
Abstract
Changes in protein metabolism are key to disease onset and progression in many neurodegenerative diseases. As a prime example, in Parkinson’s disease, folding, post-translational modification and recycling of the synaptic protein α-synuclein are clearly altered, leading to a progressive accumulation of pathogenic protein species and the formation of intracellular inclusion bodies. Altered protein folding is one of the first steps of an increasingly understood cascade in which α-synuclein forms complex oligomers and finally distinct protein aggregates, termed Lewy bodies and Lewy neurites. In neurons, an elaborated network of chaperone and co-chaperone proteins is instrumental in mediating protein folding and re-folding. In addition to their direct influence on client proteins, chaperones interact with protein degradation pathways such as the ubiquitin-proteasome-system or autophagy in order to ensure the effective removal of irreversibly misfolded and potentially pathogenic proteins. Because of the vital role of proper protein folding for protein homeostasis, a growing number of studies have evaluated the contribution of chaperone proteins to neurodegeneration. We herein review our current understanding of the involvement of chaperones, co-chaperones and chaperone-mediated autophagy in synucleinopathies with a focus on the Hsp90 and Hsp70 chaperone system. We discuss genetic and pathological studies in Parkinson’s disease as well as experimental studies in models of synucleinopathies that explore molecular chaperones and protein degradation pathways as a novel therapeutic target. To this end, we examine the capacity of chaperones to prevent or modulate neurodegeneration and summarize the current progress in models of Parkinson’s disease and related neurodegenerative disorders.
Similar content being viewed by others

Introduction
Parkinson’s disease (PD) is a common incurable neurodegenerative disease that affects around 1% of the worldwide population at age 60 years [1]. It is progressive in nature and causes a movement disorder characterized by bradykinesia, resting tremor, rigidity and postural instability along with non-motor symptoms that mainly include autonomic dysfunction and cognitive impairment [2]. No treatment with established efficacy in preventing or slowing the progression of neurodegeneration in PD is currently available and development of such treatment is of utmost importance. Progressive degeneration of neurons in defined regions of the brain and the presence of proteinaceous intracellular inclusion bodies characterize PD pathology [3]. These inclusion bodies are termed Lewy bodies and Lewy neurites and contain large amounts of ubiquitinated and phosphorylated proteins, most importantly the presynaptic protein α-synuclein [3–5]. Increased levels of α-synuclein or α-synuclein containing protein aggregates are not only a hallmark of PD but are characteristic for a whole group of neurodegenerative diseases including dementia with Lewy bodies (DLB), multiple system atrophy (MSA), Alzheimer’s disease, different forms of neurodegeneration with brain iron accumulation and others [3, 6–8]. This group of diseases can therefore be referred to as “synucleinopathies”, although overlapping pathologies (such as tau-containing neurofibrillary tangles or amyloid-β plaques) exist in many cases and may act synergistically. Strong evidence for an involvement of α-synuclein in PD is also provided by genetic studies in familial and sporadic forms of the disease. Missense mutations in the α-synuclein gene (SCNA) (A53T, A30P and E46K) [9–11] as well as gene multiplications [12–14] cause familial forms of PD, while recent genome wide association studies have revealed polymorphisms in the α-synuclein gene as risk factors for developing sporadic PD [15].
An emerging theme in many neurodegenerative diseases, including the synucleinopathies, are deficits in protein metabolism, most importantly protein folding and degradation [16–23]. Alpha-synuclein is a neuronal protein that is enriched at presynaptic terminals, where it is thought to be involved in the assembly of the SNARE (soluble NSF attachment protein receptor) machinery and vesicle release [24, 25]. Alpha-synuclein pathology in PD is believed to follow a multi-step process that starts with the misfolding of α-synuclein and progresses to the formation of increasingly complex oligomers, soluble intermediates and finally insoluble fibrils and mature aggregates [17–19]. Although α-synuclein has been classically described to have an unfolded tertiary structure and to be present as monomers that acquire an α-helical secondary structure upon binding to lipid membranes [26, 27], recent reports suggest that α-synuclein natively forms α-helically folded tetramers when isolated under non-denaturing conditions [28–30]. These results have a significant impact on future research because they add a new step to the sequence of pathological events in synucleinopathies: Events that destabilize the native α-helical tetramer conformation might precede α-synuclein misfolding and aggregation and thus compounds that preserve the native tetramers may have great therapeutic potential. It should be cautioned however that experiments from two independent laboratories have failed to confirm the presence of natively unfolded α-synuclein tetramers in PD [31, 32]. Future studies will have to decipher the exact mechanisms behind these findings and will have to explain conflicting results.
Moving downstream of simple α-synuclein misfolding, emerging evidence implicates soluble oligomeric forms of α-synuclein as the main culprit in the pathogenesis of neurodegenerative diseases associated with α-synuclein accumulation [19]. Disease causing missense mutations and multiplications of the α-synuclein gene [33] as well as oxidative stress [34], post-translational modifications such as phosphorylation [35, 36] or truncation [37, 38] and the presence of fatty acids [39–41] are known to modulate α-synuclein’s propensity to aggregate. Furthermore, levels of α-synuclein oligomers are increased in cortical tissue of patients with idiopathic PD [40] and DLB [42] compared to age-matched controls. The mechanism by which smaller soluble aggregates induce neuronal dysfunction and neurodegeneration is increasingly, albeit still incompletely, understood [19]. Using a protein-fragment complementation assay in transfected cells and viral-vector mediated rodent models of α-synuclein aggregation, oligomer formation was shown to contribute to α-synuclein’s toxic effect on neurons [43–48]. Importantly, α-synuclein oligomers are involved in key steps of the potentially prion-like propagation of neurodegeneration in PD such as exocytosis, endocytosis and seeding [19, 49–51]. Given the implications of α-synuclein oligomerization in the early stages of neurodegeneration, preventing this step is a promising approach to treat or even prevent the degenerative process associated with α-synuclein misfolding and accumulation.
Review
Molecular chaperones, co-chaperones and chaperone-mediated autophagy
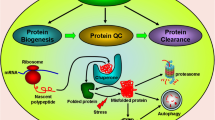
A network of highly conserved molecules, termed chaperones and co-chaperones, mediates the folding and re-folding of proteins and thus is critical for preserving the functional state and structure of client proteins [52–55]. Molecular chaperones are defined as a class of proteins that interact with, stabilize and help proteins to acquire their native conformation [52]. They are highly ubiquitous and assist the folding of newly synthesized proteins as well as the refolding of partially folded proteins into their three-dimensional structures [52, 53, 56]. In order to preserve intracellular protein homeostasis, chaperones interact with pathways of protein degradation that regulate constitutive protein turnover and the removal of misfolded proteins. Major protein degradation pathways for α-synuclein are the ubiquitin-proteasome system and the autophagy-lysosomal pathway [18, 57]. According to their molecular weight, chaperones can be classified into different groups such as Hsp60, Hsp70, Hsp90, Hsp100 and the small Hsps. Important co-chaperones, which interact with and assist chaperones in the folding of their client proteins, include for example the BAG-domain containing family (Bag1-6), the TPR-domain containing family (CHIP, Hip, Hop) and the DnaJ-domain containing co-chaperone Hsp40 [17, 22]. Cells constitutively express many chaperones (then referred to as heat shock cognates (Hsc)) and co-chaperones. However, their expression is markedly increased under environmental stress conditions, for example following hyperthermia, hypoxia, oxidative stress or exposure to toxins [52–54, 56, 58]. This stress response is triggered by the accumulation of unfolded proteins and effectively elicits chaperone expression by a signaling pathway that engages the transcription factor heat shock factor 1 (HSF-1) [54, 59, 60]. This regulatory element is part of a molecular switch that adjusts levels of chaperones to the cell’s condition. Hsp90 associates with HSF-1 in the cytosol and thus preserves its inactive monomeric state [61]. Cell stress and protein misfolding promote the dissociation of HSF-1 from Hsp90 and hence its translocation to the nucleus. At the nucleus, HSF-1 initiates the coordinated expression of Hsp70 and other heat shock proteins via heat shock response elements in the promoter regions of the respective genes [62]. Once adequate levels of chaperones have reached the cytosol, Hsp90 again associates with and inactivates HSF-1 therefore creating a dynamic Hsp90-dependent feedback loop that allows the cell to adjust to endogenous or exogenous stress [63, 64]. This feedback loop also opens opportunities to pharmacologically modulate chaperone levels, or levels of Hsp70 in particular, by applying inhibitors of Hsp90, a concept that is being increasingly investigated [17, 22].
In addition to directly folding or re-folding substrate proteins, chaperones assist many other cellular pathways for example by selecting and targeting irreversibly damaged or altered proteins for degradation. Chaperone-mediated autophagy refers to a highly-selective subtype of autophagy that utilizes chaperone proteins and lysosomal receptors to directly translocate target proteins into the lysosomal lumen, where rapid degradation takes place [65]. Target proteins carry a pentapeptide motif (KFERQ) and are thus selectively identified by the cytosolic chaperone Hsc70, a constitutively expressed member of the Hsp70 family, that facilitates delivery to the lysosomal surface [66–68]. The action of Hsc70 and its co-chaperones is crucial as the interaction with the KFERQ targeting motif confers selectivity. At the lysosomal membrane, binding of the substrate-chaperone complex to the lysosomal receptor protein LAMP-2A is followed by unfolding, multimerization of LAMP-2A, and finally translocation of the target protein [68, 69]. Lysosome-associated Hsc70, that resides within the lysosomal lumen, assists the disassembly of the LAMP-2A multimer complex after translocation and thus regenerates monomeric forms of LAMP-2A, that are again capable of substrate binding [70, 71]. The presence of lysosomal Hsc70 is a critical rate-limiting step, as, although all types of lysosomes carry the LAMP-2A receptor, only lysosomes that contain lysosomal Hsc70 show effective substrate uptake [72]. Interestingly, another chaperone, Hsp90 localizes to both the cytosolic and luminal side of the lysosomal membrane and is thought to stabilize LAMP-2A as it transitions from its monomeric form capable of substrate binding to the multimeric form that allows substrate translocation across the membrane [71]. The wide spectrum of cellular functions in which CMA is critically involved, ranging from selective protein quality control to cell-type specific functions depending on the substrate protein, emphasizes the importance of this pathway for maintaining protein homeostasis and cellular integrity, particularly in response to stress. CMA activity declines with age in many tissues [73, 74] and failure of CMA has been linked to the pathogenesis of several major neurodegenerative diseases, including the synucleinopathies (as discussed below).
Chaperones protect neurons against α-synuclein-induced toxicity
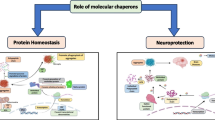
Research investigating the role of molecular chaperones in synucleinopathies followed groundbreaking work in other neurodegenerative diseases, most importantly the trinucleotide repeat expansions disorders [75–78]. First evidence for an involvement of chaperones in PD was provided by studies that identified Hsp90, Hsp70, Hsp60, Hsp40 and Hsp27 as part of Lewy bodies [79–82]. In a seminal study, Auluck et al. were able to demonstrate that Hsp70 co-expression could prevent dopaminergic cell death in a Drosophila melanogaster model of α-synuclein toxicity [81]. Furthermore interference with the endogenous chaperone system by introducing a mutation to Hsp70 could exacerbate the pathological phenotype, confirming the notion that Hsp70 is critical for maintaining α-synuclein’s folding state [81]. Based on these initial findings two pivotal hypotheses have been formulated and investigated in subsequent studies (reviewed in [17]). Firstly, Hsp70 is a critical part of the cellular mechanism that mitigates α-synuclein toxicity and secondly the sequestration of chaperones into protein aggregates results in their cellular depletion and thus subsequent loss of chaperone function may promote neurodegeneration (Figure 1).

The role of chaperones and co-chaperones in α-synuclein metabolism and pathology. As a general concept, chaperones mediate several cellular strategies that maintain protein homeostasis. In synucleinopathies, misfolded α-synuclein can be refolded, degraded, secreted or sequestered into mature aggregates such as Lewy bodies. Direct stabilization and refolding, degradation via different protein degradation pathways and sequestration into aggregates are mechanisms that are assisted or modulated by chaperones and co-chaperones. Failure of these mechanisms abolishes protein homeostasis and thus promotes α-synuclein accumulation, oligomer formation, toxicity and potentially cell-to-cell propagation of α-synuclein pathology.
Consistent with the idea that chaperones are a critical part of the response to environmental stress and protein overload, cells [83] and mice [84] treated with the mitochondrial toxins rotenone or MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) or the proteasome inhibitor lactacystin, which are often used to model dopaminergic cell degeneration, show a marked increase in chaperone levels, most importantly Hsp70. Likewise viral-vector mediated targeted overexpression of α-synuclein in the substantia nigra of mice resulted in increased mRNA levels of Hsp70, Hsp40 and Hsp27 [85]. An interesting recent study by Donmez et al. reported that SIRT1, a member of the sirtuin protein deacetylase family, deacetylates HSF-1 in the brain of A53T mutant α-synuclein mice, thus promoting the expression of Hsp70 [86]. This suggests that SIRT1 deacetylates HSF-1 and activates chaperones under stress conditions induced by the presence of mutant α-synuclein. Subsequently this mechanism leads to a suppression of α-synuclein aggregation, reduced α-synuclein-induced toxicity and extended survival in the mouse model examined [86].
Critical to novel therapeutic strategies, exogenous overexpression of Hsp70 and other chaperones has proven neuroprotective in different PD models. In cell culture models of α-synuclein aggregation and toxicity, co-expression of TorsinA (a protein with homology to Hsp104) [79], Hsp40 [79, 87], Hsp27 [88, 89], or Hsp70 [90, 91] led to reduced aggregate formation, decreased α-synuclein levels and reduced toxicity (Figure 1).
Despite these promising findings, studies evaluating different chaperones as a target of therapy in mouse models of PD provided differing results. While Klucken et al. showed that crossing of Hsp70 transgenic mice with α-synuclein transgenic mice reduced α-synuclein aggregation in vivo [91], Shimshek et al. could not confirm this finding after crossing human A53T mutant α-synuclein transgenic mice with mice overexpressing Hsp70 [92]. This argues that frank overexpression of Hsp70 alone might not have a significant impact on α-synuclein-induced toxicity in vivo. Similarly, while TorsinA was found to be a potent suppressor of α-synuclein aggregation and toxicity in cellular models [79] and in a Caenorhabditis elegans model [93], an elegant recent study using both an MPTP-induced mouse model of PD and α-synuclein transgenic mice could not detect a neuroprotective effect for overexpression of TorsinA [94].
Deciphering the molecular interaction between Hsp70 and α-synuclein, Hsp70 was found to bind α-synuclein fibrils with great affinity, through a transient and reversible interaction of Hsp70’s substrate-binding domain and the core hydrophobic region of soluble α-synuclein intermediates [95, 96]. A recent study was further able to map the exact Hsc70-α-synuclein interface, which might allow the development of an Hsc70-derived polypeptide that mimics the effects of this chaperone on α-synuclein assembly and toxicity [97]. Hsp70 was shown to promote an open conformational state that discourages interaction with other α-synuclein molecules and thus the formation of oligomers [43, 98]. Furthermore, oligomer formation of secreted extracellular α-synuclein was significantly reduced when Hsp70 was co-expressed and potentially simultaneously secreted [46], a finding that might have great implications for the propagation of α-synuclein pathology and neurodegeneration (Figure 1). A systematic investigation of the interaction of various small Hsps (αB-crystallin, Hsp27, Hsp20, HspB8, and HspB2B3) with both wild-type and mutant α-synuclein showed that all small Hsps transiently bind to the various forms of α-synuclein and inhibit mature α-synuclein fibril formation [99]. Further in vitro characterization showed that the small Hsp HspB5 can potentiate α-synuclein fiber depolymerization by several chaperones including Hsp70 and its co-chaperones [100]. Interestingly, Hsp90 has been shown to be a critical modulator of α-synuclein aggregation [101] and can bind A53T mutant α-synuclein oligomers in an ATP-independent manner to form a stable complex, thus rendering them non-toxic to cells [102].
Sequestration and depletion of chaperones into intracellular protein aggregates exacerbates neurodegeneration
Central to the idea that sequestration of chaperones into protein aggregates could result in a significant depletion is the finding that chaperone activity as well as the cell’s resistance to proteotoxic insults declines with age [18, 20] (Figure 1). This goes hand in hand with an increase in proteotoxic stress load over the lifetime of a cell, which is particularly important for post-mitotic cells like neurons [103]. As for chaperone sequestration in the PD brain, post-mortem pathological studies demonstrate, for example, the presence of αB-crystallin and Hsp27 positive neurons in PD patients but not in matched controls [104, 105]. The distribution of αB-crystallin positive neurons followed a distinct pattern and greatly overlapped with Lewy body pathology, although αB-crystallin accumulation was not exclusive to Lewy body bearing neurons [105]. Interestingly, by using a series of in vitro techniques, Waudby et al. were able to show that αB-crystallin binds along the length of α-synuclein fibrils thereby inhibiting further growth and shifting the monomer-fibril equilibrium in favor of dissociation [106]. This might explain the presence of chaperones in α-synuclein containing protein inclusions and could represent a way by which this and other chaperones limit the onset and progression of protein misfolding diseases [106]. As discussed above, a number of studies have revealed an association of several chaperones with α-synuclein pathology, thus promoting the idea that chaperones are key players in PD [79–82]. Following these reports, a number of studies have measured levels of chaperones in different brain regions in synucleinopathies. Overall, these studies revealed a correlation between levels of chaperones and detergent-soluble α-synuclein [80, 88, 107–110], consistent with data that show that chaperones mainly interact with this fraction of α-synuclein. Recent findings also suggest that Hsc70 and other proteins involved in CMA or lysosomal targeting and degradation, are significantly altered in different brain regions in PD and DLB [109, 111–115] (reviewed in [18]), supporting the concept of chaperone dysfunction in synucleinopathies (Figure 1). On a molecular level, α-synuclein oligomers were found to be capable of inhibiting the Hsp70/Hsp40 system by interacting with J-domain co-chaperones [116].
Chaperone-mediated autophagy – a link between protein-folding and degradation with implications for synucleinopathies
As discussed above, CMA is a subtype of autophagy and as such participates in the selective turnover of target proteins that contain KFERQ or KFERQ-like motifs including α-synuclein [18, 65]. Although soluble wild-type α-synuclein is a substrate of CMA [117, 118], pathogenic species of α-synuclein, such as A53T and A30P mutant α-synuclein, were found to fail translocation through the lysosomal membrane and furthermore impair degradation of other CMA substrates by binding LAMP-2A [117, 119]. Important to sporadic PD, dopamine modified wild-type α-synuclein inhibited CMA in a similar way [120]. Intriguingly, the turnover of the neuronal transcription factor MEF2D was found to depend on CMA, which was significantly disrupted by the presence of wild-type and mutant α-synuclein, leading to impaired MEF2D signaling and neurodegeneration [121]. Rat and mouse α-synuclein, containing the A53T substitution seen in familial forms of PD [122], are degraded by CMA [117, 118, 123], although this seems incongruent with findings for human A53T mutant α-synuclein [117]. Serine129 phosphorylated α-synuclein and α-synuclein oligomers are not degraded by CMA [120]. In vivo, α-synuclein transgenic mice were found to upregulate LAMP-2A, providing evidence that CMA is part of the stress response in synucleinopathies [123]. In post-mortem pathological studies, levels of CMA adapter proteins were found to be altered in both PD [109] and DLB [114, 115]. In addition, decreased levels of CMA proteins LAMP-2A and Hsc70 in PD brain samples were found to be secondary to deregulation of several microRNAs that regulate LAMP-2A and Hsc70 expression [124]. Providing further insights into the role of CMA in synucleinopathies, Malkus and Ischiropoulos recently showed that regional deficits in CMA might underlie α-synuclein aggregation and neurodegeneration in the human A53T α-synuclein transgenic mouse model [125]. CMA activity was significantly decreased in aggregation-prone regions compared to other brain regions less affected by α-synuclein pathology. Upregulation of LAMP-2A occurred in regions with developing α-synuclein inclusion bodies although this dynamic transient response was not proportional to substrate uptake or degradation [125]. Exploring the therapeutic potential of CMA in synucleinopathies, Xilouri et al. recently showed that overexpression of LAMP-2A in cell models leads to increased CMA and protection from α-synuclein-induced degeneration [126]. Interestingly, this protective effect was present even when steady-state levels of α-synuclein were unchanged, suggesting that mitigating α-synuclein induced CMA dysfunction mainly accounts for the protective properties [126]. In vivo, viral vector-mediated co-overexpression of LAMP-2A in the substantia nigra of the AAV-mediated α-synuclein overexpression mouse model of PD completely preserved nigral tyrosine hydroxylase positive neurons and restored striatal levels of dopamine [126]. Collectively, these findings highlight the important role of CMA in synucleinopathies and the potential of modulating CMA as a novel therapeutic approach.
Chaperones, endoplasmic reticulum stress and apoptosis – implications for neuroprotection in synucleinopathies
Chaperones might protect neurons by mechanisms unrelated to their chaperone function, for example by regulating key steps in programmed cell death pathways. Programmed cell death is an umbrella term that includes apoptosis (or type I cell death) and autophagic cell death (or type II cell death), both of which are implicated in progressive neurodegenerative diseases such as PD [127]. The intrinsic or mitochondrial pathway of apoptosis is of particular importance to neurodegeneration. In this pathway three distinct phases can be delineated [128]. In the pre-mitochondrial initiation phase, cells recognize danger signals and respond by activating death-inducing pathways but also pro-survival signals in an attempt to fight cellular stressors. This is followed by the integration or mitochondrial phase, in which pro- and anti-apoptotic cascades converge on mitochondria. When pro-apoptotic signals dominate, mitochondrial membrane permeabilization follows, leading to cell death if a critical number of mitochondria are affected. In the execution or post-mitochondrial phase, mitochondrial membrane permeabilization results in the breakdown of the mitochondrial transmembrane potential, respiratory chain uncoupling, ATP depletion, generation of reactive oxygen species, the release of pro-apoptotic proteins into the cytosol and finally cell death.
Along with the mitochondrial pathway of apoptosis, chaperones, such as Hsp27, Hsp70 and Hsp90, are induced in response to various cellular stressors for example DNA damage, growth factor withdrawal, hypoxia or cytotoxic drugs [128, 129]. Several chaperones have been shown to prevent apoptosis by interfering with key regulatory proteins at different stages of the mitochondrial pathway of apoptosis (see [129–131] for a detailed review). This occurs for example by inhibiting the translocation of the pro-apoptotic protein Bax to the mitochondrial membrane and subsequent prevention of membrane permeabilization and cytochrome c release, the central phenomenon in the mitochondrial apoptosis pathway [132, 133]. Other mechanisms include direct association with Apaf-1 (apoptotic peptidase activating factor 1) by Hsp70 [134–137], blockage of AIF (apoptosis inducing factor) mitochondrial release and nuclear import [136, 138–140], interaction with cytochrome c [141] or inhibition of cathepsin release from lysosomes [142].
With regard to neurotoxin-induced models of neurodegeneration, toxic effects of rotenone and MPTP were significantly ameliorated following a transient heat-shock induced overexpression of chaperones [143–145], overexpression of Hsp70 [146] or cell-penetrating peptide (TAT-Hsp70) mediated delivery of Hsp70 in cells and mice [147]. Similarly, overexpression of Hsp27 reduced 6-hydroxydopamine induced cytochrome c release and apoptosis in dopaminergic cells [148].
In addition to their influence on mitochondrial apoptosis signaling, chaperones play a pivotal role in the endoplasmic reticulum (ER)-associated stress response. Disturbance of ER function caused by dysfunction of the ubiquitin-proteasome system and/or the accumulation of misfolded proteins leads to an evolutionary conserved stress response, termed unfolded protein response (UPR) (see [149–151] for a review). This involves a global suppression of protein synthesis and the expression of specific proteins, including ER associated chaperones such as the glucose-regulated protein 78 (Grp78/Bip), in an attempt to promote cell survival. However, if protein accumulation and ER dysfunction are severe, apoptosis will be eventually triggered [152]. Important to synucleinopathies, activation of the UPR seems to be an early event in the pathogenesis of PD [153, 154] and MSA [155], a finding that can be recapitulated in diseases models in vitro and in vivo [156–161]. Hoozemans et al. found increased immunoreactivity for UPR markers, phosphorylated pancreatic-like ER kinase (PERK) and eukaryotic translation initiation factor 2α (eIF2α), in neuromelanin containing dopaminergic neurons in the substantia nigra pars compacta of post-mortem PD brain samples [153]. In addition, phosphorylated PERK co-localized with increased α-synuclein immunoreactivity in dopaminergic neurons [153]. This is in agreement with increased UPR activation in models of increased A53T mutant [156, 160] or wild-type [157, 159] and phosphorylated α-synuclein [158] expression. The ER-associated chaperone and member of the heat shock protein 70 family, Grp78/BIP is at the forefront of regulating the UPR pathways. When misfolded proteins accumulate within the ER, Grp78/Bip dissociates from the three major ER stress receptors (PERK, activating transcription factor 6 (ATF6) and inositol-requiring enzyme 1 (IRE1)) capable of initiating the UPR. In agreement with the finding that Grp78/Bip binds accumulating misfolded proteins in the ER, several studies found that Grp78/Bip forms a complex with α-synuclein in cell and animal models showing α-synuclein accumulation [159–161]. This underscores the important role of this ER chaperone in the response to increased α-synuclein misfolding and aggregation. Using A53T α-synuclein transgenic mice, Colla et al. were further able to show that α-synuclein accumulates in the ER, induces ER chaperones and sensitizes neuronal cell to ER stress induced cell death [160]. In a second elegant study, Colla et al. found that toxic α-synuclein oligomers form within the ER lumen and thus might compromise the integrity of ER membranes, hence leading to chronic ER stress [162]. Exploring the therapeutic implications of attenuating ER stress, treatment of A53T α-synuclein mice and a viral-vector mediated rat of α-synucleinopathy with Salubrinal, a pharmacological inhibitor of ER stress induced toxicity, dramatically delayed the onset of motoric symptoms and decreased accumulation of α-synuclein oligomers in vivo. Further exploring the ER-associated chaperone Grp78/Bip as a therapeutic target, Gorbatyuk et al. recently showed that overexpression of this chaperone in the substantia nigra of a viral-vector mediated rat model of synucleinopathy attenuated α-synuclein-induced neurotoxicity by reducing ER stress mediators [161].
Modulation of molecular chaperones as a novel therapeutic target in synucleinopathies
Development of neuroprotective therapies for PD and other synucleinopathies is challenging because of the slow progressive nature of these diseases, the lack of reliable biomarkers for early disease detection or disease progression and limitations of available animal models. While the available symptomatic treatment for PD patients can substantially improve motor symptoms and quality of life, there is currently no therapeutic approach that can halt or reverse neuronal degeneration in PD and other synucleinopathies. Promising novel treatment strategies that were successfully identified and evaluated in pre-clinical models include cell-based therapies (reviewed in [163]) and compounds that target different cellular pathways including mitochondrial dysfunction (reviewed in [164]), mechanisms of oxidative stress, glutamate excitotoxicity and trophic factors (reviewed in [165]) as well as altered protein metabolism (reviewed in [18]). These targets are important to many neurodegenerative diseases and research efforts will therefore not only serve patients with PD but also patients who suffer from other major diseases such as DLB, Alzheimer’s disease or Huntington’s disease. Targets in protein metabolism include misfolding and aggregation, post-translational modification and protein degradation pathways such as the ubiquitin-proteasome system and autophagy [16–18, 21, 22]. Molecular chaperones are crucially involved in protein folding and refolding and thus are promising targets that have the potential to alter early pathological changes in synucleinopathies, potentially even before significant neurodegeneration has occurred. The Hsp70 system, in particular, has emerged as a promising new target to prevent or even reverse protein misfolding and associated toxicity.
A growing number of preclinical studies have employed pharmacological compounds to upregulate chaperone expression and/or function [see [17, 22] for a detailed review]. Testing of chaperone-based therapies is not limited to PD but has been greatly influenced by research in related diseases, most importantly the trinucleotide-repeat expansion diseases [166]. Based on similarities between disease models and mechanisms, many of the compounds tested in other diseases might be promising candidates for synucleinopathies [17]. Pharmacological agents targeting molecular chaperones have mainly focused on the Hsp70 system and are categorized into three groups according to their mechanism of action: A) Hsp90 inhibitors, B) modulators of HSF-1 and C) chemical chaperones (Table 1).
Hsp90 inhibitors have received considerable attention for the treatment of advanced cancers [180]. Following drug development in oncology, an increasing number of small molecule inhibitors of Hsp90 have been investigated in neurodegenerative diseases including models of PD (Table 1A & Table 2). Besides many other effects on client proteins and associated pathways, Hsp90 inhibitors induce the activity of the transcription factor HSF-1 and thus lead to increased expression of stress-induced proteins such as Hsp70. The first compound that was investigated in PD models was Geldanamycin, a naturally occurring antibiotic of the Ansamycin family. McLean et al. found that treatment with Geldanamycin in cell culture models effectively reduced α-synuclein aggregation through increasing its clearance, leading to reduced toxicity [168]. Auluck et al. confirmed neuroprotective effects of Geldanamycin in a Drosophila melanogaster model of α-synuclein toxicity [81, 167, 169] and Shen et al. found a protective effect in the MPTP mouse model of PD [181]. Interestingly, Hsp90 also seems to be involved in the exocytosis of α-synuclein [171]. Extracellular α-synuclein, once secreted, is subject to endocytosis by adjacent cells and at least a part of the internalized α-synuclein is re-secreted, which could represent a key step in the cascade that allows cell-to-cell propagation of α-synuclein aggregates [49–51]. Liu et al. further reported that Hsp90 inhibition with Geldanamycin protects cells against extracellular α-synuclein-induced neurotoxicity by preventing re-secretion of α-synuclein [171]. Although these findings have been encouraging, the use of Geldanamycin has been limited for pharmacokinetic reasons, most importantly its poor solubility and blood–brain-barrier penetration. Other members of the Ansamycin family, like 17-AAG (Tanespimycin) and 17-DMAG (Alvespimycin), have better pharmacokinetic profiles, but other limitations [182]. Similar to Geldanamycin, 17-AAG attenuates α-synuclein toxicity, prevents oligomerization and facilitates α-synuclein clearance in cultured cells [45, 46]. Moreover, 17-AAG can effectively enhance α-synuclein clearance via macroautophagy, a potential key pathway downstream of protein misfolding [173]. Current phase I/II trials for various forms of cancer have demonstrated safety, but the use of 17-AAG in neurodegenerative diseases remains limited because of poor blood–brain-barrier permeability [180]. 17-DMAG displays better solubility but further clinical development of this compound in oncology has not been pursued due to toxicity [180, 183]. In view of these limitations, the clinical utility of all three compounds Geldanamycin, 17-AAG and 17-DMAG is questionable, despite encouraging results in disease models (Table 1A). Novel synthetic small-molecule inhibitors of Hsp90 such as SNX-2112 and derived compounds have been identified through compound library screens for scaffolds that selectively bind the ATP-binding pocket of Hsp90 and display good pharmacokinetic characteristics including blood–brain-barrier penetration. Treatment with SNX compounds in cell culture models of PD resulted in a decrease of both high-molecular weight and monomeric α-synuclein as well as a significant reduction of α-synuclein oligomerization [45] (Table 1A). Despite these promising findings, further in vivo evaluation is clearly necessary to evaluate the general prospect of Hsp90 inhibitors for the treatment of synucleinopathies.
Modulators of HSF-1 have mainly been evaluated in models of neurodegenerative diseases other than synucleinopathies. For example, Arimoclomol, a compound that prolongs the binding of HSF-1 to heat-shock-response elements and thus increases the expression of Hsp70 and other chaperones under conditions of protein overload, has shown very encouraging results in models of spinal and bulbar muscular atrophy [187] and has even reached clinical testing in amyotrophic lateral sclerosis [188, 189]. Celastrol, a compound that promotes phosphorylation of HSF-1, was found to significantly ameliorate MPTP-induced neurodegeneration in the MPTP mouse model [184] and the DJ-1A Drosophila melanogaster model of PD [190] (Table 2). Carbenoxolone (CBX), a glycyrrhizic acid derivative, was found to activate HSF-1 and to promote Hsp70 expression which can ameliorate α-synuclein aggregation in cells [174] (Table 1B).
Given the importance of HSF-1 as the master regulator of chaperone gene transcription and the limitations of global Hsp90 inhibition, small molecules that directly modulate this transcription factor are clearly advantageous. Recently, a yeast-based high-throughput screen for small molecule activators of HSF-1 identified the compound HSF1A. This compound was shown to promote HSF-1 in an Hsp90 independent manner and without the presence of proteotoxicity [191]. HSF1A-mediated Hsp70 induction reduced the de novo formation of protein aggregates and ameliorated polyglutamine-induced cytotoxicity in both a cell and Drosophila melanogaster model of Huntington’s disease [191]. Another recent sophisticated small molecule screen identified small molecule proteostasis regulators that induce HSF-1-dependent chaperone expression and importantly reduce aggregate formation and toxicity in cells and a Caenorhabditis elegans model for expression of expanded polyglutamines [192].
Compounds with direct chaperone activity, or chemical chaperones, are also being evaluated as potential therapies (Table 1C & Table 2). For example, trehalose, a disaccharide, is able to act as a chemical chaperone through direct interaction with client proteins but can also enhance protein clearance via the autophagy pathway, with beneficial effects in different models of major neurodegenerative diseases [175, 176, 185, 193–197]. The chemical chaperones 4-phenylbutyrate [179, 186], mannosylglycerate [178] and most recently mannitol [177] can significantly ameliorate α-synuclein aggregation and toxicity in a variety of PD models including yeast, Drosophila melanogaster and mouse models (Table 1C & Table 2). Given the low toxicity of most chemical chaperones tested, these compounds might be good candidates for future drug development.
Conclusions
Impaired protein metabolism is a unifying theme in neurodegenerative diseases. To prevent the formation of potentially toxic α-synuclein oligomers and aggregates, a number of exciting chaperone-based therapies are under development for use in PD. Encouraging approaches include small molecule inhibitors of Hsp90 and other strategies that target Hsp70 expression or chemical chaperones (Tables 1 & 2). Enhancing chaperone function might be able to prevent early pathological changes such as the formation of α-synuclein oligomers. With the limitations discussed above, a number of studies in disease models clearly implicate a pivotal role for chaperones and protein misfolding in the pathogenesis of PD and other synucleinopathies (Figure 1). It should be cautioned however, that despite promising results in cellular models, in vivo data are still limited. The same limitations that apply to all neuroprotective therapies on trial will also challenge testing of chaperone-based therapeutics [17]. It remains a conceptual question, whether a single agent targeted at increasing the expression of chaperone proteins will have an enduring neuroprotective effect given the presence of numerous other established disease pathways and mechanisms [17]. Approaches that employ multiple targets such as the chaperone and proteasome system or chaperones and the CMA pathway seem reasonable. With these and the specific limitations discussed above, it is now on future studies to identify novel approaches capable of preventing α-synuclein misfolding and toxicity in PD and related synucleinopathies.
Abbreviations
- DLB:
-
Dementia with Lewy bodies
- ER:
-
Endoplasmic reticulum
- Grp78/Bip:
-
Glucose-regulated protein 78
- Hsc:
-
Heat shock cognate
- HSF-1:
-
Heat shock transcription factor 1
- Hsp:
-
Heat shock protein
- LAMP-2A:
-
Lysosome-associated membrane protein type 2A
- MPTP:
-
1-methyl-4-pheny-1,2,3,6-tetrahydropyridine
- PD:
-
Parkinson’s disease
- PERK:
-
Pancreatic-like ER kinase
- SIRT1:
-
Sirtuin 1
- SNARE:
-
Soluble NSF attachment protein receptor
- TAT:
-
Trans-activator of transcription
- UPR:
-
Unfolded protein response.
References
de Lau LM, Breteler MM: Epidemiology of Parkinson’s disease. Lancet Neurol 2006, 5: 525–535. 10.1016/S1474-4422(06)70471-9
Lees AJ, Hardy J, Revesz T: Parkinson's disease. Lancet 2009, 373: 2055–2066. 10.1016/S0140-6736(09)60492-X
Dickson DW: Parkinson's disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med 2012, 2: a009258.
Stefanis L: alpha-Synuclein in Parkinson's disease. Cold Spring Harb Perspect Med 2012, 2: a009399.
Lashuel HA, Overk CR, Oueslati A, Masliah E: The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 2013, 14: 38–48.
Halliday GM, Holton JL, Revesz T, Dickson DW: Neuropathology underlying clinical variability in patients with synucleinopathies. Acta Neuropathol 2011, 122: 187–204. 10.1007/s00401-011-0852-9
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT: Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 2011, 1: a006189.
Irwin DJ, Lee VM, Trojanowski JQ: Parkinson's disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci 2013, 14: 626–636. 10.1038/nrn3549
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al.: Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 1997, 276: 2045–2047. 10.1126/science.276.5321.2045
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O: Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet 1998, 18: 106–108. 10.1038/ng0298-106
Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, et al.: The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004, 55: 164–173. 10.1002/ana.10795
Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, et al.: alpha-Synuclein locus triplication causes Parkinson's disease. Science 2003, 302: 841. 10.1126/science.1090278
Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, Agid Y, Durr A, Brice A: Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet 2004, 364: 1169–1171. 10.1016/S0140-6736(04)17104-3
Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, et al.: Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 2004, 364: 1167–1169. 10.1016/S0140-6736(04)17103-1
Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, Saad M, Simon-Sanchez J, Schulte C, Lesage S, Sveinbjornsdottir S, et al.: Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet 2011, 377: 641–649.
Dimant H, Ebrahimi-Fakhari D, McLean PJ: Molecular chaperones and co-chaperones in Parkinson disease. Neuroscientist 2012, 18: 589–601. 10.1177/1073858412441372
Ebrahimi-Fakhari D, Wahlster L, McLean PJ: Molecular Chaperones in Parkinson's Disease - Present and Future. J Parkinsons Dis 2011, 1: 299–320.
Ebrahimi-Fakhari D, Wahlster L, McLean PJ: Protein degradation pathways in Parkinson's disease: curse or blessing. Acta Neuropathol 2012, 124: 153–172. 10.1007/s00401-012-1004-6
Kalia LV, Kalia SK, McLean PJ, Lozano AM: Lang AE: alpha-Synuclein oligomers and clinical implications for Parkinson disease. Ann Neurol 2013, 73: 155–169. 10.1002/ana.23746
Ebrahimi-Fakhari D, McLean PJ, Unni VK: Alpha-synuclein's degradation in vivo: opening a new (cranial) window on the roles of degradation pathways in Parkinson disease. Autophagy 2012, 8: 281–283. 10.4161/auto.8.2.18938
Cook C, Stetler C, Petrucelli L: Disruption of protein quality control in Parkinson's disease. Cold Spring Harb Perspect Med 2012, 2: a009423.
Kalia SK, Kalia LV, McLean PJ: Molecular chaperones as rational drug targets for Parkinson's disease therapeutics. CNS Neurol Disord Drug Targets 2010, 9: 741–753. 10.2174/187152710793237386
Nixon RA: The role of autophagy in neurodegenerative disease. Nat Med 2013, 19: 983–997. 10.1038/nm.3232
Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC: Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 2005, 123: 383–396. 10.1016/j.cell.2005.09.028
Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC: Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329: 1663–1667. 10.1126/science.1195227
Davidson WS, Jonas A, Clayton DF, George JM: Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem 1998, 273: 9443–9449. 10.1074/jbc.273.16.9443
Weinreb PH, Zhen W, Poon AW, Conway KA, Lansbury PT Jr: NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry 1996, 35: 13709–13715. 10.1021/bi961799n
Bartels T, Choi JG, DJ S: alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477: 107–110. 10.1038/nature10324
Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, et al.: A soluble alpha-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A 2011, 108: 17797–17802. 10.1073/pnas.1113260108
Dettmer U, Newman AJ, Luth ES, Bartels T, Selkoe D: In vivo cross-linking reveals principally oligomeric forms of alpha-synuclein and beta-synuclein in neurons and non-neural cells. J Biol Chem 2013, 288: 6371–6385. 10.1074/jbc.M112.403311
Burre J, Vivona S, Diao J, Sharma M, Brunger AT, Sudhof TC: Properties of native brain alpha-synuclein. Nature 2013, 498: E4-E6. discussion E6–7 10.1038/nature12125
Fauvet B, Mbefo MK, Fares MB, Desobry C, Michael S, Ardah MT, Tsika E, Coune P, Prudent M, Lion N, et al.: alpha-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem 2012, 287: 15345–15364. 10.1074/jbc.M111.318949
Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT Jr: Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A 2000, 97: 571–576. 10.1073/pnas.97.2.571
Hashimoto M, Hsu LJ, Xia Y, Takeda A, Sisk A, Sundsmo M, Masliah E: Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro. Neuroreport 1999, 10: 717–721. 10.1097/00001756-199903170-00011
Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T: alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 2002, 4: 160–164.
Paleologou KE, Oueslati A, Shakked G, Rospigliosi CC, Kim HY, Lamberto GR, Fernandez CO, Schmid A, Chegini F, Gai WP, et al.: Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J Neurosci 2010, 30: 3184–3198. 10.1523/JNEUROSCI.5922-09.2010
Li W, West N, Colla E, Pletnikova O, Troncoso JC, Marsh L, Dawson TM, Jakala P, Hartmann T, Price DL, Lee MK: Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson's disease-linked mutations. Proc Natl Acad Sci U S A 2005, 102: 2162–2167. 10.1073/pnas.0406976102
Dufty BM, Warner LR, Hou ST, Jiang SX, Gomez-Isla T, Leenhouts KM, Oxford JT, Feany MB, Masliah E, Rohn TT: Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am J Pathol 2007, 170: 1725–1738. 10.2353/ajpath.2007.061232
Perrin RJ, Woods WS, Clayton DF, George JM: Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J Biol Chem 2001, 276: 41958–41962. 10.1074/jbc.M105022200
Sharon R, Bar-Joseph I, Frosch MP, Walsh DM, Hamilton JA, Selkoe DJ: The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson's disease. Neuron 2003, 37: 583–595. 10.1016/S0896-6273(03)00024-2
Karube H, Sakamoto M, Arawaka S, Hara S, Sato H, Ren CH, Goto S, Koyama S, Wada M, Kawanami T, et al.: N-terminal region of alpha-synuclein is essential for the fatty acid-induced oligomerization of the molecules. FEBS Lett 2008, 582: 3693–3700. 10.1016/j.febslet.2008.10.001
Paleologou KE, Kragh CL, Mann DM, Salem SA, Al-Shami R, Allsop D, Hassan AH, Jensen PH, El-Agnaf OM: Detection of elevated levels of soluble alpha-synuclein oligomers in post-mortem brain extracts from patients with dementia with Lewy bodies. Brain 2009, 132: 1093–1101.
Outeiro TF, Putcha P, Tetzlaff JE, Spoelgen R, Koker M, Carvalho F, Hyman BT, McLean PJ: Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS One 2008, 3: e1867. 10.1371/journal.pone.0001867
Tetzlaff JE, Putcha P, Outeiro TF, Ivanov A, Berezovska O, Hyman BT, McLean PJ: CHIP targets toxic alpha-Synuclein oligomers for degradation. J Biol Chem 2008, 283: 17962–17968. 10.1074/jbc.M802283200
Putcha P, Danzer KM, Kranich LR, Scott A, Silinski M, Mabbett S, Hicks CD, Veal JM, Steed PM, Hyman BT, McLean PJ: Brain-permeable small-molecule inhibitors of Hsp90 prevent alpha-synuclein oligomer formation and rescue alpha-synuclein-induced toxicity. J Pharmacol Exp Ther 2010, 332: 849–857. 10.1124/jpet.109.158436
Danzer KM, Ruf WP, Putcha P, Joyner D, Hashimoto T, Glabe C, Hyman BT, McLean PJ: Heat-shock protein 70 modulates toxic extracellular alpha-synuclein oligomers and rescues trans-synaptic toxicity. Faseb J 2011, 25: 326–336. 10.1096/fj.10-164624
Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, et al.: In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A 2011, 108: 4194–4199. 10.1073/pnas.1100976108
Dimant H, Kalia S, Kalia L, Zhu L, Kibuuka L, Ebrahimi-Fakhari D, McFarland N, Fan Z, Hyman B, McLean P: Direct detection of alpha synuclein oligomers in vivo. Acta Neuropathologica Communications 2013, 1: 6. 10.1186/2051-5960-1-6
Angot E, Steiner JA, Hansen C, Li JY, Brundin P: Are synucleinopathies prion-like disorders? Lancet Neurol 2010, 9: 1128–1138. 10.1016/S1474-4422(10)70213-1
Hansen C, Li JY: Beyond alpha-synuclein transfer: pathology propagation in Parkinson's disease. Trends Mol Med 2012, 18: 248–255. 10.1016/j.molmed.2012.03.002
Visanji N, Brooks P, Hazrati L-N, Lang A: The prion hypothesis in Parkinson's disease: Braak to the future. Acta Neuropathologica Communications 2013, 1: 2. 10.1186/2051-5960-1-2
Hartl FU, Bracher A, Hayer-Hartl M: Molecular chaperones in protein folding and proteostasis. Nature 2011, 475: 324–332. 10.1038/nature10317
Tyedmers J, Mogk A, Bukau B: Cellular strategies for controlling protein aggregation. Nat Rev Mol Cell Biol 2010, 11: 777–788. 10.1038/nrm2993
Gidalevitz T, Prahlad V, Morimoto RI: The stress of protein misfolding: from single cells to multicellular organisms. Cold Spring Harb Perspect Biol 2011, 3: a009704.
Finka A, Goloubinoff P: Proteomic data from human cell cultures refine mechanisms of chaperone-mediated protein homeostasis. Cell Stress Chaperones 2013, 18: 591–605. 10.1007/s12192-013-0413-3
Hartl FU, Hayer-Hartl M: Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol 2009, 16: 574–581. 10.1038/nsmb.1591
Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, Rockenstein E, Masliah E, Hyman BT, McLean PJ, Unni VK: Distinct Roles In Vivo for the Ubiquitin-Proteasome System and the Autophagy-Lysosomal Pathway in the Degradation of {alpha}-Synuclein. J Neurosci 2011, 31: 14508–14520. 10.1523/JNEUROSCI.1560-11.2011
Finka A, Mattoo RU, Goloubinoff P: Meta-analysis of heat- and chemically upregulated chaperone genes in plant and human cells. Cell Stress Chaperones 2011, 16: 15–31. 10.1007/s12192-010-0216-8
Richter K, Haslbeck M, Buchner J: The heat shock response: life on the verge of death. Mol Cell 2010, 40: 253–266. 10.1016/j.molcel.2010.10.006
Vabulas RM, Raychaudhuri S, Hayer-Hartl M, Hartl FU: Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb Perspect Biol 2010, 2: a004390.
Zou J, Guo Y, Guettouche T, Smith DF, Voellmy R: Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 1998, 94: 471–480. 10.1016/S0092-8674(00)81588-3
Mosser DD, Theodorakis NG, Morimoto RI: Coordinate changes in heat shock element-binding activity and HSP70 gene transcription rates in human cells. Mol Cell Biol 1988, 8: 4736–4744.
Morimoto RI: Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev 1998, 12: 3788–3796. 10.1101/gad.12.24.3788
Shi Y, Mosser DD, Morimoto RI: Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev 1998, 12: 654–666. 10.1101/gad.12.5.654
Kaushik S, Cuervo AM: Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 2012, 22: 407–417. 10.1016/j.tcb.2012.05.006
Dice JF: Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci 1990, 15: 305–309. 10.1016/0968-0004(90)90019-8
Chiang HL, Terlecky SR, Plant CP, Dice JF: A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 1989, 246: 382–385. 10.1126/science.2799391
Agarraberes FA, Dice JF: A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci 2001, 114: 2491–2499.
Cuervo AM, Dice JF: A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996, 273: 501–503. 10.1126/science.273.5274.501
Agarraberes FA, Terlecky SR, Dice JF: An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol 1997, 137: 825–834. 10.1083/jcb.137.4.825
Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM: The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol 2008, 28: 5747–5763. 10.1128/MCB.02070-07
Cuervo AM, Dice JF, Knecht E: A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem 1997, 272: 5606–5615. 10.1074/jbc.272.9.5606
Cuervo AM, Dice JF: Age-related decline in chaperone-mediated autophagy. J Biol Chem 2000, 275: 31505–31513.
Kiffin R, Kaushik S, Zeng M, Bandyopadhyay U, Zhang C, Massey AC, Martinez-Vicente M, Cuervo AM: Altered dynamics of the lysosomal receptor for chaperone-mediated autophagy with age. J Cell Sci 2007, 120: 782–791. 10.1242/jcs.001073
Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY: Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet 1998, 19: 148–154. 10.1038/502
Kazemi-Esfarjani P, Benzer S: Genetic suppression of polyglutamine toxicity in Drosophila. Science 2000, 287: 1837–1840. 10.1126/science.287.5459.1837
Chai Y, Koppenhafer SL, Bonini NM, Paulson HL: Analysis of the role of heat shock protein (Hsp) molecular chaperones in polyglutamine disease. J Neurosci 1999, 19: 10338–10347.
Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM: Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet 1999, 23: 425–428. 10.1038/70532
McLean PJ, Kawamata H, Shariff S, Hewett J, Sharma N, Ueda K, Breakefield XO, Hyman BT: TorsinA and heat shock proteins act as molecular chaperones: suppression of alpha-synuclein aggregation. J Neurochem 2002, 83: 846–854. 10.1046/j.1471-4159.2002.01190.x
Uryu K, Richter-Landsberg C, Welch W, Sun E, Goldbaum O, Norris EH, Pham CT, Yazawa I, Hilburger K, Micsenyi M, et al.: Convergence of heat shock protein 90 with ubiquitin in filamentous alpha-synuclein inclusions of alpha-synucleinopathies. Am J Pathol 2006, 168: 947–961. 10.2353/ajpath.2006.050770
Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM: Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science 2002, 295: 865–868. 10.1126/science.1067389
Leverenz JB, Umar I, Wang Q, Montine TJ, McMillan PJ, Tsuang DW, Jin J, Pan C, Shin J, Zhu D, Zhang J: Proteomic identification of novel proteins in cortical lewy bodies. Brain Pathol 2007, 17: 139–145. 10.1111/j.1750-3639.2007.00048.x
Zhou Y, Gu G, Goodlett DR, Zhang T, Pan C, Montine TJ, Montine KS, Aebersold RH, Zhang J: Analysis of alpha-synuclein-associated proteins by quantitative proteomics. J Biol Chem 2004, 279: 39155–39164. 10.1074/jbc.M405456200
Kuhn K, Wellen J, Link N, Maskri L, Lubbert H, Stichel CC: The mouse MPTP model: gene expression changes in dopaminergic neurons. Eur J Neurosci 2003, 17: 1–12. 10.1046/j.1460-9568.2003.02408.x
St Martin JL, Klucken J, Outeiro TF, Nguyen P, Keller-McGandy C, Cantuti-Castelvetri I, Grammatopoulos TN, Standaert DG, Hyman BT, McLean PJ: Dopaminergic neuron loss and up-regulation of chaperone protein mRNA induced by targeted over-expression of alpha-synuclein in mouse substantia nigra. J Neurochem 2007, 100: 1449–1457.
Donmez G, Arun A, Chung CY, McLean PJ, Lindquist S, Guarente L: SIRT1 protects against alpha-synuclein aggregation by activating molecular chaperones. J Neurosci 2012, 32: 124–132. 10.1523/JNEUROSCI.3442-11.2012
Fan GH, Zhou HY, Yang H, Chen SD: Heat shock proteins reduce alpha-synuclein aggregation induced by MPP + in SK-N-SH cells. FEBS Lett 2006, 580: 3091–3098. 10.1016/j.febslet.2006.04.057
Outeiro TF, Klucken J, Strathearn KE, Liu F, Nguyen P, Rochet JC, Hyman BT, McLean PJ: Small heat shock proteins protect against alpha-synuclein-induced toxicity and aggregation. Biochem Biophys Res Commun 2006, 351: 631–638. 10.1016/j.bbrc.2006.10.085
Zourlidou A: Payne Smith MD, Latchman DS: HSP27 but not HSP70 has a potent protective effect against alpha-synuclein-induced cell death in mammalian neuronal cells. J Neurochem 2004, 88: 1439–1448. 10.1046/j.1471-4159.2003.02273.x
Klucken J, Shin Y, Hyman BT, McLean PJ: A single amino acid substitution differentiates Hsp70-dependent effects on alpha-synuclein degradation and toxicity. Biochem Biophys Res Commun 2004, 325: 367–373. 10.1016/j.bbrc.2004.10.037
Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ: Hsp70 Reduces alpha-Synuclein Aggregation and Toxicity. J Biol Chem 2004, 279: 25497–25502. 10.1074/jbc.M400255200
Shimshek DR, Mueller M, Wiessner C, Schweizer T, van der Putten PH: The HSP70 molecular chaperone is not beneficial in a mouse model of alpha-synucleinopathy. PLoS One 2010, 5: e10014. 10.1371/journal.pone.0010014
Cao S, Gelwix CC, Caldwell KA, Caldwell GA: Torsin-mediated protection from cellular stress in the dopaminergic neurons of Caenorhabditis elegans. J Neurosci 2005, 25: 3801–3812. 10.1523/JNEUROSCI.5157-04.2005
Li X, Lee J, Parsons D, Janaurajs K, Standaert DG: Evaluation of TorsinA as a target for Parkinson disease therapy in mouse models. PLoS One 2012, 7: e50063. 10.1371/journal.pone.0050063
Luk KC, Mills IP, Trojanowski JQ, Lee VM: Interactions between Hsp70 and the hydrophobic core of alpha-synuclein inhibit fibril assembly. Biochemistry 2008, 47: 12614–12625. 10.1021/bi801475r
Huang C, Cheng H, Hao S, Zhou H, Zhang X, Gao J, Sun QH, Hu H, Wang CC: Heat shock protein 70 inhibits alpha-synuclein fibril formation via interactions with diverse intermediates. J Mol Biol 2006, 364: 323–336. 10.1016/j.jmb.2006.08.062
Redeker V, Pemberton S, Bienvenut W, Bousset L, Melki R: Identification of Protein Interfaces between alpha-Synuclein, the Principal Component of Lewy Bodies in Parkinson Disease, and the Molecular Chaperones Human Hsc70 and the Yeast Ssa1p. J Biol Chem 2012, 287: 32630–32639. 10.1074/jbc.M112.387530
Klucken J, Outeiro TF, Nguyen P, McLean PJ, Hyman BT: Detection of novel intracellular alpha-synuclein oligomeric species by fluorescence lifetime imaging. FASEB J 2006, 20: 2050–2057. 10.1096/fj.05-5422com
Bruinsma IB, Bruggink KA, Kinast K, Versleijen AA, Segers-Nolten IM, Subramaniam V, Kuiperij HB, Boelens W, de Waal RM, Verbeek MM: Inhibition of alpha-synuclein aggregation by small heat shock proteins. Proteins 2011, 79: 2956–2967. 10.1002/prot.23152
Duennwald ML, Echeverria A, Shorter J: Small heat shock proteins potentiate amyloid dissolution by protein disaggregases from yeast and humans. PLoS Biol 2012, 10: e1001346. 10.1371/journal.pbio.1001346
Falsone SF, Kungl AJ, Rek A, Cappai R, Zangger K: The molecular chaperone Hsp90 modulates intermediate steps of amyloid assembly of the Parkinson-related protein alpha-synuclein. J Biol Chem 2009, 284: 31190–31199. 10.1074/jbc.M109.057240
Daturpalli S, Waudby CA, Meehan S, Jackson SE: Hsp90 Inhibits alpha-Synuclein Aggregation by Interacting with Soluble Oligomers. J Mol Biol 2013, 22: 4614–4628.
Kourtis N, Tavernarakis N: Cellular stress response pathways and ageing: intricate molecular relationships. Embo J 2011, 30: 2520–2531. 10.1038/emboj.2011.162
Iwaki T, Wisniewski T, Iwaki A, Corbin E, Tomokane N, Tateishi J, Goldman JE: Accumulation of alpha B-crystallin in central nervous system glia and neurons in pathologic conditions. Am J Pathol 1992, 140: 345–356.
Braak H, Del Tredici K, Sandmann-Kiel D, Rub U, Schultz C: Nerve cells expressing heat-shock proteins in Parkinson's disease. Acta Neuropathol 2001, 102: 449–454.
Waudby CA, Knowles TP, Devlin GL, Skepper JN, Ecroyd H, Carver JA, Welland ME, Christodoulou J, Dobson CM, Meehan S: The interaction of alphaB-crystallin with mature alpha-synuclein amyloid fibrils inhibits their elongation. Biophys J 2010, 98: 843–851. 10.1016/j.bpj.2009.10.056
Cantuti-Castelvetri I, Klucken J, Ingelsson M, Ramasamy K, McLean PJ, Frosch MP, Hyman BT, Standaert DG: Alpha-synuclein and chaperones in dementia with Lewy bodies. J Neuropathol Exp Neurol 2005, 64: 1058–1066. 10.1097/01.jnen.0000190063.90440.69
Klucken J, Ingelsson M, Shin Y, Irizarry MC, Hedley-Whyte ET, Frosch M, Growdon J, McLean P, Hyman BT: Clinical and biochemical correlates of insoluble alpha-synuclein in dementia with Lewy bodies. Acta Neuropathol 2006, 111: 101–108. 10.1007/s00401-005-0027-7
Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, Caballero C, Ferrer I, Obeso JA, Schapira AH: Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol 2010, 67: 1464–1472.
Hauser MA, Li YJ, Xu H, Noureddine MA, Shao YS, Gullans SR, Scherzer CR, Jensen RV, McLaurin AC, Gibson JR, et al.: Expression profiling of substantia nigra in Parkinson disease, progressive supranuclear palsy, and frontotemporal dementia with parkinsonism. Arch Neurol 2005, 62: 917–921.
Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH: Alterations in lysosomal and proteasomal markers in Parkinson's disease: relationship to alpha-synuclein inclusions. Neurobiol Dis 2009, 35: 385–398. 10.1016/j.nbd.2009.05.023
Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, Hansen L, Adame A, Galasko D, Masliah E: Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One 2010, 5: e9313. 10.1371/journal.pone.0009313
Dehay B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Boya P, Vila M: Pathogenic lysosomal depletion in Parkinson's disease. J Neurosci 2010, 30: 12535–12544. 10.1523/JNEUROSCI.1920-10.2010
Klucken J, Poehler AM, Ebrahimi-Fakhari D, Schneider J, Nuber S, Rockenstein E, Schlotzer-Schrehardt U, Hyman BT, McLean PJ, Masliah E, Winkler J: Alpha-synuclein aggregation involves a bafilomycin A 1-sensitive autophagy pathway. Autophagy 2012, 8: 754–766. 10.4161/auto.19371
Higashi S, Moore DJ, Minegishi M, Kasanuki K, Fujishiro H, Kabuta T, Togo T, Katsuse O, Uchikado H, Furukawa Y, et al.: Localization of MAP1-LC3 in vulnerable neurons and Lewy bodies in brains of patients with dementia with Lewy bodies. J Neuropathol Exp Neurol 2011, 70: 264–280. 10.1097/NEN.0b013e318211c86a
Hinault MP, Cuendet AF, Mattoo RU, Mensi M, Dietler G, Lashuel HA, Goloubinoff P: Stable alpha-synuclein oligomers strongly inhibit chaperone activity of the Hsp70 system by weak interactions with J-domain co-chaperones. J Biol Chem 2010, 285: 38173–38182. 10.1074/jbc.M110.127753
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D: Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305: 1292–1295. 10.1126/science.1101738
Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L: Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem 2008, 283: 23542–23556. 10.1074/jbc.M801992200
Xilouri M, Vogiatzi T, Vekrellis K, Park D, Stefanis L: Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS One 2009, 4: e5515. 10.1371/journal.pone.0005515
Martinez-Vicente M, Talloczy Z, Kaushik S, Massey AC, Mazzulli J, Mosharov EV, Hodara R, Fredenburg R, Wu DC, Follenzi A, et al.: Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest 2008, 118: 777–788.
Yang Q, She H, Gearing M, Colla E, Lee M, Shacka JJ, Mao Z: Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 2009, 323: 124–127. 10.1126/science.1166088
Lavedan C: The synuclein family. Genome Res 1998, 8: 871–880.
Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, Di Monte DA: Lysosomal degradation of alpha-synuclein in vivo. J Biol Chem 2010, 285: 13621–13629. 10.1074/jbc.M109.074617
Alvarez-Erviti L, Seow Y, Schapira AH, Rodriguez-Oroz MC, Obeso JA, Cooper JM: Influence of microRNA deregulation on chaperone-mediated autophagy and alpha-synuclein pathology in Parkinson's disease. Cell Death Dis 2013, 4: e545. 10.1038/cddis.2013.73
Malkus KA, Ischiropoulos H: Regional deficiencies in chaperone-mediated autophagy underlie alpha-synuclein aggregation and neurodegeneration. Neurobiol Dis 2012, 46: 732–744. 10.1016/j.nbd.2012.03.017
Xilouri M, Brekk OR, Landeck N, Pitychoutis PM, Papasilekas T, Papadopoulou-Daifoti Z, Kirik D, Stefanis L: Boosting chaperone-mediated autophagy in vivo mitigates alpha-synuclein-induced neurodegeneration. Brain 2013, 136: 2130–2146. 10.1093/brain/awt131
Venderova K, Park DS: Programmed cell death in Parkinson's disease. Cold Spring Harb Perspect Med 2012, 2: a009365.
Galluzzi L, Blomgren K, Kroemer G: Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci 2009, 10: 481–494. 10.1038/nrn2665
Lanneau D, Brunet M, Frisan E, Solary E, Fontenay M, Garrido C: Heat shock proteins: essential proteins for apoptosis regulation. J Cell Mol Med 2008, 12: 743–761. 10.1111/j.1582-4934.2008.00273.x
Fulda S, Gorman AM, Hori O, Samali A: Cellular stress responses: cell survival and cell death. Int J Cell Biol 2010, 2010: 214074.
Benarroch EE: Heat shock proteins: multiple neuroprotective functions and implications for neurologic disease. Neurology 2011, 76: 660–667. 10.1212/WNL.0b013e31820c3119
Stankiewicz AR, Lachapelle G, Foo CP, Radicioni SM, Mosser DD: Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J Biol Chem 2005, 280: 38729–38739. 10.1074/jbc.M509497200
Gotoh T, Terada K, Oyadomari S: Mori M: hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria. Cell Death Differ 2004, 11: 390–402. 10.1038/sj.cdd.4401369
Saleh A, Srinivasula SM, Balkir L, Robbins PD, Alnemri ES: Negative regulation of the Apaf-1 apoptosome by Hsp70. Nat Cell Biol 2000, 2: 476–483. 10.1038/35019510
Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, Zamzami N, Mak T, Jaattela M, Penninger JM, Garrido C, Kroemer G: Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol 2001, 3: 839–843. 10.1038/ncb0901-839
Sabirzhanov B, Stoica BA, Hanscom M, Piao CS, Faden AI: Over-expression of HSP70 attenuates caspase-dependent and caspase-independent pathways and inhibits neuronal apoptosis. J Neurochem 2012, 123: 542–554. 10.1111/j.1471-4159.2012.07927.x
Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR: Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol 2000, 2: 469–475. 10.1038/35019501
Ruchalski K, Mao H, Li Z, Wang Z, Gillers S, Wang Y, Mosser DD, Gabai V, Schwartz JH, Borkan SC: Distinct hsp70 domains mediate apoptosis-inducing factor release and nuclear accumulation. J Biol Chem 2006, 281: 7873–7880. 10.1074/jbc.M513728200
Gurbuxani S, Schmitt E, Cande C, Parcellier A, Hammann A, Daugas E, Kouranti I, Spahr C, Pance A, Kroemer G, Garrido C: Heat shock protein 70 binding inhibits the nuclear import of apoptosis-inducing factor. Oncogene 2003, 22: 6669–6678. 10.1038/sj.onc.1206794
Matsumori Y, Hong SM, Aoyama K, Fan Y, Kayama T, Sheldon RA, Vexler ZS, Ferriero DM, Weinstein PR, Liu J: Hsp70 overexpression sequesters AIF and reduces neonatal hypoxic/ischemic brain injury. J Cereb Blood Flow Metab 2005, 25: 899–910. 10.1038/sj.jcbfm.9600080
Bruey JM, Ducasse C, Bonniaud P, Ravagnan L, Susin SA, Diaz-Latoud C, Gurbuxani S, Arrigo AP, Kroemer G, Solary E, Garrido C: Hsp27 negatively regulates cell death by interacting with cytochrome c. Nat Cell Biol 2000, 2: 645–652. 10.1038/35023595
Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Hoyer-Hansen M, Weber E, Multhoff G, Rohde M, Jaattela M: Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med 2004, 200: 425–435. 10.1084/jem.20040531
Tantucci M, Mariucci G, Taha E, Spaccatini C, Tozzi A, Luchetti E, Calabresi P, Ambrosini MV: Induction of heat shock protein 70 reduces the alteration of striatal electrical activity caused by mitochondrial impairment. Neuroscience 2009, 163: 735–740. 10.1016/j.neuroscience.2009.06.070
Quigney DJ, Gorman AM, Samali A: Heat shock protects PC12 cells against MPP + toxicity. Brain Res 2003, 993: 133–139. 10.1016/j.brainres.2003.09.004
Bush KT, Goldberg AL, Nigam SK: Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J Biol Chem 1997, 272: 9086–9092. 10.1074/jbc.272.14.9086
Dong Z, Wolfer DP, Lipp HP, Bueler H: Hsp70 gene transfer by adeno-associated virus inhibits MPTP-induced nigrostriatal degeneration in the mouse model of Parkinson disease. Mol Ther 2005, 11: 80–88.
Nagel F, Falkenburger BH, Tonges L, Kowsky S, Poppelmeyer C, Schulz JB, Bahr M, Dietz GP: Tat-Hsp70 protects dopaminergic neurons in midbrain cultures and in the substantia nigra in models of Parkinson's disease. J Neurochem 2008, 105: 853–864. 10.1111/j.1471-4159.2007.05204.x
Gorman AM, Szegezdi E, Quigney DJ, Samali A: Hsp27 inhibits 6-hydroxydopamine-induced cytochrome c release and apoptosis in PC12 cells. Biochem Biophys Res Commun 2005, 327: 801–810. 10.1016/j.bbrc.2004.12.066
Walter P, Ron D: The unfolded protein response: from stress pathway to homeostatic regulation. Science 2011, 334: 1081–1086. 10.1126/science.1209038
Wang S, Kaufman RJ: The impact of the unfolded protein response on human disease. J Cell Biol 2012, 197: 857–867. 10.1083/jcb.201110131
Korennykh A, Walter P: Structural basis of the unfolded protein response. Annu Rev Cell Dev Biol 2012, 28: 251–277. 10.1146/annurev-cellbio-101011-155826
Gorman AM, Healy SJ, Jager R, Samali A: Stress management at the ER: regulators of ER stress-induced apoptosis. Pharmacol Ther 2012, 134: 306–316. 10.1016/j.pharmthera.2012.02.003
Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W: Activation of the unfolded protein response in Parkinson's disease. Biochem Biophys Res Commun 2007, 354: 707–711. 10.1016/j.bbrc.2007.01.043
Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Scheper W: Activation of the unfolded protein response is an early event in Alzheimer's and Parkinson's disease. Neurodegener Dis 2012, 10: 212–215. 10.1159/000334536
Makioka K, Yamazaki T, Fujita Y, Takatama M, Nakazato Y, Okamoto K: Involvement of endoplasmic reticulum stress defined by activated unfolded protein response in multiple system atrophy. J Neurol Sci 2010, 297: 60–65. 10.1016/j.jns.2010.06.019
Smith WW, Jiang H, Pei Z, Tanaka Y, Morita H, Sawa A, Dawson VL, Dawson TM, Ross CA: Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet 2005, 14: 3801–3811. 10.1093/hmg/ddi396
Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, et al.: Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science 2006, 313: 324–328. 10.1126/science.1129462
Sugeno N, Takeda A, Hasegawa T, Kobayashi M, Kikuchi A, Mori F, Wakabayashi K, Itoyama Y: Serine 129 phosphorylation of alpha-synuclein induces unfolded protein response-mediated cell death. J Biol Chem 2008, 283: 23179–23188. 10.1074/jbc.M802223200
Bellucci A, Navarria L, Zaltieri M, Falarti E, Bodei S, Sigala S, Battistin L, Spillantini M, Missale C, Spano P: Induction of the unfolded protein response by alpha-synuclein in experimental models of Parkinson's disease. J Neurochem 2011, 116: 588–605. 10.1111/j.1471-4159.2010.07143.x
Colla E, Coune P, Liu Y, Pletnikova O, Troncoso JC, Iwatsubo T, Schneider BL, Lee MK: Endoplasmic reticulum stress is important for the manifestations of alpha-synucleinopathy in vivo. J Neurosci 2012, 32: 3306–3320. 10.1523/JNEUROSCI.5367-11.2012
Gorbatyuk MS, Shabashvili A, Chen W, Meyers C, Sullivan LF, Salganik M, Lin JH, Lewin AS, Muzyczka N, Gorbatyuk OS: Glucose regulated protein 78 diminishes alpha-synuclein neurotoxicity in a rat model of Parkinson disease. Mol Ther 2012, 20: 1327–1337. 10.1038/mt.2012.28
Colla E, Jensen PH, Pletnikova O, Troncoso JC, Glabe C, Lee MK: Accumulation of toxic alpha-synuclein oligomer within endoplasmic reticulum occurs in alpha-synucleinopathy in vivo. J Neurosci 2012, 32: 3301–3305. 10.1523/JNEUROSCI.5368-11.2012
de Munter JP, Lee C, Wolters E: Cell based therapy in Parkinsonism. Transl Neurodegener 2013, 2: 13. 10.1186/2047-9158-2-13
Andreux PA, Houtkooper RH, Auwerx J: Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov 2013, 12: 465–483. 10.1038/nrd4023
Meissner WG, Frasier M, Gasser T, Goetz CG, Lozano A, Piccini P, Obeso JA, Rascol O, Schapira A, Voon V, et al.: Priorities in Parkinson's disease research. Nat Rev Drug Discov 2011, 10: 377–393. 10.1038/nrd3430
Voisine C, Pedersen JS, Morimoto RI: Chaperone networks: tipping the balance in protein folding diseases. Neurobiol Dis 2010, 40: 12–20. 10.1016/j.nbd.2010.05.007
Auluck PK, Bonini NM: Pharmacological prevention of Parkinson disease in Drosophila. Nat Med 2002, 8: 1185–1186. 10.1038/nm1102-1185
McLean PJ, Klucken J, Shin Y, Hyman BT: Geldanamycin induces Hsp70 and prevents alpha-synuclein aggregation and toxicity in vitro. Biochem Biophys Res Commun 2004, 321: 665–669. 10.1016/j.bbrc.2004.07.021
Auluck PK, Meulener MC, Bonini NM: Mechanisms of Suppression of {alpha}-Synuclein Neurotoxicity by Geldanamycin in Drosophila. J Biol Chem 2005, 280: 2873–2878.
Flower TR, Chesnokova LS, Froelich CA, Dixon C, Witt SN: Heat shock prevents alpha-synuclein-induced apoptosis in a yeast model of Parkinson's disease. J Mol Biol 2005, 351: 1081–1100. 10.1016/j.jmb.2005.06.060
Liu J, Zhang JP, Shi M, Quinn T, Bradner J, Beyer R, Chen S, Zhang J: Rab11a and HSP90 regulate recycling of extracellular alpha-synuclein. J Neurosci 2009, 29: 1480–1485. 10.1523/JNEUROSCI.6202-08.2009
Emmanouilidou E, Stefanis L, Vekrellis K: Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol Aging 2010, 31: 953–968. 10.1016/j.neurobiolaging.2008.07.008
Riedel M, Goldbaum O, Schwarz L, Schmitt S, Richter-Landsberg C: 17-AAG induces cytoplasmic alpha-synuclein aggregate clearance by induction of autophagy. PLoS One 2010, 5: e8753. 10.1371/journal.pone.0008753
Kilpatrick K, Novoa JA, Hancock T, Guerriero CJ, Wipf P, Brodsky JL, Segatori L: Chemical Induction of Hsp70 Reduces alpha-Synuclein Aggregation in Neuroglioma Cells. ACS Chem Biol 2013, 7: 1460–1468.
Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC: Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem 2007, 282: 5641–5652.
Yu WB, Jiang T, Lan DM, Lu JH, Yue ZY, Wang J, Zhou P: Trehalose inhibits fibrillation of A53T mutant alpha-synuclein and disaggregates existing fibrils. Arch Biochem Biophys 2012, 523: 144–150. 10.1016/j.abb.2012.04.021
Shaltiel-Karyo R, Frenkel-Pinter M, Rockenstein E, Patrick C, Levy-Sakin M, Schiller A, Egoz-Matia N, Masliah E, Segal D, Gazit E: A blood–brain barrier (BBB) disrupter is also a potent alpha-synuclein (alpha-syn) aggregation inhibitor: a novel dual mechanism of mannitol for the treatment of Parkinson disease (PD). J Biol Chem 2013, 288: 17579–17588. 10.1074/jbc.M112.434787
Faria C, Jorge CD, Borges N, Tenreiro S, Outeiro TF, Santos H: Inhibition of formation of alpha-synuclein inclusions by mannosylglycerate in a yeast model of Parkinson's disease. Biochim Biophys Acta 1830, 2013: 4065–4072.
Ono K, Ikemoto M, Kawarabayashi T, Ikeda M, Nishinakagawa T, Hosokawa M, Shoji M, Takahashi M, Nakashima M: A chemical chaperone, sodium 4-phenylbutyric acid, attenuates the pathogenic potency in human alpha-synuclein A30P + A53T transgenic mice. Parkinsonism Relat Disord 2009, 15: 649–654. 10.1016/j.parkreldis.2009.03.002
Porter JR, Fritz CC, Depew KM: Discovery and development of Hsp90 inhibitors: a promising pathway for cancer therapy. Curr Opin Chem Biol 2010, 14: 412–420. 10.1016/j.cbpa.2010.03.019
Shen HY, He JC, Wang Y, Huang QY, Chen JF: Geldanamycin induces heat shock protein 70 and protects against MPTP-induced dopaminergic neurotoxicity in mice. J Biol Chem 2005, 280: 39962–39969. 10.1074/jbc.M505524200
Schulte TW, Neckers LM: The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother Pharmacol 1998, 42: 273–279. 10.1007/s002800050817
Kim YS, Alarcon SV, Lee S, Lee MJ, Giaccone G, Neckers L, Trepel JB: Update on Hsp90 inhibitors in clinical trial. Curr Top Med Chem 2009, 9: 1479–1492. 10.2174/156802609789895728
Cleren C, Calingasan NY, Chen J, Beal MF: Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J Neurochem 2005, 94: 995–1004. 10.1111/j.1471-4159.2005.03253.x
Casarejos MJ, Solano RM, Gomez A, Perucho J, de Yebenes JG, Mena MA: The accumulation of neurotoxic proteins, induced by proteasome inhibition, is reverted by trehalose, an enhancer of autophagy, in human neuroblastoma cells. Neurochem Int 2011, 58: 512–520. 10.1016/j.neuint.2011.01.008
Inden M, Kitamura Y, Takeuchi H, Yanagida T, Takata K, Kobayashi Y, Taniguchi T, Yoshimoto K, Kaneko M, Okuma Y, et al.: Neurodegeneration of mouse nigrostriatal dopaminergic system induced by repeated oral administration of rotenone is prevented by 4-phenylbutyrate, a chemical chaperone. J Neurochem 2007, 101: 1491–1504. 10.1111/j.1471-4159.2006.04440.x
Malik B, Nirmalananthan N, Gray AL, La Spada AR, Hanna MG, Greensmith L: Co-induction of the heat shock response ameliorates disease progression in a mouse model of human spinal and bulbar muscular atrophy: implications for therapy. Brain 2013, 136: 926–943. 10.1093/brain/aws343
Cudkowicz ME, Shefner JM, Simpson E, Grasso D, Yu H, Zhang H, Shui A, Schoenfeld D, Brown RH, Wieland S, Barber JR: Arimoclomol at dosages up to 300 mg/day is well tolerated and safe in amyotrophic lateral sclerosis. Muscle Nerve 2008, 38: 837–844. 10.1002/mus.21059
Lanka V, Wieland S, Barber J, Cudkowicz M: Arimoclomol: a potential therapy under development for ALS. Expert Opin Investig Drugs 2009, 18: 1907–1918. 10.1517/13543780903357486
Faust K, Gehrke S, Yang Y, Yang L, Beal MF, Lu B: Neuroprotective effects of compounds with antioxidant and anti-inflammatory properties in a Drosophila model of Parkinson's disease. BMC Neurosci 2009, 10: 109. 10.1186/1471-2202-10-109
Neef DW, Turski ML, Thiele DJ: Modulation of heat shock transcription factor 1 as a therapeutic target for small molecule intervention in neurodegenerative disease. PLoS Biol 2010, 8: e1000291. 10.1371/journal.pbio.1000291
Calamini B, Morimoto RI: Protein homeostasis as a therapeutic target for diseases of protein conformation. Curr Top Med Chem 2012, 12: 2623–2640.
Tanaka M, Machida Y, Niu S, Ikeda T, Jana NR, Doi H, Kurosawa M, Nekooki M, Nukina N: Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat Med 2004, 10: 148–154. 10.1038/nm985
Castillo K, Nassif M, Valenzuela V, Rojas F, Matus S, Mercado G, Court FA, van Zundert B, Hetz C: Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 2013, 9: 1308–1320. 10.4161/auto.25188
Lan DM, Liu FT, Zhao J, Chen Y, Wu JJ, Ding ZT, Yue ZY, Ren HM, Jiang YP, Wang J: Effect of trehalose on PC12 cells overexpressing wild-type or A53T mutant alpha-synuclein. Neurochem Res 2012, 37: 2025–2032. 10.1007/s11064-012-0823-0
Schaeffer V, Lavenir I, Ozcelik S, Tolnay M, Winkler DT, Goedert M: Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain 2012, 135: 2169–2177. 10.1093/brain/aws143
Kruger U, Wang Y, Kumar S, Mandelkow EM: Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging 2012, 33: 2291–2305. 10.1016/j.neurobiolaging.2011.11.009
Acknowledgements
The authors would like to thank the peer-reviewers for their valuable comments.
DE-F, L-JS and LW received funding and support by the German National Academic Foundation (Studienstiftung des Deutschen Volkes e.V.). DE-F and LW are supported by the Young Investigator Award Program at the Faculty of Medicine, Ruprecht-Karls-University Heidelberg. The funding agencies had no role in the design, preparation or writing of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no interests.
Authors’ contributions
DE-F, L-JS and LW have been involved in screening and reviewing the relevant literature, drafting the manuscript, revising it critically for important intellectual content. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Ebrahimi-Fakhari, D., Saidi, LJ. & Wahlster, L. Molecular chaperones and protein folding as therapeutic targets in Parkinson’s disease and other synucleinopathies. acta neuropathol commun 1, 79 (2013). https://doi.org/10.1186/2051-5960-1-79
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2051-5960-1-79