases demonstrating acquisition of additional chromosomal abnormalities over time. In 2008, genome-wide analysis of matched samples from diagnosis and relapse identified different patterns of genomic CNA for samples at diagnosis and relapse, with that acquired abnormalities preferentially affected cell cycle regulation and B-cell development genes [104]. They determined that only 8% of relapse clones carried identical CNA to the diagnostic clone, while almost 90% of relapse clones evolved to acquire additional or some but not all CNA from the diagnostic clone [104], highlighting that relapse in ALL is variable and complex, but often a descendent of an ancestral clone to the principal de novo leukemia clone (reviewed in [105]).
Gene sequencing studies in 2011 [106] and 2012 [107], demonstrated 20% of total pediatric ALL relapse cases [106] and 60% of high-hyperdiploid relapse cases [107] harbor mutations in CREBBP, a transcriptional co-activator and histone acetyltransferase (HAT). Mutations in CREBBP have been shown to affect the regulation of glucocorticoid responsive genes. Because glucocorticoids are the cornerstone of B-ALL therapy, CREBBP mutations are thought to influence response to treatment and influence the likelihood for relapse [108]. In this regard, a recent study identified a selective enrichment of mutations in CREBBP and other epigenetic regulators (SETD2, KDM6A, MLL2, and MSH6) in relapsed and/or chemoresistant childhood B-ALL, which could suggest that epigenetic regulation plays a central role in clonal survival and ultimately chemotherapy resistance and relapse [97]. In addition to mutations in CREBBP and SETD2, relapse-specific mutations in the NT5C2 gene have been described in childhood B-ALL [109, 110]. NT5C2 is a 5’ nucleotidase responsible for the inactivation of nucleosidase-analog drugs, therefore conferring resistance to conventional therapeutic agents such as 6-mercaptopurine (6-MP) [109].
Although the true prognostic importance of these mutations at both diagnosis and relapse is unknown, therapies aimed at modulating epigenetic regulators in B-ALL is already in clinical development, as mentioned previously. Interstingly, CREBBP has also been shown to play a role in Wnt/β-catenin signaling, a pathway critical for the self-renewal of normal hematopoietic progenitor cells. In this regard, ICG-001, a novel small-molecule modulator of Wnt/β-catenin signaling that binds to CREBBP, leads to the differentiation of pre-B ALL cells and loss of self-renewal capacity, thereby sensitizing cells to chemotherapeutic treatment [111].
Conclusions
Advances in genetic technologies have enriched our current understanding of childhood B-ALL. Although conventional karyotyping and FISH technologies play a significant role in detecting numerous recurrent abnormalities, microarray-based techniques and NGS have revealed a multitude of new molecular targets that may prove useful in the diagnosis, risk stratification, and most importantly individualized treatment of this disease.
Consent
Written informed consent was obtained from the patient’s guardian/parent/next of kin for the publication of this report and any accompanying images.
References
Howlader N, Noone AM, Krapcho M, Garshell J, Miller D, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA: SEER Cancer Statistics Review, 1975–2011. Bethesda, MD: National Cancer Institute; 1975. , based on November 2013 SEER data submission, posted to the SEER web site, April 2014 http://seer.cancer.gov/csr/1975_2011/
Gatta G, Rossi S, Foschi R, Trama A, Marcos-Gragera R, Pastore G, Peris-Bonet R, Stiller C, Capocaccia R, Group EW: Survival and cure trends for European children, adolescents and young adults diagnosed with acute lymphoblastic leukemia from 1982 to 2002. Haematologica 2013, 98: 744–752. 10.3324/haematol.2012.071597
Hunger SP, Lu X, Devidas M, Camitta BM, Gaynon PS, Winick NJ, Reaman GH, Carroll WL: Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. J Clin Oncol 2012, 30: 1663–1669. 10.1200/JCO.2011.37.8018
Kotecha RS, Gottardo NG, Kees UR, Cole CH: The evolution of clinical trials for infant acute lymphoblastic leukemia. Blood Cancer J 2014, 4: e200. 10.1038/bcj.2014.17
Margolin JF, Steuber CP, Poplack DG: Acute Lymphoblastic Leukemia. In Principles and Practice of Pediatric Oncology. 5th edition. Edited by: Pizzo PA, Poplack DG. Philadelphia, PA: Lippincott Williams and Wilkins; 2006:538–590.
Bleyer WA: Central nervous system leukemia. Pediatr Clin North Am 1988, 35: 789–814.
Inaba H, Greaves M, Mullighan CG: Acute lymphoblastic leukaemia. Lancet 2013, 381: 1943–1955. 10.1016/S0140-6736(12)62187-4
Pui CH, Robison LL, Look AT: Acute lymphoblastic leukaemia. Lancet 2008, 371: 1030–1043. 10.1016/S0140-6736(08)60457-2
WHO: classification of tumours of haematopoietic and lymphoid tissues. 4th edition. Lyon, Franc: International Agency for Research on Cancer; 2008.
Jansen MW, Corral L, van der Velden VH, Panzer-Grumayer R, Schrappe M, Schrauder A, Marschalek R, Meyer C, den Boer ML, Hop WJ, Valsecchi MG, Basso G, Biondi A, Pieters R, van Dongen JJ: Immunobiological diversity in infant acute lymphoblastic leukemia is related to the occurrence and type of MLL gene rearrangement. Leukemia 2007, 21: 633–641.
Mullighan CG: The molecular genetic makeup of acute lymphoblastic leukemia. Hematol Am Soc Hematol Educ Program 2012, 2012: 389–396.
Moorman AV: The clinical relevance of chromosomal and genomic abnormalities in B-cell precursor acute lymphoblastic leukaemia. Blood Rev 2012, 26: 123–135. 10.1016/j.blre.2012.01.001
Paulsson K, Johansson B: High hyperdiploid childhood acute lymphoblastic leukemia. Genes ChromosomCancer 2009, 48: 637–660. 10.1002/gcc.20671
Paulsson K, Forestier E, Lilljebjorn H, Heldrup J, Behrendtz M, Young BD, Johansson B: Genetic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 2010, 107: 21719–21724. 10.1073/pnas.1006981107
Moorman AV, Richards SM, Martineau M, Cheung KL, Robinson HM, Jalali GR, Broadfield ZJ, Harris RL, Taylor KE, Gibson BE, Hann IM, Hill FG, Kinsey SE, Eden TO, Mitchell CD, Harrison CJ: Outcome heterogeneity in childhood high-hyperdiploid acute lymphoblastic leukemia. Blood 2003, 102: 2756–2762. 10.1182/blood-2003-04-1128
Hussin J, Sinnett D, Casals F, Idaghdour Y, Bruat V, Saillour V, Healy J, Grenier JC, de Malliard T, Busche S, Spinella JF, Lariviere M, Gibson G, Andersson A, Holmfeldt L, Ma J, Wei L, Zhang J, Andelfinger G, Downing JR, Mullighan CG, Awadalla P: Rare allelic forms of PRDM9 associated with childhood leukemogenesis. Genome Res 2013, 23: 419–430. 10.1101/gr.144188.112
Woodward EL, Olsson ML, Johansson B, Paulsson K: Allelic variants of PRDM9 associated with high hyperdiploid childhood acute lymphoblastic leukaemia. Br J Haematol 2014. doi:10.1111/bjh.12914. [Epub ahead of print]
Heerema NA, Nachman JB, Sather HN, Sensel MG, Lee MK, Hutchinson R, Lange BJ, Steinherz PG, Bostrom B, Gaynon PS, Uckun F: Hypodiploidy with less than 45 chromosomes confers adverse risk in childhood acute lymphoblastic leukemia: a report from the children's cancer group. Blood 1999, 94: 4036–4045.
Harrison CJ, Moorman AV, Broadfield ZJ, Cheung KL, Harris RL, Reza Jalali G, Robinson HM, Barber KE, Richards SM, Mitchell CD, Eden TO, Hann IM, Hill FG, Kinsey SE, Gibson BE, Lilleyman J, Vora A, Goldstone AH, Franklin IM, Durrant J, Martineau M: Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol 2004, 125: 552–559. 10.1111/j.1365-2141.2004.04948.x
Nachman JB, Heerema NA, Sather H, Camitta B, Forestier E, Harrison CJ, Dastugue N, Schrappe M, Pui CH, Basso G, Silverman LB, Janka-Schaub GE: Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood 2007, 110: 1112–1115. 10.1182/blood-2006-07-038299
Carroll AJ, Heerema NA, Gastier-Foster JM, Astbury C, Pyatt R, Reshmi SC, Borowitz MJ, Devidas M, Linda S, Loh ML, Raetz EA, Wood B, Winick NJ, Hunger SP, Carroll WL: Masked Hypodiploidy: Hypodiploid Acute Lymphoblastic Leukemia (ALL) in Children Mimicking Hyperdiploid ALL: A Report From the Children's Oncology Group (COG) AALL03B1 Study. ASH Annu Meeting Abstr 2009, 114: 1580.
Holmfeldt L, Wei L, Diaz-Flores E, Walsh M, Zhang J, Ding L, Payne-Turner D, Churchman M, Andersson A, Chen SC, McCastlain K, Becksfort J, Ma J, Wu G, Patel SN, Heatley SL, Phillips LA, Song G, Easton J, Parker M, Chen X, Rusch M, Boggs K, Vadodaria B, Hedlund E, Drenberg C, Baker S, Pei D, Cheng C, Huether R, et al.: The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet 2013, 45: 242–252. 10.1038/ng.2532
Malkin D: Li-fraumeni syndrome. Genes Cancer 2011, 2: 475–484. 10.1177/1947601911413466
Moorman AV, Ensor HM, Richards SM, Chilton L, Schwab C, Kinsey SE, Vora A, Mitchell CD, Harrison CJ: Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol 2010, 11: 429–438. 10.1016/S1470-2045(10)70066-8
Forestier E, Heyman M, Andersen MK, Autio K, Blennow E, Borgstrom G, Golovleva I, Heim S, Heinonen K, Hovland R, Johannsson JH, Kerndrup G, Nordgren A, Rosenquist R, Swolin B, Johansson B: Outcome of ETV6/RUNX1-positive childhood acute lymphoblastic leukaemia in the NOPHO-ALL-1992 protocol: frequent late relapses but good overall survival. Br J Haematol 2008, 140: 665–672. 10.1111/j.1365-2141.2008.06980.x
Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR: AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996, 84: 321–330. 10.1016/S0092-8674(00)80986-1
Wang LC, Kuo F, Fujiwara Y, Gilliland DG, Golub TR, Orkin SH: Yolk sac angiogenic defect and intra-embryonic apoptosis in mice lacking the Ets-related factor TEL. EMBO J 1997, 16: 4374–4383. 10.1093/emboj/16.14.4374
Hiebert SW, Sun W, Davis JN, Golub T, Shurtleff S, Buijs A, Downing JR, Grosveld G, Roussell MF, Gilliland DG, Lenny N, Meyers S: The t(12;21) translocation converts AML-1B from an activator to a repressor of transcription. Mol Cell Biol 1996, 16: 1349–1355.
Aspland SE, Bendall HH, Murre C: The role of E2A-PBX1 in leukemogenesis. Oncogene 2001, 20: 5708–5717. 10.1038/sj.onc.1204592
Lu Q, Kamps MP: Heterodimerization of Hox proteins with Pbx1 and oncoprotein E2a-Pbx1 generates unique DNA-binding specifities at nucleotides predicted to contact the N-terminal arm of the Hox homeodomain–demonstration of Hox-dependent targeting of E2a-Pbx1 in vivo. Oncogene 1997, 14: 75–83. 10.1038/sj.onc.1200799
Hunger SP: Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: clinical features and molecular pathogenesis. Blood 1996, 87: 1211–1224.
Schmiegelow K, Forestier E, Hellebostad M, Heyman M, Kristinsson J, Soderhall S, Taskinen M, Nordic Society of Paediatric H, Oncology: Long-term results of NOPHO ALL-92 and ALL-2000 studies of childhood acute lymphoblastic leukemia. Leukemia 2010, 24: 345–354. 10.1038/leu.2009.251
Ribeiro RC, Abromowitch M, Raimondi SC, Murphy SB, Behm F, Williams DL: Clinical and biologic hallmarks of the Philadelphia chromosome in childhood acute lymphoblastic leukemia. Blood 1987, 70: 948–953.
Crist W, Carroll A, Shuster J, Jackson J, Head D, Borowitz M, Behm F, Link M, Steuber P, Ragab A, et al.: Philadelphia chromosome positive childhood acute lymphoblastic leukemia: clinical and cytogenetic characteristics and treatment outcome. A Pediatric Oncology Group study. Blood 1990, 76: 489–494.
Melo JV: The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood 1996, 88: 2375–2384.
Schultz KR, Bowman WP, Aledo A, Slayton WB, Sather H, Devidas M, Wang C, Davies SM, Gaynon PS, Trigg M, Rutledge R, Burden L, Jorstad D, Carroll A, Heerema NA, Winick N, Borowitz MJ, Hunger SP, Carroll WL, Camitta B: Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children's oncology group study. J Clin Oncol 2009, 27: 5175–5181. 10.1200/JCO.2008.21.2514
Slayton WB, Schultz KR, Jones T, Raetz E, Devidas M, Pulsipher MA, Loh M, Chang BH, Carroll WL, Borowitz MJ, Silverman LB, Brown VI, Winick N, Carroll AJ, Heerema NA, Gastier-Foster JM, Wood BL, Mizrahy SL, Merchant TE, Hunger SP: Continuous Dose Dasatinib Is Safe and Feasible in Combination with Intensive Chemotherapy in Pediatric Philadelphia Chromosome Positive Acute Lymphoblastic Leukemia (Ph + ALL): Children's Oncology Group (COG) Trial AALL0622. ASH Annu Meet Abstr 2012, 120: 137.
Armstrong SA, Staunton JE, Silverman LB, Pieters R, den Boer ML, Minden MD, Sallan SE, Lander ES, Golub TR, Korsmeyer SJ: MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet 2002, 30: 41–47. 10.1038/ng765
Armstrong SA, Mabon ME, Silverman LB, Li A, Gribben JG, Fox EA, Sallan SE, Korsmeyer SJ: FLT3 mutations in childhood acute lymphoblastic leukemia. Blood 2004, 103: 3544–3546. 10.1182/blood-2003-07-2441
Ansari KI, Mandal SS: Mixed lineage leukemia: roles in gene expression, hormone signaling and mRNA processing. FEBS J 2010, 277: 1790–1804. 10.1111/j.1742-4658.2010.07606.x
Chessells JM, Harrison CJ, Kempski H, Webb DK, Wheatley K, Hann IM, Stevens RF, Harrison G, Gibson BE, party MRCCLw: Clinical features, cytogenetics and outcome in acute lymphoblastic and myeloid leukaemia of infancy: report from the MRC Childhood Leukaemia working party. Leukemia 2002, 16: 776–784. 10.1038/sj.leu.2402468
Moorman AV, Raimondi SC, Pui CH, Baruchel A, Biondi A, Carroll AJ, Forestier E, Gaynon PS, Harbott J, Harms DO, Heerema N, Pieters R, Schrappe M, Silverman LB, Vilmer E, Harrison CJ: No prognostic effect of additional chromosomal abnormalities in children with acute lymphoblastic leukemia and 11q23 abnormalities. Leukemia 2005, 19: 557–563.
Biondi A, Cimino G, Pieters R, Pui CH: Biological and therapeutic aspects of infant leukemia. Blood 2000, 96: 24–33.
Pieters R, den Boer ML, Durian M, Janka G, Schmiegelow K, Kaspers GJ, van Wering ER, Veerman AJ: Relation between age, immunophenotype and in vitro drug resistance in 395 children with acute lymphoblastic leukemia–implications for treatment of infants. Leukemia 1998, 12: 1344–1348. 10.1038/sj.leu.2401129
Ramakers-van Woerden NL, Beverloo HB, Veerman AJ, Camitta BM, Loonen AH, van Wering ER, Slater RM, Harbott J, den Boer ML, Ludwig WD, Haas OA, Janka-Schaub GE, Pieters R: In vitro drug-resistance profile in infant acute lymphoblastic leukemia in relation to age, MLL rearrangements and immunophenotype. Leukemia 2004, 18: 521–529. 10.1038/sj.leu.2403253
Dyer MJ, Akasaka T, Capasso M, Dusanjh P, Lee YF, Karran EL, Nagel I, Vater I, Cario G, Siebert R: Immunoglobulin heavy chain locus chromosomal translocations in B-cell precursor acute lymphoblastic leukemia: rare clinical curios or potent genetic drivers? Blood 2010, 115: 1490–1499. 10.1182/blood-2009-09-235986
Russell LJ, Capasso M, Vater I, Akasaka T, Bernard OA, Calasanz MJ, Chandrasekaran T, Chapiro E, Gesk S, Griffiths M, Guttery DS, Haferlach C, Harder L, Heidenreich O, Irving J, Kearney L, Nguyen-Khac F, Machado L, Minto L, Majid A, Moorman AV, Morrison H, Rand V, Strefford JC, Schwab C, Tönnies H, Dyer MJ, Siebert R, Harrison CJ: Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood 2009, 114: 2688–2698. 10.1182/blood-2009-03-208397
Russell LJ, Akasaka T, Majid A, Sugimoto KJ, Loraine Karran E, Nagel I, Harder L, Claviez A, Gesk S, Moorman AV, Ross F, Mazzullo H, Strefford JC, Siebert R, Dyer MJ, Harrison CJ: t(6;14)(p22;q32): a new recurrent IGH@ translocation involving ID4 in B-cell precursor acute lymphoblastic leukemia (BCP-ALL). Blood 2008, 111: 387–391. 10.1182/blood-2007-07-092015
Chapiro E, Russell L, Radford-Weiss I, Bastard C, Lessard M, Struski S, Cave H, Fert-Ferrer S, Barin C, Maarek O, Della-Valle V, Strefford JC, Berger R, Harrison CJ, Bernard OA, Nguyen-Khac F: Overexpression of CEBPA resulting from the translocation t(14;19)(q32;q13) of human precursor B acute lymphoblastic leukemia. Blood 2006, 108: 3560–3563. 10.1182/blood-2006-03-010835
Akasaka T, Balasas T, Russell LJ, Sugimoto KJ, Majid A, Walewska R, Karran EL, Brown DG, Cain K, Harder L, Gesk S, Martin-Subero JI, Atherton MG, Brüggemann M, Calasanz MJ, Davies T, Haas OA, Hagemeijer A, Kempski H, Lessard M, Lillington DM, Moore S, Nguyen-Khac F, Radford-Weiss I, Schoch C, Struski S, Talley P, Welham MJ, Worley H, Strefford JC, et al.: Five members of the CEBP transcription factor family are targeted by recurrent IGH translocations in B-cell precursor acute lymphoblastic leukemia (BCP-ALL). Blood 2007, 109: 3451–3461. 10.1182/blood-2006-08-041012
Jaso JM, Yin CC, Lu VW, Zhao M, Abruzzo LV, You MJ, Yang Y, Luthra R, Medeiros LJ, Lu G: B acute lymphoblastic leukemia with t(14;19)(q32;p13.1) involving IGH/EPOR: a clinically aggressive subset of disease. Mod Pathol 2014, 27: 382–389. 10.1038/modpathol.2013.149
Russell LJ, De Castro DG, Griffiths M, Telford N, Bernard O, Panzer-Grumayer R, Heidenreich O, Moorman AV, Harrison CJ: A novel translocation, t(14;19)(q32;p13), involving IGH@ and the cytokine receptor for erythropoietin. Leukemia 2009, 23: 614–617. 10.1038/leu.2008.250
Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, Chen SC, Payne-Turner D, Churchman ML, Harvey RC, Chen X, Kasap C, Yan C, Becksfort J, Finney RP, Teachey DT, Maude SL, Tse K, Moore R, Jones S, Mungall K, Birol I, Edmonson MN, Hu Y, Buetow KE, Chen IM, Carroll WL, Wei L, Ma J, Kleppe M, et al.: Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell 2012, 22: 153–166. 10.1016/j.ccr.2012.06.005
Harewood L, Robinson H, Harris R, Al-Obaidi MJ, Jalali GR, Martineau M, Moorman AV, Sumption N, Richards S, Mitchell C, Harrison CJ: Amplification of AML1 on a duplicated chromosome 21 in acute lymphoblastic leukemia: a study of 20 cases. Leukemia 2003, 17: 547–553. 10.1038/sj.leu.2402849
Soulier J, Trakhtenbrot L, Najfeld V, Lipton JM, Mathew S, Avet-Loiseau H, De Braekeleer M, Salem S, Baruchel A, Raimondi SC, Raynaud SD: Amplification of band q22 of chromosome 21, including AML1, in older children with acute lymphoblastic leukemia: an emerging molecular cytogenetic subgroup. Leukemia 2003, 17: 1679–1682. 10.1038/sj.leu.2403000
Rand V, Parker H, Russell LJ, Schwab C, Ensor H, Irving J, Jones L, Masic D, Minto L, Morrison H, Ryan S, Robinson H, Sinclair P, Moorman AV, Strefford JC, Harrison CJ: Genomic characterization implicates iAMP21 as a likely primary genetic event in childhood B-cell precursor acute lymphoblastic leukemia. Blood 2011, 117: 6848–6855. 10.1182/blood-2011-01-329961
Moorman AV, Richards SM, Robinson HM, Strefford JC, Gibson BE, Kinsey SE, Eden TO, Vora AJ, Mitchell CD, Harrison CJ, Party UKMRCNCRICLW: Prognosis of children with acute lymphoblastic leukemia (ALL) and intrachromosomal amplification of chromosome 21 (iAMP21). Blood 2007, 109: 2327–2330. 10.1182/blood-2006-08-040436
Harrison CJ, Moorman AV, Schwab C, Carroll AJ, Raetz EA, Devidas M, Strehl S, Nebral K, Harbott J, Teigler-Schlegel A, Zimmerman M, Dastuge N, Baruchel A, Soulier J, Auclerc MF, Attarbaschi A, Mann G, Stark B, Cazzaniga G, Chilton L, Vandenberghe P, Forestier E, Haltrich I, Raimondi SC, Parihar M, Bourquin JP, Tchinda J, Haferlach C, Vora A, Hunger SP, et al.: An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia 2014, 28: 1015–1021. 10.1038/leu.2013.317
Li Y, Schwab C, Ryan SL, Papaemmanuil E, Robinson HM, Jacobs P, Moorman AV, Dyer S, Borrow J, Griffiths M, Heerema NA, Carroll AJ, Talley P, Bown N, Telford N, Ross FM, Gaunt L, McNally RJ, Young BD, Sinclair P, Rand V, Teixeira MR, Joseph O, Robinson B, Maddison M, Dastugue N, Vandenberghe P, Haferlach C, Stephens PJ, Cheng J, et al.: Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature 2014, 508: 98–102. 10.1038/nature13115
Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, Van Zutven LJ, Beverloo HB, Van der Spek PJ, Escherich G, Horstmann MA, Janka-Schaub GE, Kamps WA, Evans WE, Pieters R: A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol 2009, 10: 125–134. 10.1016/S1470-2045(08)70339-5
Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, Ma J, Liu W, Cheng C, Schulman BA, Harvey RC, Chen IM, Clifford RJ, Carroll WL, Reaman G, Bowman WP, Devidas M, Gerhard DS, Yang W, Relling MV, Shurtleff SA, Campana D, Borowitz MJ, Pui CH, Smith M, Hunger SP, Willman CL, Downing JR: Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 2009, 360: 470–480. 10.1056/NEJMoa0808253
Martinelli G, Iacobucci I, Storlazzi CT, Vignetti M, Paoloni F, Cilloni D, Soverini S, Vitale A, Chiaretti S, Cimino G, Papayannidis C, Paolini S, Elia L, Fazi P, Meloni G, Amadori S, Saglio G, Pane F, Baccarani M, Foà R: IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP report. J Clin Oncol 2009, 27: 5202–5207. 10.1200/JCO.2008.21.6408
van der Veer A, Waanders E, Pieters R, Willemse ME, Van Reijmersdal SV, Russell LJ, Harrison CJ, Evans WE, van der Velden VH, Hoogerbrugge PM, Van Leeuwen F, Escherich G, Horstmann MA, Mohammadi Khankahdani L, Rizopoulos D, De Groot-Kruseman HA, Sonneveld E, Kuiper RP, Den Boer ML: Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood 2013, 122: 2622–2629. 10.1182/blood-2012-10-462358
Loh ML, Zhang J, Harvey RC, Roberts K, Payne-Turner D, Kang H, Wu G, Chen X, Becksfort J, Edmonson M, Buetow KH, Carroll WL, Chen IM, Wood B, Borowitz MJ, Devidas M, Gerhard DS, Bowman P, Larsen E, Winick N, Raetz E, Smith M, Downing JR, Willman CL, Mullighan CG, Hunger SP: Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: a report from the Children's Oncology Group TARGET Project. Blood 2013, 121: 485–488. 10.1182/blood-2012-04-422691
van der Veer A, Zaliova M, Mottadelli F, De Lorenzo P, Te Kronnie G, Harrison CJ, Cave H, Trka J, Saha V, Schrappe M, Pieters R, Biondi A, Valsecchi MG, Stanulla M, den Boer ML, Cazzaniga G: IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood 2014, 123: 1691–1698. 10.1182/blood-2013-06-509794
Harvey RC, Mullighan CG, Chen IM, Wharton W, Mikhail FM, Carroll AJ, Kang H, Liu W, Dobbin KK, Smith MA, Carroll WL, Devidas M, Bowman WP, Camitta BM, Reaman GH, Hunger SP, Downing JR, Willman CL: Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 2010, 115: 5312–5321. 10.1182/blood-2009-09-245944
Harvey RC, Mullighan CG, Wang X, Dobbin KK, Davidson GS, Bedrick EJ, Chen IM, Atlas SR, Kang H, Ar K, Wilson CS, Wharton W, Murphy M, Devidas M, Carroll AJ, Borowitz MJ, Bowman WP, Downing JR, Relling M, Yang J, Bhojwani D, Carroll WL, Camitta B, Reaman GH, Smith M, Hunger SP, Willman CL: Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood 2010, 116: 4874–4884. 10.1182/blood-2009-08-239681
Perez Andreu V, Roberts KG, Harvey RC, Yang W, Cheng C, Pei D, Xu H, Gastier Foster JES, Lim JY, Chen IM, Fan Y, Devidas M, Borowitz MJ, Smith C, Neale G, Burchard EG, Torgerson DG, Klussmann FA, Villagran CR, Winick NJ, Camitta BM, Raetz E, Wood B, Yue F, Carroll WL, Larsen E, Bowman WP, Loh ML, Dean M, et al.: Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet 2013, 45: 1494–1498. 10.1038/ng.2803
Hagman J, Gutch MJ, Lin H, Grosschedl R: EBF contains a novel zinc coordination motif and multiple dimerization and transcriptional activation domains. EMBO J 1995, 14: 2907–2916.
Lengline E, Beldjord K, Dombret H, Soulier J, Boissel N, Clappier E: Successful tyrosine kinase inhibitor therapy in a refractory B-cell precursor acute lymphoblastic leukemia with EBF1-PDGFRB fusion. Haematologica 2013, 98: e146-e148. 10.3324/haematol.2013.095372
Weston BW, Hayden MA, Roberts KG, Bowyer S, Hsu J, Fedoriw G, Rao KW, Mullighan CG: Tyrosine kinase inhibitor therapy induces remission in a patient with refractory EBF1-PDGFRB-positive acute lymphoblastic leukemia. J Clin Oncol 2013, 31: e413-e416. 10.1200/JCO.2012.47.6770
Smith CA, Fan G: The saga of JAK2 mutations and translocations in hematologic disorders: pathogenesis, diagnostic and therapeutic prospects, and revised World Health Organization diagnostic criteria for myeloproliferative neoplasms. Hum Pathol 2008, 39: 795–810. 10.1016/j.humpath.2008.02.004
Mullighan CG: Molecular genetics of B-precursor acute lymphoblastic leukemia. J Clin Invest 2012, 122: 3407–3415. 10.1172/JCI61203
Mullighan CG, Zhang J, Harvey RC, Collins-Underwood JR, Schulman BA, Phillips LA, Tasian SK, Loh ML, Su X, Liu W, Devidas M, Atlas SR, Chen IM, Clifford RJ, Gerhard DS, Carroll WL, Reaman GH, Smith M, Downing JR, Hunger SP, Willman CL: JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 2009, 106: 9414–9418. 10.1073/pnas.0811761106
Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, Su X, Pui CH, Relling MV, Evans WE, Shurtleff SA, Downing JR: Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007, 446: 758–764. 10.1038/nature05690
Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, Ma J, Coustan-Smith E, Harvey RC, Willman CL, Mikhail FM, Meyer J, Carroll AJ, Williams RT, Cheng J, Heerema NA, Basso G, Pession A, Pui CH, Raimondi SC, Hunger SP, Downing JR, Carroll WL, Rabin KR: Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet 2009, 41: 1243–1246. 10.1038/ng.469
Bercovich D, Ganmore I, Scott LM, Wainreb G, Birger Y, Elimelech A, Shochat C, Cazzaniga G, Biondi A, Basso G, Cario G, Schrappe M, Stanulla M, Strehl S, Haas OA, Mann G, Binder V, Borkhardt A, Kempski H, Trka J, Bielorei B, Avigad S, Stark B, Smith O, Dastugue N, Bourquin JP, Tal NB, Green AR, Izraeli S: Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down's syndrome. Lancet 2008, 372: 1484–1492. 10.1016/S0140-6736(08)61341-0
Harrison CJ: Targeting signaling pathways in acute lymphoblastic leukemia: new insights. Hematol Am Soc Hematol Educ Program 2013, 2013: 118–125. 10.1182/asheducation-2013.1.118
Tirado CA, Chen W, Huang LJ, Laborde C, Hiemenz MC, Valdez F, Ho K, Winick N, Lou Z, Koduru P: Novel JAK2 rearrangement resulting from a t(9;22)(p24;q11.2) in B-acute lymphoblastic leukemia. Leuk Res 2010, 34: 1674–1676. 10.1016/j.leukres.2010.05.031
Ho K, Valdez F, Garcia R, Tirado CA: JAK2 Translocations in hematological malignancies: Review of the literature. J Assoc Genet Technol 2010, 36: 107–109.
Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffe M, Berthou C, Lessard M, Berger R, Ghysdael J, Bernard OA: A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science 1997, 278: 1309–1312. 10.1126/science.278.5341.1309
Tirado CA, Shabsovich D, Denicola M, Rao D, Yang L, Garcia R, Rao N: A case of pediatric B-Lymphoblastic leukemia presenting with a t(9;12)(p24;q11.2) involving JAK2 and concomitant MLL rearrangement with apparent insertion at 6q27. Biomark Res 2013, 1: 31. 10.1186/2050-7771-1-31
Cario G, Zimmermann M, Romey R, Gesk S, Vater I, Harbott J, Schrauder A, Moericke A, Izraeli S, Akasaka T, Dyer MJ, Siebert R, Schrappe M, Stanulla M: Presence of the P2RY8-CRLF2 rearrangement is associated with a poor prognosis in non-high-risk precursor B-cell acute lymphoblastic leukemia in children treated according to the ALL-BFM 2000 protocol. Blood 2010, 115: 5393–5397. 10.1182/blood-2009-11-256131
Chen IM, Harvey RC, Mullighan CG, Gastier-Foster J, Wharton W, Kang H, Borowitz MJ, Camitta BM, Carroll AJ, Devidas M, Pullen DJ, Payne-Turner D, Tasian SK, Reshmi S, Cottrell CE, Reaman GH, Bowman WP, Carroll WL, Loh ML, Winick NJ, Hunger SP, Willman CL: Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children's Oncology Group study. Blood 2012, 119: 3512–3522. 10.1182/blood-2011-11-394221
Ensor HM, Schwab C, Russell LJ, Richards SM, Morrison H, Masic D, Jones L, Kinsey SE, Vora AJ, Mitchell CD, Harrison CJ, Moorman AV: Demographic, clinical, and outcome features of children with acute lymphoblastic leukemia and CRLF2 deregulation: results from the MRC ALL97 clinical trial. Blood 2011, 117: 2129–2136. 10.1182/blood-2010-07-297135
Yoda A, Yoda Y, Chiaretti S, Bar-Natan M, Mani K, Rodig SJ, West N, Xiao Y, Brown JR, Mitsiades C, Sattler M, Kutok JL, DeAngelo DJ, Wadleigh M, Piciocchi A, Dal Cin P, Bradner JE, Griffin JD, Anderson KC, Stone RM, Ritz J, Foà R, Aster JC, Frank DA, Weinstock DM: Functional screening identifies CRLF2 in precursor B-cell acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 2010, 107: 252–257. 10.1073/pnas.0911726107
Tasian SK, Doral MY, Borowitz MJ, Wood BL, Chen IM, Harvey RC, Gastier-Foster JM, Willman CL, Hunger SP, Mullighan CG, Loh ML: Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood 2012, 120: 833–842. 10.1182/blood-2011-12-389932
Maude SL, Tasian SK, Vincent T, Hall JW, Sheen C, Roberts KG, Seif AE, Barrett DM, Chen IM, Collins JR, Mullighan CG, Hunger SP, Harvey RC, Willman CL, Fridman JS, Loh ML, Grupp SA, Teachey DT: Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood 2012, 120: 3510–3518. 10.1182/blood-2012-03-415448
Waibel M, Solomon VS, Knight DA, Ralli RA, Kim SK, Banks KM, Vidacs E, Virely C, Sia KC, Bracken LS, Collins-Underwood R, Drenberg C, Ramsey LB, Meyer SC, Takiguchi M, Dickins RA, Levine R, Ghysdael J, Dawson MA, Lock RB, Mullighan CG, Johnstone RW: Combined targeting of JAK2 and Bcl-2/Bcl-xL to cure mutant JAK2-driven malignancies and overcome acquired resistance to JAK2 inhibitors. Cell Rep 2013, 5: 1047–1059. 10.1016/j.celrep.2013.10.038
Nutt SL, Heavey B, Rolink AG, Busslinger M: Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature 1999, 401: 556–562. 10.1038/44076
Nebral K, Denk D, Attarbaschi A, Konig M, Mann G, Haas OA, Strehl S: Incidence and diversity of PAX5 fusion genes in childhood acute lymphoblastic leukemia. Leukemia 2009, 23: 134–143. 10.1038/leu.2008.306
Shah S, Schrader KA, Waanders E, Timms AE, Vijai J, Miething C, Wechsler J, Yang J, Hayes J, Klein RJ, Zhang J, Wei L, Wu G, Rusch M, Nagahawatte P, Ma J, Chen SC, Song G, Cheng J, Meyers P, Bhojwani D, Jhanwar S, Maslak P, Fleisher M, Littman J, Offit L, Rau-Murthy R, Fleischut MH, Corines M, Murali R, et al.: A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet 2013, 45: 1226–1231. 10.1038/ng.2754
Taketani T, Taki T, Sugita K, Furuichi Y, Ishii E, Hanada R, Tsuchida M, Sugita K, Ida K, Hayashi Y: FLT3 mutations in the activation loop of tyrosine kinase domain are frequently found in infant ALL with MLL rearrangements and pediatric ALL with hyperdiploidy. Blood 2004, 103: 1085–1088.
Stam RW, den Boer ML, Schneider P, Nollau P, Horstmann M, Beverloo HB, van der Voort E, Valsecchi MG, de Lorenzo P, Sallan SE, Armstrong SA, Pieters R: Targeting FLT3 in primary MLL-gene-rearranged infant acute lymphoblastic leukemia. Blood 2005, 106: 2484–2490. 10.1182/blood-2004-09-3667
Figueroa ME, Chen SC, Andersson AK, Phillips LA, Li Y, Sotzen J, Kundu M, Downing JR, Melnick A, Mullighan CG: Integrated genetic and epigenetic analysis of childhood acute lymphoblastic leukemia. J Clin Invest 2013, 123: 3099–3111. 10.1172/JCI66203
Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, Bhang HE, Taylor JE, Hu M, Englund NP, Yan F, Wang Z, Robert McDonald E 3rd, Wei L, Ma J, Easton J, Yu Z, deBeaumount R, Gibaja V, Venkatesan K, Schlegel R, Sellers WR, Keen N, Liu J, Caponigro G, Barretina J, Cooke VG, Mullighan C, Carr SA, Downing JR: Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Genet 2013, 45: 1386–1391. 10.1038/ng.2777
Mar BG, Bullinger LB, McLean KM, Grauman PV, Harris MH, Stevenson K, Neuberg DS, Sinha AU, Sallan SE, Silverman LB, Kung AL, Lo Nigro L, Ebert BL, Armstrong SA: Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat Commun 2014, 5: 3469.
Benetatos L, Vartholomatos G: MicroRNAs mark in the MLL-rearranged leukemia. Ann Hematol 2013, 92: 1439–1450. 10.1007/s00277-013-1803-4
Bernt KM, Armstrong SA: Targeting epigenetic programs in MLL-rearranged leukemias. Hematol Am Soc Hematol Educ Program 2011, 2011: 354–360. 10.1182/asheducation-2011.1.354
Liedtke M, Cleary ML: Therapeutic targeting of MLL. Blood 2009, 113: 6061–6068. 10.1182/blood-2008-12-197061
Travers J, Blagg J, Workman P: Epigenetics: Targeting leukemia on the DOT. Nat Chem Biol 2011, 7: 663–665. 10.1038/nchembio.661
Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, Pollock RM, Richon VM, Kung AL, Armstrong SA: MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 2011, 20: 66–78. 10.1016/j.ccr.2011.06.010
Rivera GK, Zhou Y, Hancock ML, Gajjar A, Rubnitz J, Ribeiro RC, Sandlund JT, Hudson M, Relling M, Evans WE, Pui CH: Bone marrow recurrence after initial intensive treatment for childhood acute lymphoblastic leukemia. Cancer 2005, 103: 368–376. 10.1002/cncr.20743
Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, Downing JR: Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 2008, 322: 1377–1380. 10.1126/science.1164266
Jan M, Majeti R: Clonal evolution of acute leukemia genomes. Oncogene 2013, 32: 135–140. 10.1038/onc.2012.48
Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, Heatley SL, Holmfeldt L, Collins-Underwood JR, Ma J, Buetow KH, Pui CH, Baker SD, Brindle PK, Downing JR: CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011, 471: 235–239. 10.1038/nature09727
Inthal A, Zeitlhofer P, Zeginigg M, Morak M, Grausenburger R, Fronkova E, Fahrner B, Mann G, Haas OA, Panzer-Grumayer R: CREBBP HAT domain mutations prevail in relapse cases of high hyperdiploid childhood acute lymphoblastic leukemia. Leukemia 2012, 26: 1797–1803. 10.1038/leu.2012.60
Tsapis M, Lieb M, Manzo F, Shankaranarayanan P, Herbrecht R, Lutz P, Gronemeyer H: HDAC inhibitors induce apoptosis in glucocorticoid-resistant acute lymphatic leukemia cells despite a switch from the extrinsic to the intrinsic death pathway. Int J Biochem Cell Biol 2007, 39: 1500–1509. 10.1016/j.biocel.2007.03.009
Meyer JA, Wang J, Hogan LE, Yang JJ, Dandekar S, Patel JP, Tang Z, Zumbo P, Li S, Zavadil J, Levine RL, Cardozo T, Hunger SP, Raetz EA, Evans WE, Morrison DJ, Mason CE, Carroll WL: Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat Genet 2013, 45: 290–294. 10.1038/ng.2558
Tzoneva G, Perez-Garcia A, Carpenter Z, Khiabanian H, Tosello V, Allegretta M, Paietta E, Racevskis J, Rowe JM, Tallman MS, Paganin M, Basso G, Hof J, Kirschner-Schwabe R, Palomero T, Rabadan R, Ferrando A: Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat Med 2013, 19: 368–371. 10.1038/nm.3078
Gang EJ, Hsieh YT, Pham J, Zhao Y, Nguyen C, Huantes S, Park E, Naing K, Klemm L, Swaminathan S, Conway EM, Pelus LM, Crispino J, Mullighan CG, McMillan M, Müschen M, Kahn M, Kim YM: Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene 2014, 33: 2169–2178. 10.1038/onc.2013.169
Acknowledgements
We apologize to the authors whose work has not been cited here, owing to space limitations. We thank the UCLA Department of Pathology and Laboratory Medicine and Dr. Scott Binder for generous support. We are also grateful for the hard work of everyone in the Histology, Flow Cytometry, and Cytogenetics Laboratories at UCLA.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors wrote, reviewed and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Woo, J.S., Alberti, M.O. & Tirado, C.A. Childhood B-acute lymphoblastic leukemia: a genetic update. Exp Hematol Oncol 3, 16 (2014). https://doi.org/10.1186/2162-3619-3-16
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2162-3619-3-16