
Abstract
Cancer stem cells (CSCs) or tumor initiating cells (TICs) contribute to tumorigenesis, metastasis, recurrence and chemoresistance. CD133, a pentaspan membrane glycoprotein, has been used as a stem cell biomarker for isolation of stem-like cells from a variety of normal and pathological tissues as well as cell lines since its discovery in 1999. Recent studies are focusing on the functionality of CD133. In this review, we summarize new insights into CD133 regulation and the involvement of CD133 in cell self-renewal, tumorigenesis, metastasis, resistance, metabolism, differentiation, autophagy, apoptosis, and regeneration.
Similar content being viewed by others


Introduction
Since CD133 was identified as a pentaspan transmembrane protein for human hematopoietic stem cells and mouse neuroepithelial cells [1–3], many studies have subsequently revealed that CD133 expression is associated with progenitor/stem cells, tumor, regeneration, differentiation, and metabolism. CD133 is one of key biomarkers for isolation and characterization of stem cells. Increasing evidence has shown that CD133 is not only a biomarker, but functions also in cell growth, development and tumor biology. Therefore, in this review, we will summarize the new functions of CD133.
CD133, also called Prominin-1, is a product of a single-copy gene on chromosome 4 (4p15.33) in human or chromosome 5 (5b3) in mice. Human CD133 is a transmembrane glycoprotein of 865 amino acids with a total molecular weight of 120 kDa. This protein consists of an N-terminal extracellular domain, five transmembrane domains with two large extracellular loops, and a 59 amino acids cytoplasmic tail [4]. It is selectively localized in microvilli and other plasma membrane protrusions [5, 6]. In general, CD133 positive and CD133 negative cells display different characters. For example, 1) CD133+ and CD133- glioma cells belong to independent cancer stem cell populations; 2) CD133+ glioma cells are derived from primordial CD133- CSCs; 3) CD133- CSCs retain their stem-like features as well as tumor initiation capacity, and can re-acquire CD133 expression in vivo; and 4) Both CD133+ and CD133- CSCs have different expression profiles in transcriptional activities and extracellular matrix molecules [7, 8].
Regulation of CD133 expression
The CD133 expression is regulated by many extracellular or intracellular factors and represents changes of cell type with particular functions [9]. Griguer, et al. revealed that hypoxia, mitochondrial dysfunction or depletion of mitochondrial DNA induced a reversible up-regulation of CD133 expression [10]. Hypoxia-induced CD133 expression is also found in human lung cancer, pancreatic cancer and glioma cells [11, 12]. Hypoxic condition increases hypoxia inducible factor 1α (HIF-1α) expression which inhibits the mammalian target of rapamycin (mTOR) C1 activity [12, 13]. Increased HIF-1α induces the expansion of the CD133+ cells [11, 12, 14]. Pharmacological inhibition of mTOR with rapamycin greatly increases both the CD133+ populations and the expression of stem cell-like genes [14, 15]. Enhancing mTOR activity by over-expressing Rheb significantly decreases CD133 expression, whereas knockdown of the mTOR yields an opposite effect [15].
Transforming growth factor β1 (TGFβ1) is identified to be capable of up-regulating CD133 expression specifically within the Huh-7 hepatocellular carcinoma (HCC) cell line in a time- and dose-dependent manner [16]. TGFβ1 inhibits DNA methyltransferases (DNMT) 1 and DNMT3β expression and subsequently induces the demethylation of promoter-1 of CD133 [16]. Analysis of Toll-like receptors (TLR) in colorectal cancer (CRC) reveals that TLR7 and 8 increase in CD133+ cells in CRCs [17]. Both TLRs and chemokines activate NF-κB signaling in cancer stem cells [18, 19]. Therefore, CD133 expression may play an important role in communication through membrane receptors.
MicroRNA (miRNA) profiling has revealed that several miRNA are involved in regulation of CD133 expression in a variety of cells. By analyzing miRNA expression profiling of CD133+ and CD133- cells from human HCC clinical specimens and cell lines, Ma, et al. has identified elevated miR-130b in CD133+ HCC TICs [20]. Forcing expression of miR-130b in CD133- cells enhances their chemoresistance, self-renewal and tumorigenicity in vivo. But upregulation of miR-125b inhibits the invasion of CD133+ primary glioblastoma cells [21]. In addition, miR-142-3p [22], miR-199b-5p [23], miR-143, miR-145 [24], and miR-150 [25] show inhibition of the colony-forming ability and tumor sphere formation of CD133+ cells. However, most of these miRNAs exhibit indirect regulation of CD133 expression. A specific miRNA targeting CD133 expression has not been identified yet.
CD133 expression is also regulated by epigenetic factors. Methylation of the CD133 promoter represses CD133 gene transcription. Demethylation of the CD133 gene has been found in a variety of human tumors including colorectal cancer [26], gastric carcinoma [27], gliomas and glioblastoma [28, 29], HCC [30], and ovarian cancer [31] and so on. TGFβ1 induced CD133 expression via demethylation of CD133 promoter-1 in Huh-7 cells [16]. Upregulation of CD133 is in CRC that exhibits a hyperactivated Ras-Raf-MEK-ERK pathway secondary to mutations in K-Ras or B-Raf [32].
CD133 in cell self-renewal and tumorigenesis
Freshly isolated CD133+ cancer cells from colorectal cancer, gallbladder carcinoma, HCC, ovarian cancer and other tumors gave rise to long-term tumor spheroids and xenograft tumors in immunodeficient mice [20, 33–35]. The underlying mechanisms involved in regulation of self-renewal in HCC may depend on the Akt/PKB and Bcl-2 pathway [36]. Using the genome-wide microarray analysis, Tang et al. revealed that a significant interleukin-8 (IL-8) signaling network was activated in CD133+ liver TICs obtained from HCC clinical samples and cell lines responsible for self-renew, tumor angiogenesis, and tumorigenesis [37]. C-terminal cytosolic domain of CD133 is phosphorylated by Src-family kinases as determined by mass spectrometry and site-directed mutagenesis. Tyrosine-828 and the nonconsensus Tyrosine-852 are the major tyrosine phosphorylation sites [38]. T-828 phosphorylation of CD133 mediates activation of PI3K/Akt pathway in glioma stem cells through interaction with p85 regulatory subunit [39]. Silencing of CD133 impairs the self-renewal and tumorigenic capacity of tumor cells [40].
Although both CD133+ and CD133- cells are capable of tumor initiation in the nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice, most of CD133+ tumor subpopulations form colonospheres in an in vitro culture and retain long-term tumorigenic capacity in a NOD/SCID serial xenotransplantation model [41]. Upstream molecules in Akt and mitogen-activated protein kinase (MAPK) pathways are preferentially activated in CD133+ colon cancer cells [42]. Ras and its downsteam effectors such as ERK, JNK, PI3K, p38K, and RalA are also significantly activated in CD133+ human primary malignant peripheral nerve sheath tumor [43]. Stemness genes, octamer biding transcription factor 3/4 (OCT4) and/or SRY-box containing gene 2 (SOX2), have been found to bind to the P1 promoter region of CD133 gene loci and ectopic OCT4 or SOX2 expression triggers the CD133P1 activity in the lung cancer cell lines N417, H358, and A549 [44]. Therefore, CD133 expression is essential for self-renewal function and tumorigenesis in certain cell types.
CD133 and metastasis
Increasing evidence indicates that a subset of tumor cells contributing to metastasis has the properties of CSCs or TICs. CD133+ cells are higher in liver metastasis than in primary colorectal tumors [45]. Compared with CD133+CXCR4- cells, CD133+CXCR4+ cancer cells have a high metastatic capacity in vitro and in vivo and undergoes epithelial-mesenchymal transitions (EMT)[45]. CD133+CD44+ cancer cells have been characterized in several highly metastatic tumors, such as CRCs [46–48], HCCs [49, 50], pancreatic cancer [51], gallbladder carcinoma [52], lung adenocarcinomas [53] and gastric cancer [54]. Immunohistochemical study of human HCC specimens reveals that the number of CD133+ CD44+ HCC cells is increased and associated with portal vein invasion [49]. In colorectal cancer with early liver metastases, co-expression of CD133 and CD44 is significantly higher when compared to those without early liver metastases [48]. Knockdown of CD133 in hepatocarcinoma PLC/PRF/5 and HCT116 cells results in decreased expressions of matrix metalloproteinase (MMP)-2, a disintegrin and metalloproteinase (ADAM)9 [55, 56]. These lead to decreased invasion as demonstrated in an in vitro system [55, 56]. In addition, chemokine CCL5 and its receptors, CCR1, CCR3 and CCR5, are found to be upregulated in CD133+ cancer stem-like cells from ovarian cancer [57]. Blocking of CCL5, CCR1 or CCR3 effectively inhibits the invasive capacity of these cells via inhibition of NF-kappaB and MMP9 secretion [57]. Therefore, CD133+ TICs may confer metastatic potential to their progenies.
CD133 and chemo- and radio-resistance
CD133 positive cells show a high degree of chemoresistance. CD133+ lung cancer cells exhibit drug resistance [58]. Isolated CD133+ CSCs from human oral squamous cell carcinoma are substantially resistant to standard chemotherapy [59]. Ectopic overexpression of CD133 in rat C6 glioma cells leads to significant reluctance to undergo apoptosis from camptothecin and doxorubicin treatments [60]. Chemoresistant CD133+ cells usually have the upregulation of ATP-binding cassette (ABC) transporter [52, 60]. Since ATP-binding cassette sub-family B member 5 (ABCB5)- mediated doxorubicin efflux [61], suppression of ABCB5 sensitizes the cells to doxorubicin uptake and apoptosis [62]. Moreover, CD133-expressing liver cancer cells following radiation exposure show higher activation of MAPK/PI3K signaling pathway and reduction in reactive oxygen species levels compared to CD133- cells. The irradiated CD133+ cell induces an increase of tumor formation in an in vivo xenograft model compared to the CD133- group, suggesting that CD133 contributes to radioresistance in HCC [63]. Treatment of unsorted HCC cells with anticancer drugs in vitro also significantly enriches the CD133+ subpopulation [36].
CD133 and metabolism
In epithelial cells, CD133 is found in microvilli, the primary cilium and the midbody [64]. This membrane protein has been found to be released from apical midbodies and the primary cilium of neuroepithelial cells as a whole or in part, into the extracellular space, yielding the CD133-enriched membrane particles found in the neural tube fluid [65]. Intriguingly, the release of these particles has been implicated in (neuro)epithelial cell differentiation [64]. CD133 is selectively associated with microvilli and largely segregated from the membrane subdomains containing placental alkaline phosphatase [66]. CD133 is also a cholesterol-interacting membrane protein responsible for the generation of plasma membrane protrusions, their lipid composition and organization as well as the membrane-to-membrane interactions [67]. Unraveling that CD133 inhibits transferrin uptake and AC133 antibody downregulates this uptake [68] further indicates the involvement of CD133 in cell metabolism.
Hexokinase II is a key enzyme in the glucolytic pathway. Its gene expression and enzymatic activity are lower in CD133+ than in CD133- hepatoma BEL-7402 [69]. Pancreatic cancer patients with low expression of hexokinase II have significantly shorter survival than those with higher expression [70]. Higher expression of hexokinase II is associated with advanced tumor grade and higher stage as well as higher mortality in HCC [71].
β-galactoside α2,6-sialyltransferase (ST6Gal-I) adds an α2-6-linked sialic acid to the N-glycans of CD133 membrane proteins that may stabilize CD133 [72]. ST6Gal-I has been reported to be upregulated in human colon cancer, induced pluripotent stem (iPS) cells and CSCs [73]. CD133 has eight N-glycosylation sites on its extracellular loops [4]. Lectin binding assay for cell surface glycan epitopes and microarray analysis for expression of N-glycan biosynthesis-related genes demonstrate that over 10% difference between CD133+ and CD133- hematopoietic stem and progenitor cells (HSPC) [74]. Biantennary complex-type N-glycans are enriched in CD133+ cells that have the overexpressed mannosyl (α-1,6-)-glycoprotein β-1,2-N-acetylglucosaminyltransferase (MGAT) 2 and underexpressed MGAT 4 [74]. Moreover, the amount of high-mannose type N-glycans and terminal α2,3-sialylation is increased in CD133+ cells [74]. N-glycosylation of CD133 is thought to be associated with cell differentiation [75] and promoted by hypoxia [76]. In addition, silencing CD133 reduces the glucose uptake [77], indicating that CD133 expression may be responsible for energic metabolism and the survival of CSCs.
Further analysis of signaling pathways in CD133+ and CD133- cells has found that freshly isolated CD133+ cells from benign prostate tissue show expression of transcripts associated with cell development, ion homeostasis and cell communication, whereas profiling of CD133- cells revealed gene transcripts related to cell proliferation and metabolism [78]. In human cord blood-derived CD133+ cells, 690 transcripts are differentially expressed in CD133+ and CD133- cells. Of these, 393 are increased and 297 are decreased in CD133+ cells in which that the highest overexpression genes are associated with metabolism, cell communication, and development [79]. Transcriptomic profiling of sorted CD133+ and CD133- cells from human glioblastoma multiforme reveals a CD133 gene expression signature composed of 214 differentially expressed genes [80]. Moreover, comparison of transcripts in CD34+ and CD133+ cells reveals that CD133+ cells have higher numbers of up-regulated genes than CD34+ cells. The uniquely expressed genes in CD34+ or CD133+ cell populations are associated with different biological processes: CD34+ cells overexpress many transcripts associate with development, while CD133+ cells express genes associated with chromatin architecture, DNA metabolism, and cell cycle [81].
CD133 and differentiation
CD133 is expressed on both CSC and differentiated tumor cells. CD133 is possibly folded as a result of differential glycosylation to mask specific epitopes [75]. Although both CD133+ and CD133- cells derived from primary glioblastomas show similar tumorigenicity in nude mice, there are 117 genes differentially expressing in these two subtypes [82]. Observation of CD133 expression in several neuroblastoma cell lines/tumor samples has shown that CD133 represses neurite extension and the expression of differentiation marker proteins, but accelerates cell proliferation, anchorage-independent colony formation and in vivo tumor formation of neuroblastoma cells [83]. Platelet-derived growth factors in the presence of a cytokine cocktail suppress ex vivo expansion of umbilical cord blood CD133+ cells and enhance their differentiation into megakaryocytic progenitor cells in a dose- and time-dependent manner [84]. Consistent with rapamycin increasing CD133 expression, mTOR inhibition severely blocks the differentiation of CD133+ to CD133- liver tumor cells [15]. Interestingly, single-cell culture experiments have revealed that CD133- liver tumor cells are capable of converting to CD133+ cells and the inhibition of mTOR signaling substantially promotes this conversion [15]. However, we should also note that CD133 expression and post-translational modification are dynamic and reversible that are dependent on cell microenvironment and physiological regulation [85].
CD133 and autophagy
Autophagy as a key homeostatic process of cytoplasmic degradation and recycling is associated with the status of tumor cells. The shift of CD133 subcellular localization from the cytoplasm to the plasma membrane leads to the alternation of its functions [40]. CD133 has been shown to affect the clathrin-endocytosis process [68]. We have found that CD133 expression promotes glucose uptake and autophagosome formation in the glucose deprivation [77]. Immunofluorescence and time-lapsed confocal techniques demonstrate co-locolization of CD133 with an autophagy marker, microtubule-associated protein light chain3 (LC3) and a lysosome marker. CD133-mediated functions are beneficial for CSC survival. Knockdown of CD133 by siRNA attenuates production of LC3-II while the expression of autophagy associated genes (Atg9, Atg5/Atg12, and beclin-1) is not affected [77]. We speculate that CD133-mediated autophagy may be involved in the membrane-mediated phagophore formation.
CD133 and apoptosis
CD133+ cell population demonstrates significant resistance to TGF β- and TNF-related apoptosis-inducing ligand (TRAIL)- induced apoptosis compared with CD133- cells [86, 87]. High expression of FLICE-like inhibitory protein (FLIP), an inhibitor of the extrinsic apoptotic pathway, in CD133+ cells is thought to be associated with the resistance to the apoptosis induced by TRAIL [88]. In addition, CD133+ population has grater resistance to staurosporine-induced apoptosis than CD133- population [89] and stress-induced apoptosis [90]. Targeting CD133 by its antibody leads to cell death via attenuation of autophagy and promotion of apoptosis in HCC cells [77].
CD133 and regeneration
CD133+ cells that are isolated from bone marrow, cord blood, and peripheral blood have been tested in both animal models and clinical trials in an attempt to repair the injured tissues with the pluripotent of CD133+ cells [91]. The cell lines derived from human endothelial progenitor cells and cord blood undergo in vitro pre-angiogenic process, form pseudovessel structures and present an accelerate angiogenesis in hypoxic conditions [92]. Cells isolated from the peripheral blood using CD133 antibodies have been shown through a mouse spinal cord injury model as being able to enhance angiogenesis, astrogliosis, axon growth and functional recovery. In contrast, the administration of CD133- cells fails to promote axon growth and functional recovery, but moderately enhances angiogenesis and astrogliosis [93]. When CD133+ cells embedded in atelocollagen gel into a silicone tube is used to bridge a 15-mm defect in the sciatic nerve of athymic rats, sciatic nerves are structurally and functionally able to regenerate within 8 weeks and the transplanted CD133+ cells are differentiated into Schwann cells [94]. In a muscle injury rat model, granulocyte colony stimulating factor-mobilized peripheral blood CD133+ cells are differentiated into endothelial and myogenic lineages [95]. In addition, autologous bone marrow-derived CD133+ stem cell therapy has been used in clinical trials for patients with chronic total occlusion and ischemia [96], myocardial infarction [97], hepatic fibrosis [98], and liver regeneration [99]. The CD133+ cells have also been used for cardiac stem cell therapy [100] and bone regeneration [101]. Better application and expansion of CD133+ cells may yield tremendous benefits for tissue engineering.
Perspective
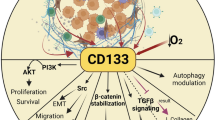
To better understand how to modulate the stem cells, particularly cancer stem cells, we have to identify specific biomarkers. Extensive studies of CD133 in different fields have provided new insights into the diverse CD133 functions (Figure 1). However, it remains a challenge to integrate the available expression, regulatory, structural, and functional data for this fascinating protein [102].

Functional outline of CD133+ cells. The number of CD133+ cells is maintained in a relative constant in bone marrow, blood, different tissues and even tumors. When cells or tissues are damaged by chemical, physical or mutational causes, CD133+ progenitor or stem cells are activated to self-renew, proliferate and differentiate in order to repair the damage. CD133+ CSCs or TICs are responsible for tumor metastasis, chemo- or radio-resistance and recurrence. CD133 expression is dynamic and reversible in response to the changes of cell microenvironment. CD133 is involved in diverse cellular processes, including glucose and transferrin uptake, autophagy, membrane-membrane interaction, and matrix metalloproteinase functions. IL-8 pathway, mTOR, PI3K and MAPK pathways are preferably activated in the CD133+ cells.
References
Miraglia S, Godfrey W, Yin AH, Atkins K, Warnke R, Holden JT, Bray RA, Waller EK, Buck DW: A novel five-transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood 1997,90(12):5013–5021.
Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, Olweus J, Kearney J, Buck DW: AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 1997,90(12):5002–5012.
Weigmann A, Corbeil D, Hellwig A, Huttner WB: Prominin, a novel microvilli-specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of non-epithelial cells. Proc Natl Acad Sci U S A 1997,94(23):12425–12430.
Shmelkov SV, St Clair R, Lyden D, Rafii S: AC133/CD133/Prominin-1. Int J Biochem Cell Biol 2005,37(4):715–719.
Corbeil D, Roper K, Fargeas CA, Joester A, Huttner WB: Prominin: a story of cholesterol, plasma membrane protrusions and human pathology. Traffic 2001,2(2):82–91.
Lardon J, Corbeil D, Huttner WB, Ling Z, Bouwens L: Stem cell marker prominin-1/AC133 is expressed in duct cells of the adult human pancreas. Pancreas 2008,36(1):e1-e6.
Campos B, Herold-Mende CC: Insight into the complex regulation of CD133 in glioma. Int J Cancer 2011,128(3):501–510.
Lottaz C, Beier D, Meyer K, Kumar P, Hermann A, Schwarz J, Junker M, Oefner PJ, Bogdahn U, Wischhusen J, et al.: Transcriptional profiles of CD133+ and CD133- glioblastoma-derived cancer stem cell lines suggest different cells of origin. Cancer Res 2010,70(5):2030–2040.
Wu Y, Wu PY: CD133 as a marker for cancer stem cells: progresses and concerns. Stem Cells Dev 2009,18(8):1127–1134.
Griguer CE, Oliva CR, Gobin E, Marcorelles P, Benos DJ, Lancaster JR Jr, Gillespie GY: CD133 is a marker of bioenergetic stress in human glioma. PLoS One 2008,3(11):e3655.
Hashimoto O, Shimizu K, Semba S, Chiba S, Ku Y, Yokozaki H, Hori Y: Hypoxia induces tumor aggressiveness and the expansion of CD133-positive cells in a hypoxia-inducible factor-1alpha-dependent manner in pancreatic cancer cells. Pathobiology 2011,78(4):181–192.
Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, Engh J, Iwama T, Kunisada T, Kassam AB, et al.: Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 2009,28(45):3949–3959.
Vadysirisack DD, Ellisen LW: mTOR activity under hypoxia. Methods Mol Biol 2012, 821: 45–58.
Matsumoto K, Arao T, Tanaka K, Kaneda H, Kudo K, Fujita Y, Tamura D, Aomatsu K, Tamura T, Yamada Y, et al.: mTOR signal and hypoxia-inducible factor-1 alpha regulate CD133 expression in cancer cells. Cancer Res 2009,69(18):7160–7164.
Yang Z, Zhang L, Ma A, Liu L, Li J, Gu J, Liu Y: Transient mTOR inhibition facilitates continuous growth of liver tumors by modulating the maintenance of CD133+ cell populations. PLoS One 2011,6(12):e28405.
You H, Ding W, Rountree CB: Epigenetic regulation of cancer stem cell marker CD133 by transforming growth factor-beta. Hepatology 2010,51(5):1635–1644.
Grimm M, Kim M, Rosenwald A, Heemann U, Germer CT, Waaga-Gasser AM, Gasser M: Toll-like receptor (TLR) 7 and TLR8 expression on CD133+ cells in colorectal cancer points to a specific role for inflammation-induced TLRs in tumourigenesis and tumour progression. Eur J Cancer 2010,46(15):2849–2857.
Storci G, Sansone P, Mari S, D'Uva G, Tavolari S, Guarnieri T, Taffurelli M, Ceccarelli C, Santini D, Chieco P, et al.: TNFalpha up-regulates SLUG via the NF-kappaB/HIF1alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J Cell Physiol 2010,225(3):682–691.
Alison MR, Lovell MJ: Liver cancer: the role of stem cells. Cell Prolif 2005,38(6):407–421.
Ma S, Tang KH, Chan YP, Lee TK, Kwan PS, Castilho A, Ng I, Man K, Wong N, To KF, et al.: miR-130b Promotes CD133(+) liver tumor-initiating cell growth and self-renewal via tumor protein 53-induced nuclear protein 1. Cell Stem Cell 2010,7(6):694–707.
Shi L, Wan Y, Sun G, Gu X, Qian C, Yan W, Zhang S, Pan T, Wang Z, You Y: Functional differences of miR-125b on the invasion of primary glioblastoma CD133-negative cells and CD133-positive cells. Neuromolecular Med 2012,14(4):303–316.
Bissels U, Wild S, Tomiuk S, Hafner M, Scheel H, Mihailovic A, Choi YH, Tuschl T, Bosio A: Combined characterization of microRNA and mRNA profiles delineates early differentiation pathways of CD133+ and CD34+ hematopoietic stem and progenitor cells. Stem Cells 2011,29(5):847–857.
Garzia L, Andolfo I, Cusanelli E, Marino N, Petrosino G, De Martino D, Esposito V, Galeone A, Navas L, Esposito S, et al.: MicroRNA-199b-5p impairs cancer stem cells through negative regulation of HES1 in medulloblastoma. PLoS One 2009,4(3):e4998.
Huang S, Guo W, Tang Y, Ren D, Zou X, Peng X: miR-143 and miR-145 inhibit stem cell characteristics of PC-3 prostate cancer cells. Oncol Rep 2012,28(5):1831–1837.
Zhang J, Luo N, Luo Y, Peng Z, Zhang T, Li S: microRNA-150 inhibits human CD133-positive liver cancer stem cells through negative regulation of the transcription factor c-Myb. Int J Oncol 2012,40(3):747–756.
Hibi K, Sakata M, Kitamura YH, Sakuraba K, Shirahata A, Goto T, Mizukami H, Saito M, Ishibashi K, Kigawa G, et al.: Demethylation of the CD133 gene is frequently detected in advanced colorectal cancer. Anticancer Res 2009,29(6):2235–2237.
Hibi K, Sakata M, Kitamura YH, Sakuraba K, Shirahata A, Goto T, Mizukami H, Saito M, Ishibashi K, Kigawa G, et al.: Demethylation of the CD133 gene is frequently detected in early gastric carcinoma. Anticancer Res 2010,30(4):1201–1203.
Tabu K, Sasai K, Kimura T, Wang L, Aoyanagi E, Kohsaka S, Tanino M, Nishihara H, Tanaka S: Promoter hypomethylation regulates CD133 expression in human gliomas. Cell Res 2008,18(10):1037–1046.
Yi JM, Tsai HC, Glockner SC, Lin S, Ohm JE, Easwaran H, James CD, Costello JF, Riggins G, Eberhart CG, et al.: Abnormal DNA methylation of CD133 in colorectal and glioblastoma tumors. Cancer Res 2008,68(19):8094–8103.
Zhang C, Xu Y, Zhao J, Fan L, Jiang G, Li R, Ling Y, Wu M, Wei L: Elevated Expression of the Stem Cell Marker CD133 Associated with Line-1 Demethylation in Hepatocellular Carcinoma. Ann Surg Oncol 2011,18(8):2373–2380.
Baba T, Convery PA, Matsumura N, Whitaker RS, Kondoh E, Perry T, Huang Z, Bentley RC, Mori S, Fujii S, et al.: Epigenetic regulation of CD133 and tumorigenicity of CD133+ ovarian cancer cells. Oncogene 2009,28(2):209–218.
Kemper K, Versloot M, Cameron K, Colak S, DeSousa EMelo F, DeJong JH, Bleackley J, Vermeulen L, Versteeg R, Koster J: Mutations in the Ras-Raf Axis underlie the prognostic value of CD133 in colorectal cancer. Clin Cancer Res 2012,18(11):3132–3141.
Fang DD, Kim YJ, Lee CN, Aggarwal S, McKinnon K, Mesmer D, Norton J, Birse CE, He T, Ruben SM, et al.: Expansion of CD133(+) colon cancer cultures retaining stem cell properties to enable cancer stem cell target discovery. Br J Cancer 2010,102(8):1265–1275.
Shi CJ, Gao J, Wang M, Wang X, Tian R, Zhu F, Shen M, Qin RY: CD133(+) gallbladder carcinoma cells exhibit self-renewal ability and tumorigenicity. World J Gastroenterol 2011,17(24):2965–2971.
Kusumbe AP, Mali AM, Bapat SA: CD133-expressing stem cells associated with ovarian metastases establish an endothelial hierarchy and contribute to tumor vasculature. Stem Cells 2009,27(3):498–508.
Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY: CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008,27(12):1749–1758.
Tang KH, Ma S, Lee TK, Chan YP, Kwan PS, Tong CM, Ng IO, Man K, To KF, Lai PB, et al.: CD133(+) liver tumor-initiating cells promote tumor angiogenesis, growth, and self-renewal through neurotensin/interleukin-8/CXCL1 signaling. Hepatology 2012,55(3):807–820.
Boivin D, Labbe D, Fontaine N, Lamy S, Beaulieu E, Gingras D, Beliveau R: The stem cell marker CD133 (prominin-1) is phosphorylated on cytoplasmic tyrosine-828 and tyrosine-852 by Src and Fyn tyrosine kinases. Biochemistry 2009,48(18):3998–4007.
Wei Y, Jiang Y, Zou F, Liu Y, Wang S, Xu N, Xu W, Cui C, Xing Y, Cao B, et al.: Activation of PI3K/Akt pathway by CD133-p85 interaction promotes tumorigenic capacity of glioma stem cells. Proc Natl Acad Sci U S A 2013,110(17):6829–6834.
Neuzil J, Stantic M, Zobalova R, Chladova J, Wang X, Prochazka L, Dong L, Andera L, Ralph SJ: Tumour-initiating cells vs. cancer 'stem' cells and CD133: what's in the name? Biochem Biophys Res Commun 2007,355(4):855–859.
Shmelkov SV, Butler JM, Hooper AT, Hormigo A, Kushner J, Milde T, St Clair R, Baljevic M, White I, Jin DK, et al.: CD133 expression is not restricted to stem cells, and both CD133+ and CD133- metastatic colon cancer cells initiate tumors. J Clin Invest 2008,118(6):2111–2120.
Wang YK, Zhu YL, Qiu FM, Zhang T, Chen ZG, Zheng S, Huang J: Activation of Akt and MAPK pathways enhances the tumorigenicity of CD133+ primary colon cancer cells. Carcinogenesis 2010,31(8):1376–1380.
Borrego-Diaz E, Terai K, Lialyte K, Wise AL, Esfandyari T, Behbod F, Mautner VF, Spyra M, Taylor S, Parada LF, et al.: Overactivation of Ras signaling pathway in CD133+ MPNST cells. J Neurooncol 2012,108(3):423–434.
Iida H, Suzuki M, Goitsuka R, Ueno H: Hypoxia induces CD133 expression in human lung cancer cells by up-regulation of OCT3/4 and SOX2. Int J Oncol 2012,40(1):71–79.
Zhang SS, Han ZP, Jing YY, Tao SF, Li TJ, Wang H, Wang Y, Li R, Yang Y, Zhao X, et al.: CD133+CXCR4+ colon cancer cells exhibit metastatic potential and predict poor prognosis of patients. BMC Med 2012, 10: 85.
Chen KL, Pan F, Jiang H, Chen JF, Pei L, Xie FW, Liang HJ: Highly enriched CD133(+)CD44(+) stem-like cells with CD133(+)CD44(high) metastatic subset in HCT116 colon cancer cells. Clin Exp Metastasis 2011,28(8):751–763.
Haraguchi N, Ohkuma M, Sakashita H, Matsuzaki S, Tanaka F, Mimori K, Kamohara Y, Inoue H, Mori M: CD133+CD44+ population efficiently enriches colon cancer initiating cells. Ann Surg Oncol 2008,15(10):2927–2933.
Huang X, Sheng Y, Guan M: Co-expression of stem cell genes CD133 and CD44 in colorectal cancers with early liver metastasis. Surg Oncol 2012,21(2):103–107.
Hou Y, Zou Q, Ge R, Shen F, Wang Y: The critical role of CD133(+)CD44(+/high) tumor cells in hematogenous metastasis of liver cancers. Cell Res 2012,22(1):259–272.
Bellizzi A, Sebastian S, Ceglia P, Centonze M, Divella R, Manzillo EF, Azzariti A, Silvestris N, Montemurro S, Caliandro C, et al.: Co-expression of CD133(+)/CD44(+) in human colon cancer and liver metastasis. J Cell Physiol 2013,228(2):408–415.
Wang D, Zhu H, Zhu Y, Liu Y, Shen H, Yin R, Zhang Z, Su Z: CD133(+)/CD44(+)/Oct4(+)/Nestin(+) stem-like cells isolated from Panc-1 cell line may contribute to multi-resistance and metastasis of pancreatic cancer. Acta Histochem 2013,115(4):349–356.
Shi C, Tian R, Wang M, Wang X, Jiang J, Zhang Z, Li X, He Z, Gong W, Qin R: CD44+ CD133+ population exhibits cancer stem cell-like characteristics in human gallbladder carcinoma. Cancer Biol Ther 2010,10(11):1182–1190.
Okudela K, Woo T, Mitsui H, Tajiri M, Masuda M, Ohashi K: Expression of the potential cancer stem cell markers, CD133, CD44, ALDH1, and beta-catenin, in primary lung adenocarcinoma–their prognostic significance. Pathol Int 2012,62(12):792–801.
Wakamatsu Y, Sakamoto N, Oo HZ, Naito Y, Uraoka N, Anami K, Sentani K, Oue N, Yasui W: Expression of cancer stem cell markers ALDH1, CD44 and CD133 in primary tumor and lymph node metastasis of gastric cancer. Pathol Int 2012,62(2):112–119.
Kohga K, Tatsumi T, Takehara T, Tsunematsu H, Shimizu S, Yamamoto M, Sasakawa A, Miyagi T, Hayashi N: Expression of CD133 confers malignant potential by regulating metalloproteinases in human hepatocellular carcinoma. J Hepatol 2010,52(6):872–879.
Zhang M, Liu Y, Feng H, Bian X, Zhao W, Yang Z, Gu B, Li Z: CD133 Affects the Invasive Ability of HCT116 Cells by Regulating TIMP-2. Am J Pathol 2013,182(2):565–576.
Long H, Xie R, Xiang T, Zhao Z, Lin S, Liang Z, Chen Z, Zhu B: Autocrine CCL5 signaling promotes invasion and migration of CD133+ ovarian cancer stem-like cells via NF-kappaB-mediated MMP-9 upregulation. Stem Cells 2012,30(10):2309–2319.
Damdinsuren B, Nagano H, Kondo M, Natsag J, Hanada H, Nakamura M, Wada H, Kato H, Marubashi S, Miyamoto A, et al.: TGF-beta1-induced cell growth arrest and partial differentiation is related to the suppression of Id1 in human hepatoma cells. Oncol Rep 2006,15(2):401–408.
Zhang Q, Shi S, Yen Y, Brown J, Ta JQ, Le AD: A subpopulation of CD133(+) cancer stem-like cells characterized in human oral squamous cell carcinoma confer resistance to chemotherapy. Cancer Lett 2010,289(2):151–160.
Angelastro JM, Lame MW: Overexpression of CD133 promotes drug resistance in C6 glioma cells. Mol Cancer Res 2010,8(8):1105–1115.
Frank NY, Margaryan A, Huang Y, Schatton T, Waaga-Gasser AM, Gasser M, Sayegh MH, Sadee W, Frank MH: ABCB5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res 2005,65(10):4320–4333.
Cheung ST, Cheung PF, Cheng CK, Wong NC, Fan ST: Granulin-Epithelin Precursor and ATP-Dependent Binding Cassette (ABC)B5 Regulate Liver Cancer Cell Chemoresistance. Gastroenterology 2011,140(1):344–355. e342
Piao LS, Hur W, Kim TK, Hong SW, Kim SW, Choi JE, Sung PS, Song MJ, Lee BC, Hwang D, et al.: CD133(+) liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett 2012,315(2):129–137.
Corbeil D, Marzesco AM, Wilsch-Brauninger M, Huttner WB: The intriguing links between prominin-1 (CD133), cholesterol-based membrane microdomains, remodeling of apical plasma membrane protrusions, extracellular membrane particles, and (neuro)epithelial cell differentiation. FEBS Lett 2010,584(9):1659–1664.
Dubreuil V, Marzesco AM, Corbeil D, Huttner WB, Wilsch-Brauninger M: Midbody and primary cilium of neural progenitors release extracellular membrane particles enriched in the stem cell marker prominin-1. J Cell Biol 2007,176(4):483–495.
Roper K, Corbeil D, Huttner WB: Retention of prominin in microvilli reveals distinct cholesterol-based lipid micro-domains in the apical plasma membrane. Nat Cell Biol 2000,2(9):582–592.
Corbeil D, Marzesco AM, Fargeas CA, Huttner WB: Prominin-1: a distinct cholesterol-binding membrane protein and the organisation of the apical plasma membrane of epithelial cells. Subcell Biochem 2010, 51: 399–423.
Bourseau-Guilmain E, Griveau A, Benoit JP, Garcion E: The Importance of the Stem Cell Marker Prominin-1/CD133 in the Uptake of Transferrin and in Iron Metabolism in Human Colon Cancer Caco-2 Cells. PLoS One 2011,6(9):e25515.
Gong L, Cui Z, Yu X, Wei Y, Peng J, Leng X: Hexokinase II in CD133+ and CD133- hepatoma BEL-7402 Cells. Pathol Oncol Res 2012,18(2):377–381.
Lyshchik A, Higashi T, Hara T, Nakamoto Y, Fujimoto K, Doi R, Imamura M, Saga T, Togashi K: Expression of glucose transporter-1, hexokinase-II, proliferating cell nuclear antigen and survival of patients with pancreatic cancer. Cancer Invest 2007,25(3):154–162.
Kwee SA, Hernandez B, Chan O, Wong L: Choline kinase alpha and hexokinase-2 protein expression in hepatocellular carcinoma: association with survival. PLoS One 2012,7(10):e46591.
Zhou F, Cui C, Ge Y, Chen H, Li Q, Yang Z, Wu G, Sun S, Chen K, Gu J, et al.: Alpha2,3-Sialylation regulates the stability of stem cell marker CD133. J Biochem 2010,148(3):273–280.
Swindall AF, Londono-Joshi AI, Schultz MJ, Fineberg N, Buchsbaum DJ, Bellis SL: ST6Gal-I protein expression is upregulated in human epithelial tumors and correlates with stem cell markers in normal tissues and colon cancer cell lines. Cancer Res 2013,73(7):2368–2378.
Hemmoranta H, Satomaa T, Blomqvist M, Heiskanen A, Aitio O, Saarinen J, Natunen J, Partanen J, Laine J, Jaatinen T: N-glycan structures and associated gene expression reflect the characteristic N-glycosylation pattern of human hematopoietic stem and progenitor cells. Exp Hematol 2007,35(8):1279–1292.
Kemper K, Sprick MR, de Bree M, Scopelliti A, Vermeulen L, Hoek M, Zeilstra J, Pals ST, Mehmet H, Stassi G, et al.: The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res 2010,70(2):719–729.
Lehnus KS, Donovan LK, Huang X, Zhao N, Warr TJ, Pilkington GJ, An Q: CD133 glycosylation is enhanced by hypoxia in cultured glioma stem cells. Int J Oncol 2013,42(3):1011–1017.
Chen H, Luo Z, Dong L, Tan Y, Yang J, Feng G, Wu M, Li Z, Wang H: CD133/prominin-1-mediated autophagy and glucose uptake beneficial for hepatoma cell survival. PLoS One 2013,8(2):e56878.
Shepherd CJ, Rizzo S, Ledaki I, Davies M, Brewer D, Attard G, de Bono J, Hudson DL: Expression profiling of CD133+ and CD133- epithelial cells from human prostate. Prostate 2008,68(9):1007–1024.
Jaatinen T, Hemmoranta H, Hautaniemi S, Niemi J, Nicorici D, Laine J, Yli-Harja O, Partanen J: Global gene expression profile of human cord blood-derived CD133+ cells. Stem Cells 2006,24(3):631–641.
Yan X, Ma L, Yi D, Yoon JG, Diercks A, Foltz G, Price ND, Hood LE, Tian Q: A CD133-related gene expression signature identifies an aggressive glioblastoma subtype with excessive mutations. Proc Natl Acad Sci U S A 2011,108(4):1591–1596.
Hemmoranta H, Hautaniemi S, Niemi J, Nicorici D, Laine J, Yli-Harja O, Partanen J, Jaatinen T: Transcriptional profiling reflects shared and unique characters for CD34+ and CD133+ cells. Stem Cells Dev 2006,15(6):839–851.
Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ, Aigner L, Brawanski A, Bogdahn U, Beier CP: CD133(+) and CD133(−) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res 2007,67(9):4010–4015.
Takenobu H, Shimozato O, Nakamura T, Ochiai H, Yamaguchi Y, Ohira M, Nakagawara A, Kamijo T: CD133 suppresses neuroblastoma cell differentiation via signal pathway modification. Oncogene 2011,30(1):97–105.
Aghideh AN, Kheirandish M, Abolghasemi H, Gharehbaghian A: Platelet growth factors suppress ex vivo expansion and enhance differentiation of umbilical cord blood CD133+ stem cells to megakaryocyte progenitor cells. Growth Factors 2010,28(6):409–416.
Feng JM, Miao ZH, Jiang Y, Chen Y, Li JX, Tong LJ, Zhang J, Huang YR, Ding J: Characterization of the conversion between CD133 (+) and CD133 (−) cells in colon cancer SW620 cell line. Cancer Biol Ther 2012, 13: 14.
Ding W, Mouzaki M, You H, Laird JC, Mato J, Lu SC, Rountree CB: CD133+ liver cancer stem cells from methionine adenosyl transferase 1A-deficient mice demonstrate resistance to transforming growth factor (TGF)-beta-induced apoptosis. Hepatology 2009,49(4):1277–1286.
Zobalova R, Stantic M, Prokopova K, Dong LF, Neuzil J: Cancer cells with high expression of CD133 exert FLIP upregulation and resistance to TRAIL-induced apoptosis. Biofactors 2008,34(3):231–235.
Zobalova R, McDermott L, Stantic M, Prokopova K, Dong LF, Neuzil J: CD133-positive cells are resistant to TRAIL due to up-regulation of FLIP. Biochem Biophys Res Commun 2008,373(4):567–571.
Elsaba TM, Martinez-Pomares L, Robins AR, Crook S, Seth R, Jackson D, McCart A, Silver AR, Tomlinson IP, Ilyas M: The stem cell marker CD133 associates with enhanced colony formation and cell motility in colorectal cancer. PLoS One 2010,5(5):e10714.
Zobalova R, Prokopova K, Stantic M, Stapelberg M, Dong LF, Ralph SJ, Akporiaye E, Neuzil J: The potential role of CD133 in immune surveillance and apoptosis: a mitochondrial connection? Antioxid Redox Signal 2011,15(12):2989–3002.
Nishimura-Sakurai Y, Sakamoto N, Mogushi K, Nagaie S, Nakagawa M, Itsui Y, Tasaka-Fujita M, Onuki-Karakama Y, Suda G, Mishima K, et al.: Comparison of HCV-associated gene expression and cell signaling pathways in cells with or without HCV replicon and in replicon-cured cells. J Gastroenterol 2010,45(5):523–536.
Paprocka M, Krawczenko A, Dus D, Kantor A, Carreau A, Grillon C, Kieda C: CD133 positive progenitor endothelial cell lines from human cord blood. Cytometry A 2011,79(8):594–602.
Kamei N, Kwon SM, Alev C, Nakanishi K, Yamada K, Masuda H, Ishikawa M, Kawamoto A, Ochi M, Asahara T: Ex-vivo expanded human blood-derived CD133(+) cells promote repair of injured spinal cord. J Neurol Sci 2013,328(1–2):41–50.
Kijima Y, Ishikawa M, Sunagawa T, Nakanishi K, Kamei N, Yamada K, Tanaka N, Kawamata S, Asahara T, Ochi M: Regeneration of peripheral nerve after transplantation of CD133+ cells derived from human peripheral blood. J Neurosurg 2009,110(4):758–767.
Shi M, Ishikawa M, Kamei N, Nakasa T, Adachi N, Deie M, Asahara T, Ochi M: Acceleration of skeletal muscle regeneration in a rat skeletal muscle injury model by local injection of human peripheral blood-derived CD133-positive cells. Stem Cells 2009,27(4):949–960.
Adler DS, Lazarus H, Nair R, Goldberg JL, Greco NJ, Lassar T, Laughlin MJ, Das H, Pompili VJ: Safety and efficacy of bone marrow-derived autologous CD133+ stem cell therapy. Front Biosci 2011, 3: 506–514.
Ahmadi H, Baharvand H, Ashtiani SK, Soleimani M, Sadeghian H, Ardekani JM, Mehrjerdi NZ, Kouhkan A, Namiri M, Madani-Civi M, et al.: Safety analysis and improved cardiac function following local autologous transplantation of CD133(+) enriched bone marrow cells after myocardial infarction. Curr Neurovasc Res 2007,4(3):153–160.
Nikeghbalian S, Pournasr B, Aghdami N, Rasekhi A, Geramizadeh B, Hosseini Asl SM, Ramzi M, Kakaei F, Namiri M, Malekzadeh R, et al.: Autologous transplantation of bone marrow-derived mononuclear and CD133(+) cells in patients with decompensated cirrhosis. Arch Iran Med 2011,14(1):12–17.
AmEsch JS, Knoefel WT, Klein M, Ghodsizad A, Fuerst G, Poll LW, Piechaczek C, Burchardt ER, Feifel N, Stoldt V, et al.: Portal application of autologous CD133+ bone marrow cells to the liver: a novel concept to support hepatic regeneration. Stem Cells 2005,23(4):463–470.
Ceballos MP, Parody JP, Alvarez Mde L, Ingaramo PI, Carnovale CE, Carrillo MC: Interferon-alpha2b and transforming growth factor-beta1 treatments on HCC cell lines: Are Wnt/beta-catenin pathway and Smads signaling connected in hepatocellular carcinoma? Biochem Pharmacol 2011,82(11):1682–1691.
Cao Y, Zhang W, Gao X, Zhang G, Falzon M, Townsend CM Jr, Hellmich MR, Ko TC: PTHrP is a novel mediator for TGF-beta-induced apoptosis. Regul Pept 2013, 184: 40–46.
Ueno T, Hashimoto O, Kimura R, Torimura T, Kawaguchi T, Nakamura T, Sakata R, Koga H, Sata M: Relation of type II transforming growth factor-beta receptor to hepatic fibrosis and hepatocellular carcinoma. Int J Oncol 2001,18(1):49–55.
Acknowledgments
The author is supported by fundings from the National Natural Science Foundation of China (81041070 and 81272427 to Z Li), National Key Basic Research Program of China (2013CB910500 to Z Li) and the startup fund from Eastern Hepatobiliary Surgery Hospital and the 10th People’s Hospital.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares that he has no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Li, Z. CD133: a stem cell biomarker and beyond. Exp Hematol Oncol 2, 17 (2013). https://doi.org/10.1186/2162-3619-2-17
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2162-3619-2-17