
Abstract
MicroRNAs (miRNAs or miRs) are a family of small non-coding RNA species that have been implicated in the control of many fundamental cellular and physiological processes such as cellular differentiation, proliferation, apoptosis and stem cell maintenance. miRNAs regulate gene expression by the sequence-selective targeting of mRNAs, leading to translational repression or mRNA degradation. Some microRNAs have been categorized as “oncomiRs” as opposed to “tumor suppressor miRs” Modulating the miRNA activities may provide exciting opportunities for cancer therapy. This review highlights the latest discovery of miRNAs involved in carcinogenesis as well as the potential applications of miRNA regulations in cancer treatment. Several studies have demonstrated the feasibility of restoring tumor suppressive miRNAs and targeting oncogenic miRNAs for cancer therapy using in vivo model systems.
Similar content being viewed by others

Review
Introduction
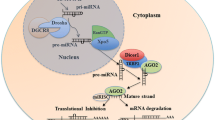
In 1993, Lee and coworkers described that a small non-coding RNA was able to regulate the expression and function of another protein-coding RNA [1]. This change in paradigms has made a profound impact in our current understanding of gene regulation. miRNAs are usually 18–25 nucleotides long and highly conserved during evolution (reviewed in [2]) miRNA genes are generally transcribed by RNA polymerase II into long primary transcripts, up to several kilobases (pri-miRNA) [3]. These are subsequently processed in the nucleus by a microprocessor complex, which contains the RNase III enzyme Drosha [4] and DGCR8 [5] to become the so-called pre-miR (around 70 nucleotides long). These precursors are exported by exportin 5 and a RanGTP [6, 7] from the nucleus to the cytoplasm where they are bound to the RNase III enzyme Dicer and the RNA-induced silencing complex (RISC) [8]. RISC is composed of the transactivation-responsive RNA-binding protein (TRBP) and Argonaute 2 (Ago2) [9, 10]. Ago2 cleaves the pre-miRNA 12 nucleotides from its 3’ end and then the Dicer cleaves the Ago2-cleaved precursor miRNA into the double-stranded miRNA [11]. While the active mature strand is retained in RISC, the passenger strand is removed and degraded [12] miRNAs mainly bind to the 3’ untranslated region (UTR) of their target mRNAs. However, recent studies have reported that miRNAs also bind to 5’UTR [13], or open reading frame (ORF) [14] of the target mRNA. By binding to their target mRNA, miRNAs regulate the translation of proteins from mRNA or degrade the mRNA itself [9–11]. Depending on the degree of homology to their 3’UTR target sequence, miRNAs induce translational repression or degradation of mRNAs. miRNAs modulate gene expression impacting all cell functions, including apoptosis, proliferation, cell cycle, differentiation, stem cell maintenance and metabolism [15]. It is estimated that more than 1000 miRNAs are transcribed and that 30% of the human genome is under miRNA regulation, one miRNA being able to modulate post-transcriptionally hundreds of downstream genes.
MicroRNAs and cancer
More than half of the miRNAs genes are located in cancer-associated genomic regions or in fragile sites. Specific miRNA signatures have been associated with distinct subsets of solid tumors and hematological malignancies [15] miRNAs can act as tumor suppressors when their function loss can initiate or contribute to the malignant transformation of a normal cell. The loss of function of a miRNA could be due to several mechanisms, including genomic deletion, mutation, epigenetic silencing, and/or miRNA processing alterations [16–19]. On the other hand, miRNAs can act as oncogenic microRNAs by targeting mRNAs encoding tumor suppressor proteins.
The let-7 family of miRNAs is a typical tumor suppressor and is therefore downregulated in many tumors, including lung and breast cancer [20, 21]. Many of the let-7 family members are located in fragile genomic areas associated with lung, breast, and cervical cancer [22]. Furthermore, let-7 family members functionally inhibit the mRNAs of well-characterized oncogenes, such as RAS [23, 24], HMGA2 [25], and c-Myc [26]. The miR-29 family comprises three isoforms arranged in two clusters: miR-29b-1/miR-29a in chromosome 7q32 and miR-29b-2/miR-29c in chromosome 1q23. miR-29 family members have been shown to b downregulated in chronic lymphatic leukemia (CLL), acute myeloid leukemia (AML), lung cancer, breast cancer, and cholangiocarcinoma [17, 20, 21, 27, 28].
miR-155 was one of the first described oncogenic miRNAs [29, 30] and it is highly expressed in a variety of tumors [17, 20, 21, 28–31]. The miR-155 gene is located in chromosome 21q23 embedded in a host noncoding RNA named the B cell integration cluster (BIC) [32]. BIC is known to cooperate with c-Myc in oncogenesis. Another widely expressed miRNA in hematopoietic and solid tumors is miRNA-21 [17, 28, 31, 33–35] miR-21 targets several tumor suppressor genes such as phosphatase and tensin homolog (PTEN) [34], programmed cell death 4 (PDCD4) [36], and tropomyosin 1 (TPM1) [37]. The miR-17-92 cluster (miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, miR-92-1) is located at 13q31.3 in a region that is frequently amplified in follicular lymphoma and diffuse large B cell lymphoma [38]. Members of the miR-17-92 cluster are highly expressed in a variety of solid tumors and hematological malignancies [39], Interestingly, the miR-17-92 cluster is transactivated by c-Myc, a frequently activated oncogene in cancer [40].
Recently, miRNAs have been found to foster tumor progression by the mediation of inflammation processes through regulation of components of the innate immune system. Two recent studies described the miRNAs miR-21 and miR-29a to serve as ligands for Toll-like receptor (TLR) activation. Fabbri et al. showed that tumor-originating extracellular miRNA could bind to murine TLR7 and human TLR8 to cause a proinflammatory response leading to tumor progression both in vitro and in vivo [41]. In a separate study, Lehmann et al. showed that extracellular let-7 could activate TLR7 to induce neurodegeneration [42]. These off-target effects might be overcome by chemical modifications and improved delivery systems as discussed in one of the subsequent paragraphs.
Targeting microRNAs in cancer
General aspects of miRNA therapeutics
Every miRNA has multiple target sites in different genes (on average about 500 for each miRNA family). Reciprocally, about two third of all mRNAs have one or more evolutionarily conserved sequences that are predicted to interact with miRNAs [43–46]. The rationale for using miRNAs as therapeutic agents is based on the two following criteria. (1) miRNA expression is dysregulated in cancer compared to normal tissues and (2) the cancer phenotype can be changed by targeting miRNA expression [16, 20, 21, 24, 28, 47–50]. Compared to other strategies, miRNA-based therapeutics have several advantages, as for example the fact that miRNAs as therapeutic agents have the ability to target multiple genes, frequently in the context of a network. The challenges for microRNA-based are the same as the challenges for small interfering RNA therapeutics and include issues of delivery, potential off-target effects and safety. One of the major obstacles for the use of miRNA therapeutics is the tissue-specific delivery [51, 52]. Moreover, the fact that one miRNA targets multiple genes is also a drawback as the potential off-target effects may cause toxic phenotypes [51, 53]. The fact that some biological functions of miRNAs may be partially redundant, or cell-type dependent, is another relevant issue in the development of miRNA therapeutics [54]. Although successful delivery is an obstacle to effective miRNA-based therapeutics, new findings from recent trials and the rapid advances in systemic drug delivery systems provide an optimistic perspective on the progress in this field [55].
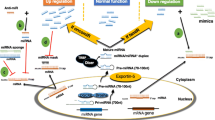
In general, miRNA therapeutic approaches can be divided into two different categories: (1) miRNA inhibition therapy when the target miRNA is overexpressed and (2) miRNA replacement therapy when the miRNA is repressed. Therapeutic targeting of microRNAs can be accomplished either by direct inhibition or replacement of miRNAs or by targeting specific genes and therefore regulating the expression of specific miRNAs. For this purpose small-interfering RNAs (siRNAs) and genetically encoded expression vectors encoding small hairpin RNAs (shRNAs) are used [56].
microRNA modifications
There are different hurdles to develop miRNA-based treatment approaches. One is that RNAs in general have low stability in vivo. Thus, miRNA introduced into mice via the tail vein is cleared from the circulatory system within 30 minutes [57]. Unmodified RNAs undergo degradation by RNases and then rapid renal excretion [58]. Therefore, the plasma half-life of RNAs needs to be increased for clinical use of miRNA-based therapeutic approaches. An improvement could be reached by higher miRNA stability or by protection from RNases. By using chemically modified oligonucleotides the stability of the antisense sequences is augmented [59, 60]. In the following established chemical modifications are listed: locked nucleic acid (LNA) oligonucleotides [61], phosphorothioate containing oligonucleotides [62], 2’–O-methyl-(2’–O-Me) or 2’–O-methoxyethyl-oligonucleotides (2’–O-MOE) [63], peptide nucleic acids (PNA) [64], fluorine derivatives (FANA and 2’-F) and other chemical modifications [65]. LNAs are oligonucleotides complementary to the sequence of the targeted mature miRNA, but biochemically modified to reduce the risk of degradation by cellular RNases. LNAs have an extra bridge connecting the 2’ oxygen and the 4’ carbon. PNAs are uncharged oligonucleotides analogs in which the sugar-phosphate backbone is replaced by an achiral structure consisting of N-(2-aminoethyl)-glycine units [66–70]. These molecules form a double helix by hybridization with complementary DNA and RNA [66, 67] PNAs have a higher affinity to RNA than to DNA and are resistant to DNases and proteases [67]. This technology has been investigated in different studies [70, 71] PNA targeting miR-221 has shown to specifically interact with miR-221 expressed in aggressive breast cancer cell lines [72] PNA-anti-miR-21 has recently been shown to reduce breast cancer metastasis in an animal model [73]. It is well established that cancers are often driven by deregulation of various miRNAs or families of miRNAs. Therefore, methods to silence multiple miRNAs have been investigated. Tiny 8-mer LNAs with a phosphorothioate backbone to enhance stability have been developed [74].
miRNA delivery systems
Beside chemical modification of miRNAs to avoid rapid degradation and excretion, the development of improved delivery systems leads to enhanced stability and more precise delivery of miRNAs. miRNAs can be conjugated to a cholesterol moiety, increasing stability in the circulation and facilitating cell entry [75]. A further mechanism of protection is to enclose miRNA mimics or LNAs into nanoparticles to form micelle-like structures. Liposome-nanoparticles are phospholipid structures that are capable of incorporating various types of nucleic acids and charged small molecules, such as microRNAs, siRNAs, shRNAs, plasmid DNA, and protein, within the aqueous core of the liposome [76]. The major drawback of liposomes are nonspecific uptake and induction of immune response [77]. Polycationic liposome-hyaluronic acid (LPH) nanoparticles have also been used as miRNA carriers [78]. Using LPH particles as a carrier for miR-34a significantly reduced lung metastases in a murine melanoma model [76]. It has been shown that systemic administration of positively charged lipid nanoparticles in vivo is toxic and stimulates inflammatory response by elevating both Th1 and Th17 cytokines and interferon responsive genes [79]. Clearly, a complete understanding of the best liposomal design for delivery of therapeutic substances is still evolving. It is possible that with nucleic acid delivery the use of a neutral lipid, such as 1,2-Dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC) will have several advantages [80]. Other delivery systems used for microRNAs are polyethylenimine (PEI)-based systems [81–83], dendrimers [84–86], poly(lactide-co-glycolide) (PLGA) particles [87, 88], protamine [89], atelocollagen [90–92], as well as inorganic materials (e.g. gold [93, 94] and silica-based nanoparticles [95]).
Another hurdle in the design and application of miRNA therapeutics is to ensure tumor-specific delivery. Due to the fact that most miRNAs target many different mRNAs, off-target effects are a substantial problem. Targeted delivery to specific tissues can be achieved by binding tumor-specific ligands to nanoparticles, which can be directed to tumor cells via active or passive targeting. Active targeting is achieved by conjugation with different compounds that have a specific affinity to tumors. As an example, cancer cell receptors (EGFR, HER-2) or hyaluronic acid could be used [96–98]. Hyaluronic acid is a polysaccharide that binds to the cancer stem cell marker CD44, which is overexpressed in various tumor cells [99].
miRNA inhibition therapy
Oncogenic microRNAs could be therapeutically targeted by repression and therefore inhibition of the interaction between miRNA and mRNA. A simple method to inhibit miRNAs is the use of oligonucleotides complementary to the mature miRNA (antagomiRs). These oligonucleotides disrupt the miRISC complex and therefore prevent the degradation of the mRNA which can then be translated. microRNA sponges have been developed to inhibit the activity of miRNA families sharing a common seed sequence. miRNA sponges work with multiple complementary 3’UTR mRNA sites of a specific miRNA and saturate the miRISC complex repressing the activity toward natural mRNA [100]. A major drawback of miRNA sponges is the limited homogeneity of transcripts expression and therefore miRNA sponges could lead to serious side effects [101]. Another approach to more specifically inhibit the miRNA function is the use of miRNA masks which are complementary to the binding sites in the 3’UTR of the target mRNA [15]. This method allows a more specific inhibition of the mRNA targeted by a specific miRNA.
microRNA replacement therapy
miRNA replacement therapy aims at substitution of tumor suppressive miRNAs expressed at lower levels by using oligonucleotide mimics containing the same sequence as the mature endogenous miRNA. As double stranded miRNA mimics have a much higher potency as single stranded miRNA mimics they are most often used [102]. The guide strand contains a sequence identical to the mature miRNA and the passenger strand sequence is complementary to the mature miRNA. Additionally to miRNA mimics containing the same sequence as the endogenous miRNA, synthetic miRNA precursor mimics with longer sequences are used [103].
MicroRNA therapeutics
Using a luciferase reporter assay to screen small molecule libraries for a compound that could inhibit the expression of specific oncogenic miRNAs has recently been successful.
OncomiRs
The expression of microRNA miR-122 is confined to the liver, where it constitutes 70% of the total miRNA population [3]. Within the liver, miR-122 has been implicated in cholesterol and lipid metabolism, and was identified as a regulator for systematic iron homeostasis [7, 8]. Moreover, miR-122 has also been demonstrated to be necessary for the replication and infectious production of hepatitis C virus (HCV). Binding of miR-122 to the 5′ noncoding region of the HCV genome upregulates expression, causing accumulation of viral RNA in liver cells [9]. HCV infection is one of the major causes of liver disease worldwide, including cirrhosis and hepatocellular carcinoma [9]. The essential interaction between miR-122 and HCV suggests that miR-122 could be an excellent therapeutic target for the treatment of HCV infections. Anti-miR-122 is the only miRNA-based treatment tested in human beings so far. In 2010, data from a drug trial of an intravenously delivered anti-miR-122 LNA in chimpanzees were reported [104]. Anti-miR-122 LNA given to chronically infected chimpanzees once a week for 12 weeks led to a reduction in viral load in the serum and the liver. Based on these results, a phase 1 trial in 77 healthy volunteers demonstrated the safety of anti-miR-122 application in humans. In the subsequent phase 2 trial the safety and efficacy of the treatment was confirmed [105]. The discovery of small molecule inhibitors of miR-122 function demonstrates a novel approach to inhibit HCV replication in liver cells [10]. A very recent publication describes the development of an assay for the discovery of small molecule regulators of miR-122, and ultimately HCV therapeutics [106].
miR-21 is an oncogene and therefore frequently highly expressed in solid tumors and hematological malignancies [21, 107–116]. Inhibition of miR-21 resulted in reduced cell proliferation accompanied by increased apoptosis in breast and glioblastoma cell lines [117, 118]. Again by performing luciferase reporter assay an inhibitor of miR-21 has been identified. This agent was able to inhibit miR-21 expression and elicit antitumoral effects [119]. miR-21 transfection leads to the downregulation of PTEN and increased signaling through the PI3K-AKT pathway [34].
Members of the miRNA-29 family (miR-29a, miR-29b, and miR-29c) are known to be highly expressed in normal tissues and downregulated in different types of cancer, including neuroblastoma, sarcoma, glioma, high-risk chronic lymphatic leukemia (CLL), invasive breast cancer, cholangiocarcinoma and lung cancer.(35–40) miR-29a has been shown to reduce invasiveness and proliferation of human carcinoma cell lines.(41) The miR-29 family members also target DNA methyltransferases (DNMT3A and DNMT3B), and can thereby restore patterns of DNA methylation and expression of silenced tumor suppressor genes.(31) We recently showed that inhibition of endogenous miR-29b by stable transduction of a lentiviral vector containing an antisense nucleotide in human lung cancer cells caused increase of inhibitor of differentiation 1 (ID1) and Matrix-Metalloproteinase-9 (MMP9), and enhanced matrigel invasion [120]. On the contrary side, stable overexpression of miR-29b caused decrease of ID1 and MMP9 and significantly decreased invasion [120]. In a further study we observed a reciprocal association between miR-381 and ID1 in lung cancer cell lines and primary adenocarcinomas [121]. Our results also provide first evidence that ectopic expression of miR-381 reduced ID1 mRNA and protein levels, and significantly decreased lung cancer cell migration and invasion.(reviewed in [122]).
The use of antagomiRs against miR-10b in an animal model of breast tumor-bearing mice was associated with reduced metastasis, both in vitro and in vivo[123]. The silencing of miR-10b with antagomiRs significantly decrease dmiR-10b levels and increased the levels of a functionally important miR-10b target, Hoxd10. The use of this antagomiRs in mice bearing highly metastatic breast cancer cells did not reduce primary mammary tumor growth but markedly suppressed the formation of lung metastases. The therapy was well tolerated by mice.
miR-155 was found to be overexpressed in different types of solid cancers as well as lymphomas [20, 124–133] miR-155 is a negative prognostic factor in pancreatic and lung cancer patients [20, 131]. In malignant glioma the downregulation of the GABA-A receptor was shown to correlate with the grade of the tumor. The knockdown of miR-155 involves the re-expression of GABRA 1 protein in vivo and therefore controlling proliferation and signaling pathways regulated by the GABA-A receptor [134].
The inhibition of the MYC-driven miR-9 using a miRNA sponge could reduce the development of lung metastases in a breast cancer mouse model [135]. On the other hand, the inhibition of the tumor suppressive miR-31 with sponge miRNAs in a breast cancer model induced the development of lung metastases [136].
Tumor suppressor miRNAs
The let-7 family is one of the best described tumor suppressor miRNAs [24, 137–141] and is frequently downregulated in tumor tissue [142]. In xenograft models, tumor burden was reduced by intratumoral delivery of let-7b [143]. By intranasal delivery of let-7a using lentivirus in a lung cancer xenograft model the tumor burden was significantly reduced [57]. One emerging concept for miRNA regulation is based on functional polymorphisms in the target mRNA 3’UTR interfering with miRNA binding and function. Target polymorphisms in the 3’UTR of KRAS interfere with the function of let-7 and are associated with outcomes in breast and lung cancer [144, 145].
The miR-34 family has been reported as direct p53 transcriptional target. Overexpression of miR-34 family member induces apoptosis and cell cycle arrest [146, 147]. The correlation between downregulation of miR-34 and various tumor types has been demonstrated [148–151] miR-34 incorporated in a lipid-based particle was able to block tumor growth in a mouse model of non-small cell lung cancer [57] miR-34a accumulated in the tumor tissue, resulting in downregulation of its direct targets. Similar results were obtained in a second study of non-small cell lung cancer with the delivery of miR-34a or let-7 mimics [57]. Based on these results, miR-34 as a liposomal miR-34 mimic (MRX34, Mirna Therapeutics Inc.) is investigated in clinical trials [152].
miR-16 conjugated to atelocollagen has been shown to reduce bone metastases in a xenografts model of prostate cancer [153]. Atelocollagen is a collagen solubilized by protease with similar physical properties to those of natural, insolubilized collagen [92].
Conclusions
microRNAs represent critical regulators of tumor cell differentiation, proliferation, cell cycle progression, invasion and metastasis. Based on microRNA arrays various miRNAs have been described as oncogenes or tumor suppressors and many of them are used for diagnosis and as prognostic or predictive tools [122].
Emerging evidence suggests that inhibition of overexpressed oncogenic miRNAs or substitution of tumor suppressive miRNAs could become a novel treatment strategy in cancer therapy. The optimization of the stability of miRNAs, the improvement in delivery systems and targeted drug delivery as well as the understanding and control of off-target effects of miRNA therapeutics are challenges for the future development.
References
Lee RC, Feinbaum RL, Ambros V: The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993, 75: 843-854. 10.1016/0092-8674(93)90529-Y.
Garzon R, Calin GA, Croce CM: MicroRNAs in Cancer. Annu Rev Med. 2009, 60: 167-179. 10.1146/annurev.med.59.053006.104707.
Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN: MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23: 4051-4060. 10.1038/sj.emboj.7600385.
Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN: The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003, 425: 415-419. 10.1038/nature01957.
Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ: Processing of primary microRNAs by the Microprocessor complex. Nature. 2004, 432: 231-235. 10.1038/nature03049.
Bohnsack MT, Czaplinski K, Gorlich D: Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA. 2004, 10: 185-191. 10.1261/rna.5167604.
Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U: Nuclear export of microRNA precursors. Science. 2004, 303: 95-98. 10.1126/science.1090599.
Hutvagner G, Zamore PD: A microRNA in a multiple-turnover RNAi enzyme complex. Science. 2002, 297: 2056-2060. 10.1126/science.1073827.
Hammond SM, Bernstein E, Beach D, Hannon GJ: An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature. 2000, 404: 293-296. 10.1038/35005107.
Chendrimada TP, Gregory RI, Kumaraswamy E, Norman J, Cooch N, Nishikura K, Shiekhattar R: TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005, 436: 740-744. 10.1038/nature03868.
Diederichs S, Haber DA: Dual role for argonautes in microRNA processing and posttranscriptional regulation of microRNA expression. Cell. 2007, 131: 1097-1108. 10.1016/j.cell.2007.10.032.
Bartel DP: MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004, 116: 281-297. 10.1016/S0092-8674(04)00045-5.
Orom UA, Nielsen FC, Lund AH: MicroRNA-10a binds the 5'UTR of ribosomal protein mRNAs and enhances their translation. Mol Cell. 2008, 30: 460-471. 10.1016/j.molcel.2008.05.001.
Qin W, Shi Y, Zhao B, Yao C, Jin L, Ma J, Jin Y: miR-24 regulates apoptosis by targeting the open reading frame (ORF) region of FAF1 in cancer cells. PLoS One. 2010, 5: e9429-10.1371/journal.pone.0009429.
Garzon R, Marcucci G, Croce CM: Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010, 9: 775-789. 10.1038/nrd3179.
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K: Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002, 99: 15524-15529. 10.1073/pnas.242606799.
Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, Iorio MV, Visone R, Sever NI, Fabbri M: A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005, 353: 1793-1801. 10.1056/NEJMoa050995.
Nakamura T, Canaani E, Croce CM: Oncogenic All1 fusion proteins target Drosha-mediated microRNA processing. Proc Natl Acad Sci USA. 2007, 104: 10980-10985. 10.1073/pnas.0704559104.
Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, Jones PA: Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006, 9: 435-443. 10.1016/j.ccr.2006.04.020.
Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, Stephens RM, Okamoto A, Yokota J, Tanaka T: Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006, 9: 189-198. 10.1016/j.ccr.2006.01.025.
Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M: MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65: 7065-7070. 10.1158/0008-5472.CAN-05-1783.
Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, Croce CM: Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci USA. 2004, 101: 2999-3004. 10.1073/pnas.0307323101.
Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ: RAS is regulated by the let-7 microRNA family. Cell. 2005, 120: 635-647. 10.1016/j.cell.2005.01.014.
Akao Y, Nakagawa Y, Naoe T: let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol Pharm Bull. 2006, 29: 903-906. 10.1248/bpb.29.903.
Lee YS, Dutta A: The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007, 21: 1025-1030. 10.1101/gad.1540407.
Sampson VB, Rong NH, Han J, Yang Q, Aris V, Soteropoulos P, Petrelli NJ, Dunn SP, Krueger LJ: MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007, 67: 9762-9770. 10.1158/0008-5472.CAN-07-2462.
Mott JL, Kobayashi S, Bronk SF, Gores GJ: mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene. 2007, 26: 6133-6140. 10.1038/sj.onc.1210436.
Garzon R, Volinia S, Liu CG, Fernandez-Cymering C, Palumbo T, Pichiorri F, Fabbri M, Coombes K, Alder H, Nakamura T: MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood. 2008, 111: 3183-3189. 10.1182/blood-2007-07-098749.
Kluiver J, Poppema S, de Jong D, Blokzijl T, Harms G, Jacobs S, Kroesen BJ, van den Berg A: BIC and miR-155 are highly expressed in Hodgkin, primary mediastinal and diffuse large B cell lymphomas. J Pathol. 2005, 207: 243-249. 10.1002/path.1825.
Metzler M, Wilda M, Busch K, Viehmann S, Borkhardt A: High expression of precursor microRNA-155/BIC RNA in children with Burkitt lymphoma. Genes Chromosomes Cancer. 2004, 39: 167-169. 10.1002/gcc.10316.
Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M: A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006, 103: 2257-2261. 10.1073/pnas.0510565103.
Tam W, Hughes SH, Hayward WS, Besmer P: Avian bic, a gene isolated from a common retroviral site in avian leukosis virus-induced lymphomas that encodes a noncoding RNA, cooperates with c-myc in lymphomagenesis and erythroleukemogenesis. J Virol. 2002, 76: 4275-4286. 10.1128/JVI.76.9.4275-4286.2002.
Garzon R, Garofalo M, Martelli MP, Briesewitz R, Wang L, Fernandez-Cymering C, Volinia S, Liu CG, Schnittger S, Haferlach T: Distinctive microRNA signature of acute myeloid leukemia bearing cytoplasmic mutated nucleophosmin. Proc Natl Acad Sci USA. 2008, 105: 3945-3950. 10.1073/pnas.0800135105.
Meng F, Henson R, Wehbe-Janek H, Ghoshal K, Jacob ST, Patel T: MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology. 2007, 133: 647-658. 10.1053/j.gastro.2007.05.022.
Ciafre SA, Galardi S, Mangiola A, Ferracin M, Liu CG, Sabatino G, Negrini M, Maira G, Croce CM, Farace MG: Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem Biophys Res Commun. 2005, 334: 1351-1358. 10.1016/j.bbrc.2005.07.030.
Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH: Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem. 2008, 283: 1026-1033. 10.1074/jbc.M707224200.
Zhu S, Si ML, Wu H, Mo YY: MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol Chem. 2007, 282: 14328-14336. 10.1074/jbc.M611393200.
Ota A, Tagawa H, Karnan S, Tsuzuki S, Karpas A, Kira S, Yoshida Y, Seto M: Identification and characterization of a novel gene, C13orf25, as a target for 13q31-q32 amplification in malignant lymphoma. Cancer Res. 2004, 64: 3087-3095. 10.1158/0008-5472.CAN-03-3773.
Mendell JT: miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008, 133: 217-222. 10.1016/j.cell.2008.04.001.
O'Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT: c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005, 435: 839-843. 10.1038/nature03677.
Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, Lovat F, Fadda P, Mao C, Nuovo GJ: MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci USA. 2012, 109: E2110-E2116. 10.1073/pnas.1209414109.
Lehmann SM, Kruger C, Park B, Derkow K, Rosenberger K, Baumgart J, Trimbuch T, Eom G, Hinz M, Kaul D: An unconventional role for miRNA: let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nat Neurosci. 2012, 15: 827-835. 10.1038/nn.3113.
Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB: Prediction of mammalian microRNA targets. Cell. 2003, 115: 787-798. 10.1016/S0092-8674(03)01018-3.
Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N: Combinatorial microRNA target predictions. Nat Genet. 2005, 37: 495-500. 10.1038/ng1536.
Betel D, Wilson M, Gabow A, Marks DS, Sander C: The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008, 36: D149-D153.
Friedman RC, Farh KK, Burge CB, Bartel DP: Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19: 92-105.
Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y: Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004, 64: 3753-3756. 10.1158/0008-5472.CAN-04-0637.
Motoyama K, Inoue H, Nakamura Y, Uetake H, Sugihara K, Mori M: Clinical significance of high mobility group A2 in human gastric cancer and its relationship to let-7 microRNA family. Clin Cancer Res. 2008, 14: 2334-2340. 10.1158/1078-0432.CCR-07-4667.
Yang N, Kaur S, Volinia S, Greshock J, Lassus H, Hasegawa K, Liang S, Leminen A, Deng S, Smith L: MicroRNA microarray identifies Let-7i as a novel biomarker and therapeutic target in human epithelial ovarian cancer. Cancer Res. 2008, 68: 10307-10314. 10.1158/0008-5472.CAN-08-1954.
Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J, Song E: let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007, 131: 1109-1123. 10.1016/j.cell.2007.10.054.
Aagaard L, Rossi JJ: RNAi therapeutics: principles, prospects and challenges. Adv Drug Deliv Rev. 2007, 59: 75-86. 10.1016/j.addr.2007.03.005.
Zhao X, Pan F, Holt CM, Lewis AL, Lu JR: Controlled delivery of antisense oligonucleotides: a brief review of current strategies. Expert Opin Drug Deliv. 2009, 6: 673-686. 10.1517/17425240902992894.
Dias N, Stein CA: Antisense oligonucleotides: basic concepts and mechanisms. Mol Cancer Ther. 2002, 1: 347-355.
Concepcion CP, Han YC, Mu P, Bonetti C, Yao E, D'Andrea A, Vidigal JA, Maughan WP, Ogrodowski P, Ventura A: Intact p53-dependent responses in miR-34-deficient mice. PLoS Genet. 2012, 8: e1002797-10.1371/journal.pgen.1002797.
Cho WC: MicroRNAs as therapeutic targets and their potential applications in cancer therapy. Expert Opin Ther Targets. 2012, 16: 747-759. 10.1517/14728222.2012.696102.
Rao DD, Vorhies JS, Senzer N, Nemunaitis J: siRNA vs. shRNA: similarities and differences. Adv Drug Deliv Rev. 2009, 61: 746-759. 10.1016/j.addr.2009.04.004.
Trang P, Wiggins JF, Daige CL, Cho C, Omotola M, Brown D, Weidhaas JB, Bader AG, Slack FJ: Systemic delivery of tumor suppressor microRNA mimics using a neutral lipid emulsion inhibits lung tumors in mice. Mol Ther. 2011, 19: 1116-1122. 10.1038/mt.2011.48.
Czauderna F, Fechtner M, Dames S, Aygun H, Klippel A, Pronk GJ, Giese K, Kaufmann J: Structural variations and stabilising modifications of synthetic siRNAs in mammalian cells. Nucleic Acids Res. 2003, 31: 2705-2716. 10.1093/nar/gkg393.
Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M: Silencing of microRNAs in vivo with 'antagomirs'. Nature. 2005, 438: 685-689. 10.1038/nature04303.
Krutzfeldt J, Kuwajima S, Braich R, Rajeev KG, Pena J, Tuschl T, Manoharan M, Stoffel M: Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007, 35: 2885-2892. 10.1093/nar/gkm024.
Wahlestedt C, Salmi P, Good L, Kela J, Johnsson T, Hokfelt T, Broberger C, Porreca F, Lai J, Ren K: Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc Natl Acad Sci USA. 2000, 97: 5633-5638. 10.1073/pnas.97.10.5633.
Crooke ST, Graham MJ, Zuckerman JE, Brooks D, Conklin BS, Cummins LL, Greig MJ, Guinosso CJ, Kornbrust D, Manoharan M: Pharmacokinetic properties of several novel oligonucleotide analogs in mice. J Pharmacol Exp Ther. 1996, 277: 923-937.
Yoo BH, Bochkareva E, Bochkarev A, Mou TC, Gray DM: 2'–O-methyl-modified phosphorothioate antisense oligonucleotides have reduced non-specific effects in vitro. Nucleic Acids Res. 2004, 32: 2008-2016. 10.1093/nar/gkh516.
Hyrup B, Nielsen PE: Peptide nucleic acids (PNA): synthesis, properties and potential applications. Bioorg Med Chem. 1996, 4: 5-23. 10.1016/0968-0896(95)00171-9.
Pallan PS, Greene EM, Jicman PA, Pandey RK, Manoharan M, Rozners E, Egli M: Unexpected origins of the enhanced pairing affinity of 2'-fluoro-modified RNA. Nucleic Acids Res. 2011, 39: 3482-3495. 10.1093/nar/gkq1270.
Nielsen PE, Egholm M, Berg RH, Buchardt O: Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science. 1991, 254: 1497-1500. 10.1126/science.1962210.
Demidov VV, Kuhn H, Lavrentieva-Smolina IV, Frank-Kamenetskii MD: Peptide nucleic acid-assisted topological labeling of duplex dna. Methods. 2001, 23: 123-131. 10.1006/meth.2000.1113.
Gambari R: Peptide-nucleic acids (PNAs): a tool for the development of gene expression modifiers. Curr Pharm Des. 2001, 7: 1839-1862. 10.2174/1381612013397087.
Karkare S, Bhatnagar D: Promising nucleic acid analogs and mimics: characteristic features and applications of PNA, LNA, and morpholino. Appl Microbiol Biotechnol. 2006, 71: 575-586. 10.1007/s00253-006-0434-2.
Soomets U, Hallbrink M, Langel U: Antisense properties of peptide nucleic acids. Front Biosci. 1999, 4: D782-D786. 10.2741/Soomets.
Nielsen PE: Targeting double stranded DNA with peptide nucleic acid (PNA). Curr Med Chem. 2001, 8: 545-550. 10.2174/0929867003373373.
Brognara E, Fabbri E, Aimi F, Manicardi A, Bianchi N, Finotti A, Breveglieri G, Borgatti M, Corradini R, Marchelli R, Gambari R: Peptide nucleic acids targeting miR-221 modulate p27Kip1 expression in breast cancer MDA-MB-231 cells. Int J Oncol. 2012, 41: 2119-2127.
Yan LX, Wu QN, Zhang Y, Li YY, Liao DZ, Hou JH, Fu J, Zeng MS, Yun JP, Wu QL: Knockdown of miR-21 in human breast cancer cell lines inhibits proliferation, in vitro migration and in vivo tumor growth. Breast Cancer Res. 2011, 13: R2-10.1186/bcr2803.
Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, Fu C, Lindow M, Stenvang J, Straarup EM: Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet. 2011, 43: 371-378. 10.1038/ng.786.
van Solingen C, Seghers L, Bijkerk R, Duijs JM, Roeten MK, van Oeveren-Rietdijk AM, Baelde HJ, Monge M, Vos JB, de Boer HC: Antagomir-mediated silencing of endothelial cell specific microRNA-126 impairs ischemia-induced angiogenesis. J Cell Mol Med. 2009, 13: 1577-1585. 10.1111/j.1582-4934.2008.00613.x.
Chen Y, Zhu X, Zhang X, Liu B, Huang L: Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol Ther. 2010, 18: 1650-1656. 10.1038/mt.2010.136.
Lv H, Zhang S, Wang B, Cui S, Yan J: Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release. 2006, 114: 100-109. 10.1016/j.jconrel.2006.04.014.
Medina OP, Zhu Y, Kairemo K: Targeted liposomal drug delivery in cancer. Curr Pharm Des. 2004, 10: 2981-2989. 10.2174/1381612043383467.
Kedmi R, Ben-Arie N, Peer D: The systemic toxicity of positively charged lipid nanoparticles and the role of Toll-like receptor 4 in immune activation. Biomaterials. 2010, 31: 6867-6875. 10.1016/j.biomaterials.2010.05.027.
Peer D: Immunotoxicity derived from manipulating leukocytes with lipid-based nanoparticles. Adv Drug Deliv Rev. 2012, 64: 1738-1748. 10.1016/j.addr.2012.06.013.
de Fougerolles A, Vornlocher HP, Maraganore J, Lieberman J: Interfering with disease: a progress report on siRNA-based therapeutics. Nat Rev Drug Discov. 2007, 6: 443-453. 10.1038/nrd2310.
Ibrahim AF, Weirauch U, Thomas M, Grunweller A, Hartmann RK, Aigner A: MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res. 2011, 71: 5214-5224. 10.1158/0008-5472.CAN-10-4645.
Park TG, Jeong JH, Kim SW: Current status of polymeric gene delivery systems. Adv Drug Deliv Rev. 2006, 58: 467-486. 10.1016/j.addr.2006.03.007.
Ren Y, Zhou X, Mei M, Yuan XB, Han L, Wang GX, Jia ZF, Xu P, Pu PY, Kang CS: MicroRNA-21 inhibitor sensitizes human glioblastoma cells U251 (PTEN-mutant) and LN229 (PTEN-wild type) to taxol. BMC Cancer. 2010, 10: 27-10.1186/1471-2407-10-27.
Dutta T, Jain NK, McMillan NA, Parekh HS: Dendrimer nanocarriers as versatile vectors in gene delivery. Nanomedicine. 2010, 6: 25-34. 10.1016/j.nano.2009.05.005.
Hong S, Bielinska AU, Mecke A, Keszler B, Beals JL, Shi X, Balogh L, Orr BG, Baker JR, Banaszak Holl MM: Interaction of poly(amidoamine) dendrimers with supported lipid bilayers and cells: hole formation and the relation to transport. Bioconjug Chem. 2004, 15: 774-782. 10.1021/bc049962b.
Babar IA, Cheng CJ, Booth CJ, Liang X, Weidhaas JB, Saltzman WM, Slack FJ: Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc Natl Acad Sci USA. 2012, 109: E1695-E1704. 10.1073/pnas.1201516109.
Cheng CJ, Saltzman WM: Polymer nanoparticle-mediated delivery of microRNA inhibition and alternative splicing. Mol Pharm. 2012, 9: 1481-1488.
Suh JS, Lee JY, Choi YS, Chong PC, Park YJ: Peptide-mediated intracellular delivery of miRNA-29b for osteogenic stem cell differentiation. Biomaterials. 2013, 34: 4347-4359. 10.1016/j.biomaterials.2013.02.039.
Tazawa H, Tsuchiya N, Izumiya M, Nakagama H: Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci USA. 2007, 104: 15472-15477. 10.1073/pnas.0707351104.
Matsuyama H, Suzuki HI, Nishimori H, Noguchi M, Yao T, Komatsu N, Mano H, Sugimoto K, Miyazono K: miR-135b mediates NPM-ALK-driven oncogenicity and renders IL-17-producing immunophenotype to anaplastic large cell lymphoma. Blood. 2011, 118: 6881-6892. 10.1182/blood-2011-05-354654.
Minakuchi Y, Takeshita F, Kosaka N, Sasaki H, Yamamoto Y, Kouno M, Honma K, Nagahara S, Hanai K, Sano A: Atelocollagen-mediated synthetic small interfering RNA delivery for effective gene silencing in vitro and in vivo. Nucleic Acids Res. 2004, 32: e109-10.1093/nar/gnh093.
Ghosh R, Singh LC, Shohet JM, Gunaratne PH: A gold nanoparticle platform for the delivery of functional microRNAs into cancer cells. Biomaterials. 2013, 34: 807-816. 10.1016/j.biomaterials.2012.10.023.
Crew E, Tessel MA, Rahman S, Razzak-Jaffar A, Mott D, Kamundi M, Yu G, Tchah N, Lee J, Bellavia M, Zhong CJ: MicroRNA conjugated gold nanoparticles and cell transfection. Anal Chem. 2012, 84: 26-29. 10.1021/ac202749p.
Bitar A, Ahmad NM, Fessi H, Elaissari A: Silica-based nanoparticles for biomedical applications. Drug Discov Today. 2012, 17: 1147-1154. 10.1016/j.drudis.2012.06.014.
Wu X, Liu H, Liu J, Haley KN, Treadway JA, Larson JP, Ge N, Peale F, Bruchez MP: Immunofluorescent labeling of cancer marker Her2 and other cellular targets with semiconductor quantum dots. Nat Biotechnol. 2003, 21: 41-46.
Nida DL, Rahman MS, Carlson KD, Richards-Kortum R, Follen M: Fluorescent nanocrystals for use in early cervical cancer detection. Gynecol Oncol. 2005, 99: S89-S94. 10.1016/j.ygyno.2005.07.050.
Choi KY, Yoon HY, Kim JH, Bae SM, Park RW, Kang YM, Kim IS, Kwon IC, Choi K, Jeong SY: Smart nanocarrier based on PEGylated hyaluronic acid for cancer therapy. ACS Nano. 2011, 5: 8591-8599. 10.1021/nn202070n.
Orian-Rousseau V: CD44, a therapeutic target for metastasising tumours. Eur J Cancer. 2010, 46: 1271-1277. 10.1016/j.ejca.2010.02.024.
Ebert MS, Neilson JR, Sharp PA: MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007, 4: 721-726. 10.1038/nmeth1079.
Zhang Y: Wang Z. 2013, Progress in microRNA delivery. J Control Release: Gemeinhart RA
Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD: Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003, 115: 199-208. 10.1016/S0092-8674(03)00759-1.
Terasawa K, Shimizu K, Tsujimoto G: Synthetic Pre-miRNA-Based shRNA as Potent RNAi Triggers. J Nucleic Acids. 2011, 2011: 131579-
Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Orum H: Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science. 2010, 327: 198-201. 10.1126/science.1178178.
Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y: Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013, 368: 1685-1694. 10.1056/NEJMoa1209026.
Tripp VT, Young DD: Discovery of Small Molecule Modifiers of microRNAs for the Treatment of HCV Infection. Methods Mol Biol. 2014, 1103: 153-163. 10.1007/978-1-62703-730-3_12.
Folini M, Gandellini P, Longoni N, Profumo V, Callari M, Pennati M, Colecchia M, Supino R, Veneroni S, Salvioni R: miR-21: an oncomir on strike in prostate cancer. Mol Cancer. 2010, 9: 12-10.1186/1476-4598-9-12.
Lakomy R, Sana J, Hankeova S, Fadrus P, Kren L, Lzicarova E, Svoboda M, Dolezelova H, Smrcka M, Vyzula R: MiR-195, miR-196b, miR-181c, miR-21 expression levels and O-6-methylguanine-DNA methyltransferase methylation status are associated with clinical outcome in glioblastoma patients. Cancer Sci. 2011, 102: 2186-2190. 10.1111/j.1349-7006.2011.02092.x.
Selaru FM, Olaru AV, Kan T, David S, Cheng Y, Mori Y, Yang J, Paun B, Jin Z, Agarwal R: MicroRNA-21 is overexpressed in human cholangiocarcinoma and regulates programmed cell death 4 and tissue inhibitor of metalloproteinase 3. Hepatology. 2009, 49: 1595-1601. 10.1002/hep.22838.
Li J, Huang H, Sun L, Yang M, Pan C, Chen W, Wu D, Lin Z, Zeng C, Yao Y: MiR-21 indicates poor prognosis in tongue squamous cell carcinomas as an apoptosis inhibitor. Clin Cancer Res. 2009, 15: 3998-4008. 10.1158/1078-0432.CCR-08-3053.
Feber A, Xi L, Luketich JD, Pennathur A, Landreneau RJ, Wu M, Swanson SJ, Godfrey TE, Litle VR: MicroRNA expression profiles of esophageal cancer. J Thorac Cardiovasc Surg. 2008, 135: 255-260. 10.1016/j.jtcvs.2007.08.055. discussion 260
Chan SH, Wu CW, Li AF, Chi CW, Lin WC: miR-21 microRNA expression in human gastric carcinomas and its clinical association. Anticancer Res. 2008, 28: 907-911.
Connolly E, Melegari M, Landgraf P, Tchaikovskaya T, Tennant BC, Slagle BL, Rogler LE, Zavolan M, Tuschl T, Rogler CE: Elevated expression of the miR-17-92 polycistron and miR-21 in hepadnavirus-associated hepatocellular carcinoma contributes to the malignant phenotype. Am J Pathol. 2008, 173: 856-864. 10.2353/ajpath.2008.080096.
Markou A, Tsaroucha EG, Kaklamanis L, Fotinou M, Georgoulias V, Lianidou ES: Prognostic value of mature microRNA-21 and microRNA-205 overexpression in non-small cell lung cancer by quantitative real-time RT-PCR. Clin Chem. 2008, 54: 1696-1704. 10.1373/clinchem.2007.101741.
Dillhoff M, Liu J, Frankel W, Croce C, Bloomston M: MicroRNA-21 is overexpressed in pancreatic cancer and a potential predictor of survival. J Gastrointest Surg. 2008, 12: 2171-2176. 10.1007/s11605-008-0584-x.
Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, Bowman ED, Yanaihara N, Yuen ST, Chan TL, Kwong DL, Au GK: MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008, 299: 425-436.
Mei M, Ren Y, Zhou X, Yuan XB, Han L, Wang GX, Jia Z, Pu PY, Kang CS, Yao Z: Downregulation of miR-21 enhances chemotherapeutic effect of taxol in breast carcinoma cells. Technol Cancer Res Treat. 2010, 9: 77-86.
Corsten MF, Miranda R, Kasmieh R, Krichevsky AM, Weissleder R, Shah K: MicroRNA-21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell delivered S-TRAIL in human gliomas. Cancer Res. 2007, 67: 8994-9000. 10.1158/0008-5472.CAN-07-1045.
Gumireddy K, Young DD, Xiong X, Hogenesch JB, Huang Q, Deiters A: Small-molecule inhibitors of microrna miR-21 function. Angew Chem Int Ed Engl. 2008, 47: 7482-7484. 10.1002/anie.200801555.
Rothschild SI, Tschan MP, Federzoni EA, Jaggi R, Fey MF, Gugger M, Gautschi O: MicroRNA-29b is involved in the Src-ID1 signaling pathway and is dysregulated in human lung adenocarcinoma. Oncogene. 2012, 31: 4221-4232. 10.1038/onc.2011.578.
Rothschild SI, Tschan MP, Jaggi R, Fey MF, Gugger M, Gautschi O: MicroRNA-381 represses ID1 and is deregulated in lung adenocarcinoma. J Thorac Oncol. 2012, 7: 1069-1077. 10.1097/JTO.0b013e31824fe976.
Rothschild SI: Epigenetic therapy in lung cancer - role of microRNAs. Front Oncol. 2013, 3: 158-
Ma L, Reinhardt F, Pan E, Soutschek J, Bhat B, Marcusson EG, Teruya-Feldstein J, Bell GW, Weinberg RA: Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat Biotechnol. 2010, 28: 341-347. 10.1038/nbt.1618.
Hui AB, Lenarduzzi M, Krushel T, Waldron L, Pintilie M, Shi W, Perez-Ordonez B, Jurisica I, O'Sullivan B, Waldron J: Comprehensive MicroRNA profiling for head and neck squamous cell carcinomas. Clin Cancer Res. 2010, 16: 1129-1139. 10.1158/1078-0432.CCR-09-2166.
Xie Q, Chen X, Lu F, Zhang T, Hao M, Wang Y, Zhao J, McCrae MA, Zhuang H: Aberrant expression of microRNA 155 may accelerate cell proliferation by targeting sex-determining region Y box 6 in hepatocellular carcinoma. Cancer. 2012, 118: 2431-2442. 10.1002/cncr.26566.
Du ZM, Hu LF, Wang HY, Yan LX, Zeng YX, Shao JY, Ernberg I: Upregulation of MiR-155 in nasopharyngeal carcinoma is partly driven by LMP1 and LMP2A and downregulates a negative prognostic marker JMJD1A. PLoS One. 2011, 6: e19137-10.1371/journal.pone.0019137.
Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E, Dahlberg JE: Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci USA. 2005, 102: 3627-3632. 10.1073/pnas.0500613102.
Nikiforova MN, Tseng GC, Steward D, Diorio D, Nikiforov YE: MicroRNA expression profiling of thyroid tumors: biological significance and diagnostic utility. J Clin Endocrinol Metab. 2008, 93: 1600-1608. 10.1210/jc.2007-2696.
Chen J, Wang BC, Tang JH: Clinical significance of microRNA-155 expression in human breast cancer. J Surg Oncol. 2011, 106: 260-266.
Shibuya H, Iinuma H, Shimada R, Horiuchi A, Watanabe T: Clinicopathological and prognostic value of microRNA-21 and microRNA-155 in colorectal cancer. Oncology. 2010, 79: 313-320. 10.1159/000323283.
Greither T, Grochola LF, Udelnow A, Lautenschlager C, Wurl P, Taubert H: Elevated expression of microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated with poorer survival. Int J Cancer. 2010, 126: 73-80. 10.1002/ijc.24687.
Philippidou D, Schmitt M, Moser D, Margue C, Nazarov PV, Muller A, Vallar L, Nashan D, Behrmann I, Kreis S: Signatures of microRNAs and selected microRNA target genes in human melanoma. Cancer Res. 2010, 70: 4163-4173. 10.1158/0008-5472.CAN-09-4512.
White NM, Bao TT, Grigull J, Youssef YM, Girgis A, Diamandis M, Fatoohi E, Metias M, Honey RJ, Stewart R: miRNA profiling for clear cell renal cell carcinoma: biomarker discovery and identification of potential controls and consequences of miRNA dysregulation. J Urol. 2011, 186: 1077-1083. 10.1016/j.juro.2011.04.110.
Poltronieri P, D'Urso PI, Mezzolla V, D'Urso OF: Potential of anti-cancer therapy based on anti-miR-155 oligonucleotides in glioma and brain tumours. Chem Biol Drug Des. 2013, 81: 79-84. 10.1111/cbdd.12002.
Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Muth D, Teruya-Feldstein J, Reinhardt F, Onder TT, Valastyan S: miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010, 12: 247-256.
Valastyan S, Reinhardt F, Benaich N, Calogrias D, Szasz AM, Wang ZC, Brock JE, Richardson AL, Weinberg RA: A pleiotropically acting microRNA, miR-31, inhibits breast cancer metastasis. Cell. 2009, 137: 1032-1046. 10.1016/j.cell.2009.03.047.
Esquela-Kerscher A, Trang P, Wiggins JF, Patrawala L, Cheng A, Ford L, Weidhaas JB, Brown D, Bader AG, Slack FJ: The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell Cycle. 2008, 7: 759-764. 10.4161/cc.7.6.5834.
Ricarte-Filho JC, Fuziwara CS, Yamashita AS, Rezende E, da-Silva MJ, Kimura ET: Effects of let-7 microRNA on cell growth and differentiation of papillary thyroid cancer. Transl Oncol. 2009, 2: 236-241.
Dong Q, Meng P, Wang T, Qin W, Wang F, Yuan J, Chen Z, Yang A, Wang H: MicroRNA let-7a inhibits proliferation of human prostate cancer cells in vitro and in vivo by targeting E2F2 and CCND2. PLoS One. 2010, 5: e10147-10.1371/journal.pone.0010147.
Lee ST, Chu K, Oh HJ, Im WS, Lim JY, Kim SK, Park CK, Jung KH, Lee SK, Kim M, Roh JK: Let-7 microRNA inhibits the proliferation of human glioblastoma cells. J Neurooncol. 2011, 102: 19-24. 10.1007/s11060-010-0286-6.
Yu CC, Chen YW, Chiou GY, Tsai LL, Huang PI, Chang CY, Tseng LM, Chiou SH, Yen SH, Chou MY: MicroRNA let-7a represses chemoresistance and tumourigenicity in head and neck cancer via stem-like properties ablation. Oral Oncol. 2011, 47: 202-210. 10.1016/j.oraloncology.2010.12.001.
Barh D, Malhotra R, Ravi B, Sindhurani P: MicroRNA let-7: an emerging next-generation cancer therapeutic. Curr Oncol. 2010, 17: 70-80.
Trang P, Medina PP, Wiggins JF, Ruffino L, Kelnar K, Omotola M, Homer R, Brown D, Bader AG, Weidhaas JB, Slack FJ: Regression of murine lung tumors by the let-7 microRNA. Oncogene. 2009, 29: 1580-1587.
Chin LJ, Ratner E, Leng S, Zhai R, Nallur S, Babar I, Muller RU, Straka E, Su L, Burki EA: A SNP in a let-7 microRNA complementary site in the KRAS 3' untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008, 68: 8535-8540. 10.1158/0008-5472.CAN-08-2129.
Paranjape T, Heneghan H, Lindner R, Keane FK, Hoffman A, Hollestelle A, Dorairaj J, Geyda K, Pelletier C, Nallur S: A 3'-untranslated region KRAS variant and triple-negative breast cancer: a case–control and genetic analysis. Lancet Oncol. 2011, 12: 377-386. 10.1016/S1470-2045(11)70044-4.
He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D: A microRNA component of the p53 tumour suppressor network. Nature. 2007, 447: 1130-1134. 10.1038/nature05939.
He L, He X, Lowe SW, Hannon GJ: microRNAs join the p53 network–another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007, 7: 819-822. 10.1038/nrc2232.
Gallardo E, Navarro A, Vinolas N, Marrades RM, Diaz T, Gel B, Quera A, Bandres E, Garcia-Foncillas J, Ramirez J, Monzo M: miR-34a as a prognostic marker of relapse in surgically resected non-small-cell lung cancer. Carcinogenesis. 2009, 30: 1903-1909. 10.1093/carcin/bgp219.
O'Day E, Lal A: MicroRNAs and their target gene networks in breast cancer. Breast Cancer Res. 2010, 12: 201-10.1186/bcr2484.
Corney DC, Hwang CI, Matoso A, Vogt M, Flesken-Nikitin A, Godwin AK, Kamat AA, Sood AK, Ellenson LH, Hermeking H, Nikitin AY: Frequent downregulation of miR-34 family in human ovarian cancers. Clin Cancer Res. 2010, 16: 1119-1128. 10.1158/1078-0432.CCR-09-2642.
Yamakuchi M, Ferlito M, Lowenstein CJ: miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci USA. 2008, 105: 13421-13426. 10.1073/pnas.0801613105.
Bader AG: miR-34 - a microRNA replacement therapy is headed to the clinic. Front Genet. 2012, 3: 120-
Takeshita F, Patrawala L, Osaki M, Takahashi RU, Yamamoto Y, Kosaka N, Kawamata M, Kelnar K, Bader AG, Brown D, Ochiya T: Systemic delivery of synthetic microRNA-16 inhibits the growth of metastatic prostate tumors via downregulation of multiple cell-cycle genes. Mol Ther. 2010, 18: 181-187. 10.1038/mt.2009.207.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.